
Abstract
Newborn screening for galactosemia yields a high number of false-positive results. Confirmatory DNA testing for unknown galactosemia mutations on the initial positive sample using novel techniques of mutation detection tenders itself to reduce the recall rate. The potential benefits of confirmatory DNA testing, however, could be offset by the detection of a high percentage of galactosemia carriers, Duarte/galactosemia compound heterozygotes, and infants with benign sequence changes in the galactose-1-phosphate uridyltransferase (GALT) gene among infants with a positive biochemical screening test. Our aim was to determine the frequency and allelic distribution of all sequence changes in the GALT gene in 110 newborns with a positive total galactose screening test among 43,688 Austrian newborns screened consecutively. We found that only 20 of the 110 probands carried at least one known or novel candidate galactosemia mutation (one galactosemia homozygote, 7 Duarte/galactosemia compounds, 12 carriers) as judged by denaturing gradient gel electrophoresis and cleavage fragment length polymorphism analysis. Four novel galactosemia candidate mutations (Q9H, A46fsdelCAGCT, M129T, L342I) were identified. Sixty-seven probands had no detectable sequence changes and 23 carried only the benign Duarte or Los Angeles variant alleles or silent mutations. We conclude that a rapid and automatable confirmation test for unknown GALT mutations, e.g. on a high-density oligonucleotide array basis, has the potential to lower the recall rate of galactosemia screening in our population by about five-fold from 0.25 to 0.046%. Further research, however, will be required before the development of such a test can be advocated.
Similar content being viewed by others

Main
Classical galactosemia (McKusick 230400) is an autosomal recessive inborn error of carbohydrate metabolism caused by a deficiency of GALT occurring with an incidence of 1:40,000–60,000 in Caucasian populations (1). Newborn screening and dietary galactose restriction prevent the life-threatening neonatal symptoms of galactosemia. Despite dietary treatment, the majority of affected children develop long-term complications including mental retardation, speech deficits, and ovarian failure.
Galactosemia screening by microbiological or biochemical tests for elevated blood galactose/galactose-1-P and/or reduced GALT activity yields a high rate of false positives (2). Only 1 in about 190 infants recalled in galactosemia screening programs across the United States will actually have galactosemia (3). Because of the potentially rapid and fatal course of the disease, newborns with positive test results have to be recalled urgently. This causes substantial anxiety among the families concerned (4) and increases the workload of the screening centers.
DNA confirmatory testing for common mutations on the original dried blood spot with a positive initial biochemical screening result has been used to reduce the number of false positives in screening programs for cystic fibrosis (5) and hemoglobinopathies (6). Although two common mutations, Q188R and K285N, account for approximately 69–88% of galactosemia alleles in Caucasian populations, over 170 different rare sequence changes have been described so far (7–11). This considerable allelic heterogeneity renders a confirmatory test for common mutations impossible without compromising sensitivity. Recently, however, several methods for high-throughput detection of unknown point mutations with high sensitivity and specificity have been developed (12–16). These novel techniques should allow the development of a confirmatory test for all sequence changes in the GALT gene. Additionally, other technical advances like direct PCR amplification from Guthrie cards without DNA extraction or the development of microchip-based PCR encourage a wider application of DNA tests in newborn screening (17–19). The potential benefits of confirmatory DNA testing for all mutations in the GALT gene, however, could be offset by the detection of a substantial number of carriers, Duarte variant/galactosemia (D/G) compound heterozygotes, and newborns with benign sequence changes in the GALT gene among infants with a positive biochemical screening test (20, 21).
The present study was designed to determine the frequency and the allelic distribution of all sequence changes occurring in the coding region and flanking intronic sequences of the GALT gene in a representative group of Austrian newborns with elevated total blood galactose in the initial newborn screening test. Based on this information, we estimated the effect on the recall rate achievable by using a confirmatory DNA test in a galactosemia screening strategy.
METHODS
Study design.
To obtain an unbiased and representative estimate of the frequency and the allelic distribution of GALT mutations, our study included all Austrian newborns screened for galactosemia at the National Newborn Screening Laboratory (Department Pediatrics, University of Vienna) within a period of 5 mo. Our laboratory provides general newborn screening for the entire country with a coverage of about 99% of all newborns. Dried blood specimens were collected on d 3–5 of life and were tested for total galactose using the Quantase Phe/Gal screening assay (Bio-Rad, Hercules, CA, U.S.A.) (22). Samples from infants with total galactose >12.0 mg/dL were retested. All infants with galactose >12.0 mg/dL in the retested sample were recalled for a repeat specimen. Infants with initial blood galactose >20.0 mg/dL were contacted urgently for a repeat dried blood specimen and for quantitative determination of GALT activity in erythrocyte lysates (23) (normal values: 27.8 ± 6 μmol/h/g Hb). Genomic DNA from the original dried blood spots of all infants with a positive initial galactosemia screening result was analyzed for sequence changes in the 11 exons and flanking intronic sequences of the GALT gene. The study was carried out on coded samples as required by the Austrian Gene Technology Act, and approved by the ethics review board of the Vienna General Hospital. The analysts had no knowledge of the results of the repeat total galactose test or the clinical follow up. The results of DNA analysis were not used for diagnosis or therapy and no information was released to the parents.
Probands.
Among 43,688 newborns screened during the study period, 110 (0.25%) had a positive total galactose screening result (total galactose >12.0 mg/dL). All 110 were available for follow-up studies. In the follow-up examinations, one newborn with classical galactosemia and three D/G compound heterozygotes were identified by standard laboratory procedures.
DNA extraction.
A 3-mm diameter disc was punched from the coded dried blood specimen. The discs were incubated in 1 mL of sterile aqua dest for 10 min at room temperature and centrifuged at 16,000 ×g for 3 min at room temperature. The supernate was discarded and 200 μL of Chelex 100 particles (Promega, Madison, WI, U.S.A.) were added to the pellet (24). The suspension was incubated at 56°C for 20 min, vortexed briefly, and incubated at 100°C for 8 min. After centrifugation at 16,000 ×g for 3 min, the supernate was transferred to a Microcon-50 concentrator (Millipore, Bedford, MA, U.S.A.) and centrifuged for 4 min at 16,000 ×g. The DNA was recovered from the ultrafilter by addition of 50 μL sterile aqua dest.
Denaturing gradient gel electrophoresis/Cleavase fragment length polymorphism analysis.
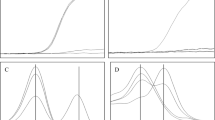
The 11 exons of the GALT gene and the flanking intronic sequences were amplified by PCR as described previously (10). DGGE of exons 2–11 of the GALT gene was performed under the conditions reported earlier for DNA samples extracted from whole blood (10) (Fig. 1). To evaluate the sensitivity of the procedure using DNA amplified from dried blood, blood spots from 14 D/G compound heterozygotes with previously sequenced mutations (10; Item C, unpublished results) were analyzed. The Duarte allele was identified by the sequence changes N314D, IVS4–27G>C and IVS5–24G>A as described previously (10). All 28 alleles (10 Q188R, 1 S143L, 1 M142 K, 1 R201H, 1 K285N, and 14 Duarte) were detected by DGGE from dried blood spots. The specificity of the method was tested on dried blood samples from 20 healthy infants. Thirty-eight wild-type alleles and 2 Duarte alleles [expected frequency: 9.5%(25)], but no false positives were found. Exon 1, which has a melting temperature too high for the DGGE method used here, was scanned for mutations using the cleavage patterns generated by a structure specific thermostable endonuclease, Cleavase 1 (26) (Cleavase Fragment Length Polymorphism Scanning Kit, CFLPScan®, Roche Molecular Chemicals, Mannheim Germany). Twenty-five microliters of PCR product was incubated with 25 units of Cleavase I for 15 min at 37°C in 100 mM MOPS pH 7.5, 0.5% Tween 20, 0.5% Nonidet P-40. The digestion products were mixed with sample buffer, heated for 2 min at 95°C, and separated on a 8% polyacrylamide gel containing 7 M urea. The CFLP assay resolved the known GALT mutation in this exon, D28Y (10), from a dried blood sample (Fig. 2).

Mutation scanning of the human GALT gene by DGGE using DNA amplified from dried blood. Representative results for exon 6 and flanking intronic sequences showing the DGGE patterns of the common mutation Q188R, the novel silent mutation I170I, and the intronic sequence change IVS5–24G>A associated with the Duarte allele. Lanes 1, 2, 5, 6, 8, 9, 11–15, 17, 19: N/N;lane 3: I170I/N;lane 4: Q188R/N;lanes 7, 10, 16: IVS5–24G>A/N;lane 18: IVS5–24G>A/Q188R.

Mutation scanning of exon 1 of the human GALT gene by cleavage fragment length polymorphism analysis using DNA amplified from dried blood spots. Lanes 1, 2: N/N;lane 3: D28Y/N.
Identification of mutations.
Mutations in exons 2–11 previously analyzed in this laboratory (10) were identified by mixing the respective PCR product with a control sample containing the known mutation followed by heteroduplex formation and DGGE as described by Guldberg et al.(27). Novel sequence changes were verified by double-stranded automated cycle sequencing of PCR products on an ABI 370 sequencer (Applied Biosystems, Foster City, CA, U.S.A.). PCR cloning of exon 2 was performed in one patient using the PCR-TRAP cloning system (GenHunter, Nashville, TN, U.S.A.). A colony PCR method was used to screen for plasmids containing a DNA insert as described by the manufacturer and the resulting plasmid DNA was used for sequencing.
Restriction analysis.
All samples with a normal genotype according to DGGE/CFLPA (67 of 110) were analyzed for Q188R and N314D by restriction digestion to confirm the absence of common GALT mutations in these infants. A 256-bp fragment of exon 6 and a 166-bp fragment of exon 10 were amplified by PCR using the following primers: exon 6: 5′-AAGCTTTGGTTCTGGGGAGT-3′, 5′-TTTCCTCTGTCCCATCCATT-3′; exon 10: 5′-TCTCTCCCCACTGTCTCTCTTC-3′, 5′-CTGAGCCTGAGCAAGCATTT-3′. Amplifications were performed in a mixture containing 2.5 μL DNA extracted from dried blood, 10 mM Tris-HCl, pH 8.3, 50 mM KCl, 2.5 mM MgCl2, 0.2 mM of each deoxynucleotide triphosphate, 0.8 pM of each primer, 24.5 μL distilled water, and 5 U AmpliTaq Gold Polymerase (Applied Biosystems). Samples were processed through an initial denaturation for 5 min followed by 40 cycles consisting of 1 min at 94°C (denaturation), 45 s at 59°C (annealing), 1 min at 72°C (extension), and a final extension at 72°C for 10 min. For detection of the Q188R mutation, the exon 6 fragment was digested with Hpa II (7), resulting in two fragments of 158 bp and 98 bp in the presence of the mutation. For detection of N314D, the exon 10 fragment was digested with Ava II (28), resulting in two fragments of 100 bp and 66 bp in the presence of the mutation. The fragments were separated on 2.5% agarose gels.
RESULTS
Frequency of sequence changes in the GALT gene in newborns with a positive screening test.
Twenty of the 110 newborns with a positive galactosemia screening test either had a known galactosemia allele, a new candidate mutation, or other sequence changes possibly associated with galactosemia as judged by DGGE/CFLP analysis and sequencing (Table 1). This amounts to 0.046% of the total population of 43,688 screened. The only patient with classical galactosemia identified in this study was compound heterozygous for two rare mutations. Seven other newborns with a positive screening test were D/G compound heterozygotes, and the remaining 12 were galactosemia carriers. Twenty-three newborns did not harbor sequence changes associated with classical galactosemia, but had alleles coding for the benign Duarte (D) or Los Angeles (LA) variants of galactosemia or for silent mutations, or alleles with intronic sequence changes. The remaining 67 infants had a normal genotype as judged by DGGE/CFLPA. As an additional confirmation of the absence of GALT mutations in these infants, we amplified fragments of exon 6 and 10 from all 67 samples with a normal genotype according to DGGE/CFLPA and analyzed them for the common mutations Q188R and N314D by restriction digestion with Hpa II and Ava II, respectively. Restriction digestion showed the presence of a wild-type restriction pattern in all cases and thus confirmed the absence of the common mutations Q188R and N314D in these samples (data not shown).
Biochemical and clinical follow up investigations.
The biochemical and clinical follow-up investigations performed in the routine screening process were consistent with the findings of mutation analysis (Table 1). The patient carrying the two rare mutations, M129T and W154X, was a Chinese boy who presented with total galactose of 63 mg/dL in the initial screening test. No residual GALT activity was detectable in his erythrocyte lysate and he was diagnosed with classical galactosemia. The other family members declined determination of GALT activity. None of the 90 infants with the genotypes D/D, D/N, D/T253T, LA/N, I1701I/N, IVS4+7A>G/N, or N/N had elevated galactose at their follow-up examinations.
As expected, DGGE/CFLPA detected significantly more galactosemia carriers than the routine biochemical follow-up process (n = 12 versus 1, χ2 = 9.89, p < 0.05, df = 1). By contrast, the number of D/G compound heterozygotes identified by DGGE/CFLPA and by routine follow-up did not differ significantly. (n = 7 versus 3, χ2 = 1.68, p = 0.20, df = 1).
Distribution of GALT alleles in newborns with a positive screening test.
In the group of 110 newborns with a positive screening test, 21 known or candidate galactosemia alleles were found (Table 2). Of these, 13 carried the common mutations Q188R or K285N. To calculate the percentage of rare alleles occurring in Austrian infants with a positive galactosemia screening test, we excluded the two mutations M129T and W154X identified in a Chinese patient. The two L195P alleles identified in this study were the first among 342 mutant GALT alleles characterized in our laboratory so far (10; Item C, unpublished data). Therefore, L195P was considered as rare, although it has been described in several European populations (11). Six of the remaining 19 alleles (31%) harbored rare mutations. This percentage of rare mutations was higher than in a group of Austrian galactosemia patients (12%) studied earlier (10) (χ2 = 4.2, df = 1, p = 0.04). Seven novel sequence changes in the GALT gene were detected. The novel missense mutation M129T in exon 5 was found in the only patient with classical galactosemia identified in our study in conjunction the rare mutation W154X (29) (Fig. 3). Two other novel candidate missense mutations, L342I and Q9H, were identified in D/G compounds. GALT activity in these patients was reduced to 12% and 34% of normal, respectively. A 5-bp deletion affecting nucleotides 442–446 in exon 2 was found in a heterozygote with a GALT activity of 38% of normal values and elevated galactose at the follow-up examination (Fig. 3, Table 1). Sequencing of PCR products produced ambiguous results and therefore exon 2 was cloned for sequence analysis. The deletion, A46fsdelCAGCT, results in a frameshift with A46 as the first affected amino acid. The new reading frame terminates at a stop codon at position 67. Finally, two silent mutations (I170I and T253T) and an intronic sequence change (IVS4+7A>G) were identified.

DGGE patterns of the novel candidate galactosemia mutations A46fsdelCAGCT, M129T, and W154X. (Panel a) DGGE of exon 2. Lane 1: A46fsdelCAGCT;lane 2: N/N. (Panel b) DGGE of exon 5. Lane 1: M129T/W154X;lane 2: N/N.
DISCUSSION
Our study in Austrian newborns with a positive screening test for elevated total blood galactose shows that only 19% of these infants carry a known or candidate galactosemia mutation. Most of these infants are carriers and about one third of them are D/G compound heterozygotes. It appears unlikely that a significant number of sequence changes were missed, as the DGGE method used here has detected 238 of 242 expected galactosemia and Duarte mutations analyzed in our laboratory so far (10; Item C, unpublished data). Moreover, restriction analysis confirmed the absence of common mutations in the infants with a normal genotype according to DGGE/CFLPA.
Some of the infants with high blood galactose in the absence of sequence changes in the GALT gene are likely to harbor mutations in genes encoding other enzymes of galactose metabolism, such as galactokinase or UDP-galactose 4 epimerase (21, 30). Moreover, portal-systemic shunts, delayed maturation of liver function, and, in some cases, overt liver dysfunction have been described in infants with transient galactosemia identified by newborn screening (30, 31). In D/G compound heterozygotes, and probably also in galactosemia carriers, the raised blood galactose can be explained by reduced galactose tolerance (30, 32). The identification of four carriers of the LA allele with elevated blood galactose, however, is intriguing, because the LA allele is characterized by increased GALT activity (33).
The infants with a positive newborn screening test showed a higher percentage of rare GALT alleles than a group of Austrian patients with classical galactosemia studied earlier (10). This may be due to the selection of probands on the basis of a single positive screening test versus a diagnosis of galactosemia. The detection of seven novel sequence changes, four of which may be considered as candidate galactosemia mutations, confirms the molecular heterogeneity of galactosemia. This illustrates that a confirmatory test only for the most common mutations is not suitable for galactosemia screening. The only patient with classical galactosemia identified in this study was compound heterozygous for two rare alleles, like two other unrelated Austrian galactosemia patients studied earlier (10). The risk of missing a patient by a test for common mutations can be estimated as 0.23–5.1% (95% confidence interval) based on the frequency of rare mutations in our population (12% of all galactosemia alleles, 95% confidence interval: 4.8–22.6%, n = 60 alleles) (10).
The low frequency of sequence changes in the GALT gene in newborns with a positive screening test suggests that a confirmatory DNA test for unknown GALT mutations has the potential to lower the recall rate of galactosemia screening by about five-fold from 0.25 to 0.046% and to increase the positive predictive value from 0.9 to 4.8% in our population. This calculation assumes that 19% of all infants with a positive screening test carry at least one known or candidate galactosemia mutation and are recalled. For purposes of comparison, the positive predictive value of nationwide galactosemia screening in the United States has recently been reported as 0.53%(3). The increase in specificity achievable by a confirmatory DNA test may render galactosemia screening about as specific as cystic fibrosis screening by the immunoreactive trypsin/DNA test protocol (5). Currently, two major strategies are used to lower the recall rate of galactosemia screening. One is to use a high cut-off level for total blood galactose (21). Galactose accumulation, however, depends on the ingestion of an adequate amount of milk. Taking into account the current practice of early newborn discharge, a high cut-off level for blood galactose must be viewed with caution. A second strategy is to combine a total galactose screening test with a determination of GALT activity (Beutler test) on the same specimen (19). Beutler test results, however, are affected by several variables like ambient temperature and humidity, or blood transfusions (34, 35). Confirmatory DNA testing is not affected by feeding status or other external factors, which may be of considerable advantage. Measurement of hexose monophosphates in dried blood spots by tandem mass spectrometry has been reported recently, and may become an interesting novel strategy to improve biochemical galactosemia screening (36). The expected increase in specificity, however, awaits confirmation in prospective studies.
In addition to lowering the recall rate, a confirmatory DNA test on the initial blood spot could permit an earlier diagnosis of galactosemia than conventional follow-up procedures. In Austria, dried blood samples are currently collected on d 3–5 of life. The result of the first screening test becomes available around d 7, and the result on a repeat specimen around d 15. Using microarray technology, the result of a confirmatory DNA analysis on the initial blood sample could be available on the day after the initial screening test.
In our study, 12 infants or 0.03% of the screened cohort were identified as galactosemia carriers by DGGE/CFLPA. Although the number of carriers identified is small, carrier detection clearly is an undesirable side effect. In addition to galactosemia carriers, D/G heterozygotes need to be recalled, because N314D has been found in cis with additional mutations causing classical galactosemia (37–39), which may have gone undetected. In our group of infants with a positive galactosemia screening test, DGGE/CFLPA did not identify significantly more D/G compound heterozygotes than the routine biochemical follow-up procedure, but the numbers of infants studied here may be too small to detect a possible difference. D/G compound heterozygotes identified by their genotype will require genetic counseling. As long as the question of whether these infants benefit from treatment remains unresolved, DNA testing bears the risk of incorrectly labeling them as affected by an inherited disease (20).
Another concern is that the high costs to be expected may offset the potential overall benefit of confirmatory DNA testing on the initial sample, although the expenses for collecting a repeat specimen would be avoided. The reagent costs of sequencing a gene of comparable size using a commercial oligonucleotide microarray are quoted as $190 US per sample at present (40). For comparison, the costs of a combined determination of total galactose and GALT activity in a dried blood sample amount to only about $5 US in our laboratory.
Finally, a suitable rapid and automatable confirmatory DNA test for all sequence changes in the GALT gene has not yet been developed. The DGGE/CFPLA technique used in this study is quite sensitive, but it cannot readily be automated for routine application. In the context of the Human Genome Project, several novel methods for high-throughput identification of unknown point mutations, such as high-density oligonucleotide arrays, microchip capillary electrophoresis, MALDI-TOF mass spectrometry, or automated SSCP-based capillary electrophoresis have been established (12–16). Among these, hybridization to high-density oligonucleotide arrays has already successfully been applied to the identification of unknown heterozygous mutations even in large genes. For example, the detection of unknown heterozygous mutations in the entire 9.17-kb sequence of the large ataxia-telangiectasia mutated gene distributed over 61 coding exons has been achieved with excellent sensitivity and few positive calls by this technique (12). Similarly, ligation to generic complete 8- or 9-mer arrays allowed the detection of 100% of mutations in a 1.2-kb target sequence (16). Moreover, an oligonucleotide microarray interrogating each of 1262 bases in exons 2–11 of the TP53 tumor suppressor gene (41) has recently become commercially available. Thus, the development of a test for unknown mutations in the GALT gene comprising 1400 bp of coding sequence in 11 exons appears technically feasible.
In summary, our results indicate that only a small part of infants with a positive biochemical galactosemia screening test carry galactosemia mutations. Our findings suggest that a suitable confirmatory DNA test has the potential to reduce the burden of false positives in galactosemia screening considerably. Further research into the application of the new generation of mutation detection tools in galactosemia newborn screening will be required before the development of a routine confirmatory DNA test can be advocated.
Abbreviations
- GALT:
-
galactose-1-phosphate uridyltransferase
- D:
-
Duarte variant allele
- G:
-
galactosemia allele
- LA:
-
Los Angeles variant allele
- DGGE:
-
denaturing gradient gel electrophoresis
- CFLPA:
-
cleavage fragment length polymorphism analysis
- SSCP:
-
single-strand conformation polymorphism
- MOPS:
-
3-(N-morpholino) propanesulfonic acid
- MALDI-TOF:
-
matrix-assisted laser desorption ionization–time of flight
References
Segal S 1995 Disorders of galactose metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Metabolic and Molecular Basis of Inherited Disease. McGraw-Hill, New York, 967–1000
Webster D, Allen DM 1993 Laboratory methods for galactosemia testing in newborn screening. In: Therell BL (ed) Laboratory Methods for Neonatal Screening. American Public Health Association, Washington, DC, 77–114
Kwon C, Farrell PM 2000 The magnitude and challenge of false-positive newborn screening test results. Arch Pediatr Adolesc Med 154: 714–718
Sorenson JR, Levy HL, Mangione TW, Sepe SJ 1984 Parental response to repeat testing of infants with ‘false-positive' results in a newborn screening program. Pediatrics 73: 183–187
Wilcken B, Wiley V, Sherry G, Bayliss U 1995 Neonatal screening for cystic fibrosis: a comparison of two strategies for case detection in 1.2 million babies. J Pediatr 127: 965–970
Descartes M, Huang Y, Zhang YH, McCabe LL, Gibbs R, Therrell BL Jr, McCabe ER 1992 Genotypic confirmation from the original dried blood specimens in a neonatal hemoglobinopathy screening program. Pediatr Res 31: 217–221
Leslie ND, Immerman EB, Flach JE, Florez M, Fridovich-Keil JL, Elsas LJ 1992 The human galactose-1-phosphate uridyltransferase gene. Genomics 14: 474–480
Tyfield L, Reichardt J, Fridovich-Keil J, Croke DT, Elsas LJ, Strobl W, Kozak L, Coskun T, Novelli G, Okano Y, Zekanowski C, Shin Y, Boleda MD 1999 Classical galactosemia and mutations at the galactose-1-phosphate uridyltransferase (GALT) gene. Hum Mutat 13: 417–430
Ng WG, Xu Y-K, Kaufman FR, Donnell GN, Wolff J, Allen RJ, Koritala S, Reichardt JKV 1994 Biochemical and molecular studies in 132 patients with galactosemia. Hum Genet 94: 359–363
Greber-Platzer S, Guldberg P, Scheibenreiter S, Item C, Schuller E, Patel N, Strobl W 1997 Molecular heterogeneity of classical and Duarte galactosemia: mutation analysis by denaturing gradient gel electrophoresis. Hum Genet 10: 49–57
The Galactose-1-Phosphate Uridyl Transferase Mutation Analysis Database Home Page. Available at: http://www.ich.bris.ac.uk/galtdb./ Accessed January 2002
Hacia JG, Sun B, Hunt N, Edgemon K, Mosbrook D, Robbins C, Fodor SP, Tagle DA, Collins FS 1998 Strategies for mutational analysis of the large multiexon ATM gene using high-density oligonucleotide arrays. Genome Res 8: 1245–1258
Schmalzing D, Belenky A, Novotny MA, Koutny L, Salas-Solano O, El-Difrawy S, Adourian A, Matsudaira P, Ehrlich D 2000 Microchip electrophoresis: a method for high-speed SNP detection. Nucleic Acids Res 28: E43
Fu DJ, Tang K, Braun A, Reuter D, Darnhofer-Demar B, Little DP 1998 Sequencing exons 5 to 8 of the p53 gene by MALDI-TOF mass spectrometry. Nat Biotechnol 16: 381–384
Ren J 2000 High-throughput single-strand conformation polymorphism analysis by capillary electrophoresis. J Chromatogr B Biomed Sci Appl 741: 115–128
Gunderson KL, Huang XC, Morris MS, Lipshutz RJ, Lockhart DJ, Chee MS 1998 Mutation detection by ligation to complete n-mer DNA arrays. Genome Res 8: 1142–1153
Caggana M, Conroy JM, Pass KA 1998 Rapid, efficient method for multiplex amplification from filter paper. Hum Mutat 11: 404–409
Kopp MU, Mello AJ, Manz A 1998 Chemical amplification: continuous-flow PCR on a chip. Science 280: 1046–1048
McCabe ER McCabe LL 1999 State-of-the-art for DNA technology in newborn screening. Acta Paediatr Suppl 432: 58–60 1999
Gitzelmann R, Bosshard NU 1995 Partial deficiency of galactose-1-phosphate uridyltransferase. Eur J Pediatr 154: ( suppl 2), S40–S44
Schweitzer S 1995 Newborn mass screening for galactosemia. Eur J Pediatr 154: ( suppl 2), S37
Diepenbrock F, Heckler R, Schickling H, Engelhard T, Bock D, Sander J 1992 Colorimetric determination of galactose and galactose-1-phosphate from dried blood. Clin Biochem 25: 37–39
Shin YS 1991 Galactose metabolites and disorders of galactose metabolism. In: Hommes FA (ed) Techniques in Diagnostic Human Biochemical Genetics. New-York, Wiley-Liss, 267–284
Walsh PS, Metzger DA, Higuchi R 1991 Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10: 506–513
Podskarbi T, Bieger W, Shin Y 1997 Allelic heterogeneity of the galactose-1-phosphate uridyltransferase gene: biochemical and clinical phenotypes. Eur J Clin Chem Clin Biochem 35: A27
Rossetti S, Englisch S, Bresin E, Pignatti PF, Turco AE 1997 Detection of mutations in human genes by a new rapid method: cleavage fragment length polymorphism analysis (CFLPA). Mol Cell Probes 11: 155–160
Guldberg P, Guttler F 1993 A simple method for identification of point mutations using denaturing gradient gel electrophoresis. Nucleic Acids Res. Nucleic Acids Res 21: 2261–2262
Elsas LJ, Dembure PP, Langley S, Paulk EM, Hjelm LN, Fridovich-Keil J 1994 A common mutation associated with the Duarte galactosemia allele. Am J Hum Genet 54: 1030–1036
Elsas LJ, Lai K 1998 The molecular biology of galactosemia. Genet Med 1: 40–48
Ono H, Mawatari H, Mizoguchi N, Eguchi T, Sakura N, Hamakawa M 1999 Transient galactosemia detected by neonatal mass screening. Pediatr Int 41: 281–284
Ono H, Mawatari H, Mizoguchi N, Eguchi T, Sakura N, Hamakawa M 2000 Delay of liver maturation as a cause of transient neonatal galactosemia. Pediatr Int 42: 61–63
Schwarz HP, Zuppinger KA, Zimmerman A, Dauwalder H, Scherz R, Bier DM 1982 Galactose intolerance in individuals with double heterozygosity for Duarte variant and galactosemia. J Pediatr 100: 704–709
Bergren WG, Donnell GN 1973 A new variant of galactose-1-phosphate uridyltransferase in man: the Los Angeles variant. Ann Hum Genet 37: 1–8
Shih VE, Levy HI, Karolkewicz V, Houghton S, Efron ML, Isselbacher KJ, Beutler E, MacCready RA 1971 Galactosemia screening of newborns in Massachusetts. N Engl J Med 284: 753–757
Frazier DM, Clemons EH, Kirkman HN 1992 Minimizing false positive diagnoses in newborn screening for galactosemia. Biochem Med Metab Biol 48: 199–211
Jensen UG, Brandt NJ, Christensen E, Skovby F, Norgaard-Pedersen B, Simonsen H 2001 Neonatal screening for galactosemia by quantitative analysis of hexose monophosphates using tandem mass spectrometry: a retrospective study. Clin Chem 47: 1364–1372
Schuster V, Podskarbi T, Ottensmeier H, Haubner M, Shin YS 1998 Simultaneous occurrence of various mutations and polymorphisms in cis and in trans of the galactose-1-phosphate uridyltransferase gene in a Turkish family with classical galactosemia. J Mol Med 76: 715–719
Elsas LJ, Langley S, Steele E, Evinger J, Fridovich-Keil JL, Brown A, Singh R, Fernhoff P, Hjelm LN, Dembure PP 1995 Galactosemia: a strategy to identify new biochemical phenotypes and molecular genotypes. Am J Hum Genet 56: 630–639
Lin HC, Kirby LT, Ng WG, Reichardt JK 1994 On the molecular nature of the Duarte variant of galactose-1-phosphate uridyltransferase (GALT). Hum Genet 93: 167–169
Wikman FP, Lu ML, Thykjaer T, Olesen SH, Andersen LD, Cordon-Cardo C, Orntoft TF 2000 Evaluation of the performance of a p53 sequencing microarray chip using 140 previously sequenced bladder tumor samples. Clin Chem 46: 1523–1525
Wen WH, Bernstein L, Lescallett J, Beazer-Barclay Y, Sullivan-Halley J, White M, Press MF Comparison of TP53 mutations identified by oligonucleotide microarray and conventional DNA sequence analysis. Cancer Res 60: 2716–2722
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was supported, in part, by a research grant from SHS Gesellschaft für klinische Ernährung mbH, Heilbronn, Germany.
Rights and permissions
About this article
Cite this article
Item, C., Hagerty, B., Mühl, A. et al. Mutations at the Galactose-1-P-Uridyltransferase Gene in Infants with a Positive Galactosemia Newborn Screening Test. Pediatr Res 51, 511–516 (2002). https://doi.org/10.1203/00006450-200204000-00018
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200204000-00018
This article is cited by
-
Clinical profile and molecular characterization of Galactosemia in Brazil: identification of seven novel mutations
BMC Medical Genetics (2016)
-
Appropriateness of newborn screening for classic galactosaemia: a systematic review
Journal of Inherited Metabolic Disease (2016)
-
Early and severe indirect hyperbilirubinemia as a manifestation of galactosemia
Journal of Perinatology (2010)
-
Galactosaemia in a Brazilian population: High incidence and cost–benefit analysis
Journal of Inherited Metabolic Disease (2009)
-
Low allelic heterogeneity in a sample of Mexican patients with classical galactosaemia
Journal of Inherited Metabolic Disease (2008)
