
Abstract
Congenital disorders of glycosylation (CDGs) are a rapidly growing group of inherited disorders caused by defects in the synthesis and processing of the asparagine(ASN)-linked oligosaccharides of glycoproteins. The first CDG patients were described in 1980. Fifteen years later, a phosphomannomutase deficiency was found as the basis of the most frequent type, CDG-Ia. In recent years several novel types have been identified. The N-glycosylation pathway is highly conserved from yeast to human, and the rapid progress in this field can largely be attributed to the systematic application of the knowledge of yeast mutants. Up to now, eight diseases have been characterized, resulting from enzyme or transport defects in the cytosol, endoplasmic reticulum, or Golgi compartment. CDGs affect all organs and particularly the CNS, except for CDG-Ib, which is mainly a hepatic-intestinal disease.
Similar content being viewed by others


Main
The biosynthesis of asparagine (ASN)-linked oligosaccharides of glycoproteins proceeds along an extended pathway, spread over the cytosol, the ER, and the Golgi compartment. In the cytosol and the ER, numerous glycosyltransferases stepwise attach monosaccharides to a growing LLO. The sugar donors are either nucleotides or dolichol phosphate-linked sugars. The completed basic glycan (Glc3 Man9GlcNAc2) is transferred en bloc within the ER to an asparagine residue of a nascent protein, through the oligosaccharyltransferase complex (Fig. 1). In the ER and the Golgi compartment, several glycosidases and glycosyltransferases remodel the oligosaccharide chain into a more complex structure by removal of glucose and mannose residues and the addition of GlcNAc, galactose, fucose, and sialic acid residues (1).

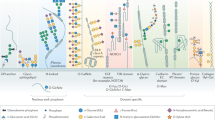
Simplified scheme of the cytosolic and ER part of the N-glycosylation pathway. The pathway begins in the cytoplasm with the synthesis from glucose of the activated sugar precursor GDP-Man. Key enzymes in this pathway are: (1) phosphoglucose isomerase; (2) MPI; (3) PMM; and (4) GDP-Man synthase. Dolichol phosphate mannose and dolichol phosphate glucose are synthesized from GDP-Man and UDP-Glc (reactions 5 and 15, respectively). The two dolichol phosphate sugar precursors are then flipped from the cytoplasmic side of the ER membrane to the luminal side. On the cytoplasmic side of the ER membrane, one molecule of GlcNAc-1-phosphate is transferred from UDP-GlcNAc to dolichol phosphate to form dolichol pyrophosphate GlcNAc, followed by addition of GlcNAc to form dolichol pyrophosphate (GlcNAc)2 (reaction 6). Five Man residues are then added from GDP-Man to form dolicholpyrophosphate(GlcNAc)2(Man)5 (reaction 7), which then flips to the luminal side (reaction 8) and serves as an acceptor for four Man residues derived from dolichol phosphate mannose (reaction 9). Three Glc residues are then added from dolichol phosphate glucose (reactions 10, 11, and 12). The Glc3Man9GlcNAc2 oligosaccharide is transferred from dolichol pyrophosphate to an asparagine residue of the nascent glycoprotein to initiate protein N-glycosylation by the action of oligosaccharyltransferase (reaction 13). The three Glc residues are removed by glycosidases, and the protein proceeds to the Golgi apparatus for further processing. CDG types Ia, Ib, Ic, Id, and Ie are caused by defective enzymes at steps 3, 2, 10, 9, and 5, respectively.
Glycoconjugates play many critical roles in metabolism (2), e.g. in cell recognition and adhesion, cell migration, protease resistance, host defense, and antigenicity. Therefore it is obvious that hypoglycosylation of proteins leads to variable and in general severe and ubiquitous problems in affected individuals. In human, eight inherited diseases are known to date to be caused by defects in the synthesis of N-glycans. Following recent recommendations (3, 4), congenital disorders of glycosylation (CDG) are divided into two groups (Table 1). CDG-I comprises defects in the assembly of the LLO chain and its transfer to the protein. Known enzyme defects are located to the cytosol (CDG-Ia, CDG-Ib) and the ER (CDG-Ic, CDG-Id, and CDG-Ie). CDG-II refers to defects in the processing of the protein-bound glycans, either late in the ER (CDG-IIb) or in the Golgi compartment (CDG-IIa and CDG-IIc). It is convention that untyped cases are labeled CDG-x until they are fully characterized.
IEF of serum transferrin remains the most powerful screening test for CDG, although not all types can be detected by this assay. A type 1 IEF pattern (observed in CDG-I) is characterized by a decrease of tetrasialotransferrin and an increase of di- and asialotransferrin bands, whereas a type 2 pattern (observed in CDG-II) shows in addition an increase of tri- and monosialotransferrin.
This review presents highlights of the known and newly discovered congenital disorders of N-linked glycosylation and summarizes diagnostic and therapeutic approaches.
Clinical Presentations
CDG-Ia (PMM deficiency).
An estimated 300 patients are known worldwide with this most frequent type of CDG. Often patients can be diagnosed in the neonatal or early infantile period on the basis of typical clinical features (inverted nipples and fat pads) in addition to strabismus, muscular hypotonia, failure to thrive, and elevated transaminases (5, 6). As a rule there is osteopenia. A very common feature is cerebellar hypoplasia, which can usually be documented at or shortly after birth. There is a substantial childhood mortality of approximately 25%, owing to severe infections or organ failure (7, 8).
At a later age, the impairment of the neurologic system becomes more evident with a variable degree of mental retardation, cerebellar dysfunction, and retinitis pigmentosa. Some children experience seizures or exhibit stroke-like episodes. In adults, nonprogressive ataxia, stable mental retardation, and peripheral neuropathy mainly characterize the disease. The large majority of patients are wheelchair bound. Adult female patients present as a rule with hypergonadotropic hypogonadism (9, 10). Thanks to the broadened screening for CDG, the number of patients with a less typical presentation is increasing, including children with nearly normal psychomotor development.
CDG-Ia is a result of PMM 2 (EC 5.4.2.8) deficiency (11), and the gene is localized on chromosome 16p13 (12, 13).
CDG-Ib (MPI deficiency).
Although MPI (EC 5.3.1.8) catalyzes the reaction one step upstream of PMM, the clinical presentation of this disease is very different from the PMM deficiency and the other CDG types. Some 20 patients are known. The classic clinical presentation is protein-losing enteropathy, congenital hepatic fibrosis, and coagulopathy without overt neurologic manifestations (14–18). Other presentations are persistent vomiting (15) and hypoglycemia (18). Early diagnosis is essential, because patients can be successfully treated with oral mannose (see “Treatment”).
CDG-Ic (ALG 6 deficiency).
ALG 6 deficiency (CDG-Ic) causes mainly a neurologic disorder that is in general milder than CDG-Ia (19, 20). Features of CDG-Ia, such as cerebellar hypoplasia, polyneuropathy, fat pads, and inverted nipples, are missing (21, 22). In two of 20 patients the disease had a fatal outcome because of severe coagulopathy and hormonal disturbances, combined with untreatable seizures (unpublished data). This type of CDG is probably underdiagnosed because of the absence of typical morphologic features and cerebellar hypoplasia. The enzyme, ALG 6, catalyzes the attachment of the first glucose to the LLO intermediate Man9GlcNAc2-PP-dolichol in the ER.
CDG-Id (ALG 3 deficiency).
Thus far, only one patient has been described with an ALG 3 deficiency (23). This child was first reported in 1995 as CDG “type IV”(24). The boy presented with microcephaly, severe epilepsy, and nearly absent psychomotor development. The IEF of serum transferrin (type 1 pattern) shows no increase of asialotransferrin. The defect is in the ALG 3 that transfers mannose from dolichol-phosphate mannose to the LLO intermediate Man5GlcNAc2-PP-dolichol.
CDG-Ie (dolichol-P-mannose synthase 1 deficiency).
Two groups have reported on four severely handicapped patients (in three families) with minimal psychomotor development, microcephaly, absent visual contact, and severe epilepsy. Brain imaging revealed delayed myelination in two of them. As in the previous types, IEF of serum transferrin showed a type 1 pattern with only little or no increase of asialotransferrin. This clinical picture is caused by a dolichol-P-mannose synthase deficiency (EC 2.4.1.83), and mutations in DPM1, the catalytic subunit of dolichol-P-mannose synthase, have been identified (25, 26).
CDG-IIa (N-acetyl-glucosaminyltransferase II deficiency).
When in the early 1990s an Iranian girl and a Belgian boy were reported with severe psychomotor retardation but without peripheral neuropathy or cerebellar hypoplasia, it became clear that CDGs are a heterogeneous group of disorders. Up to now, four children in three families have been diagnosed as CDG-IIa (27, 28). They showed craniofacial dysmorphy, stereotypic hands movement, and psychomotor retardation to a variable degree. Glycan structural studies revealed a similar monoantennary N-acetyllactosamine type glycan as found in patients with congenital dyserythropoietic anemia type II (29, 30). This suggested an N-acetylglucosaminyltransferase II (EC 2.4.1.143) deficiency, which was demonstrated in monocytes and fibroblasts (31, 32). Mutations have been found in the MGAT2 gene (33).
CDG-IIb (glucosidase I deficiency).
Very recently, a different type of CDG was identified in a child with severe developmental delay, muscular hypotonia, recurrent edema, seizures, and peculiar dysmorphy, including retrognathia, high arched palate, and overlapping of fingers. It results from defective glucosidase that, in the ER, reacts in the first step of the processing of the glycan structure, after its transfer to the protein. Interestingly, IEF of transferrin was normal, whereas the IEF of serum hexosaminidase was slightly abnormal. The defect was found after oligosaccharide analysis of urine revealed the presence of a tetrasaccharide [Glc(α1–2)Glc(α1–3)Glc (α1–3)Man](34).
CDG-IIc (GDP-fucose transporter deficiency).
Three patients have been reported (35, 36); they exhibited craniofacial dysmorphism, severe psychomotor retardation, hypotonia, and growth retardation. A typical feature was recurrent infections with marked leukocytosis. Neutrophils of these patients lack sialyl-Lewis x, a fucose-containing carbohydrate ligand of the selectin family of cell adhesion molecules. Sialyl-Lewis x is necessary for the recruitment of neutrophils to the infection focus. Other fucose-containing carbohydrate sequences, such as A, B, O, and Lewis A blood groups, are also absent. Mutations in the gene for a specific GDP-fucose transporter were identified as the underlying causes of this so-called leukocyte adhesion deficiency type II (37, 38).
CDG-x.
The number of patients with a CDG not belonging to the known types is steadily increasing. They undoubtedly harbor different defects in the numerous other steps of the N- and O-glycosylation pathways. Their clinical presentations are very variable: hydrops fetalis, dysmorphy, and early death after intractable seizures (39); severe hypotonia, cataracts, failure to thrive, absent psychomotor development, progressive microcephaly, and death in status epilepticus (40); oligohydramnion, dysmorphy, hypotonia, seizures, cerebellar hypoplasia, and severe thrombocytopenia (41); oligohydramnion, hypotonia, diarrhea, vomiting, ascites, demineralization of distal bones, and tubulopathia (42); dysmorphy, muscular hypotonia, and infantile spasms (43).
Table 2 summarizes either the most common or most characteristic symptoms of the known defects. Note that for most of the disorders only very few patients are known, making a comprehensive clinical description premature.
DIAGNOSIS
In view of the extremely broad clinical spectrum of known CDG patients, it is recommended to consider CDG in any unexplained multisystem disorder. Figure 2 proposes a stepwise diagnostic flowchart for the detection and characterization of CDG patients.

Proposed diagnostic flowchart for congenital disorders of N-linked glycosylation (CDG). *The use of EDTA plasma can cause artifacts as a result of iron chelation.
IEF of serum transferrin.
Transferrin, one of the predominant serum glycoproteins, has two N-glycosylation sites. Hence, most transferrin molecules carry two biantennary chains with terminal sialic acid residues, and tetrasialotransferrin is the main serum sialotransferrin. Deficient synthesis of N-glycans results in a deficient incorporation of sialic acid, the terminal negatively charged sugar. The molecules acquire a more positive charge, which causes a cathodal shift in the IEF pattern of transferrin. In the so-called type 1 pattern (the most frequent), there is an increase of di- and asialotransferrin and a decrease of tetrasialotransferrin, whereas the type 2 pattern is a combination of the type 1 pattern and an increase of trisialotransferrin and, but not always, monosialotransferrin. One has to make sure that the abnormal pattern is not the result of a transferrin protein variant by performing IEF of the parents' transferrin. Preincubation of the sample with neuraminidase is another means to differentiate between CDG and protein variants. It is worthwhile performing IEF of other glycoproteins (e.g. hexosaminidase, thyroxine-binding globulin, or α1- antitrypsin) to document that there is a generalized glycosylation defect in the patients (44).
Because IEF is based on the deficiency of sialic acid, fucose defects cannot be picked up. In patients suspected of a fucose defect, the blood groups should be determined: they show the Bombay blood group (38). Also, the IEF of serum transferrin was normal in the one reported patient with CDG-IIb (34) and can be normal in patients with CDG-Ia (45).
Carbohydrate-deficient transferrins can also be observed in uncontrolled fructosemia and galactosemia and in chronic alcohol ingestion (46–50). The cathodal shift on IEF disappears with efficient treatment.
Enzymatic measurements.
PMM and MPI activity are most readily measured in fibroblasts or leukocytes (11). Regarding PMM activity, leukocytes seem to be more reliable than fibroblasts, because a high residual activity has been observed in the fibroblasts of some CDG-Ia patients, whereas the leukocyte values were always in the clearly abnormal range (51). Thus, patients with slightly decreased or low normal values of PMM in fibroblasts might still harbor mutations in PMM2. Especially in the case of a clinical picture that strongly suggests CDG-Ia, it is worthwhile to look for PMM2 mutations.
Mutation studies.
The PMM2 gene was cloned in 1997 (13), and more than 50 different mutations have been identified (13, 52–56). There is a clear predominance of missense mutations. The R141H mutation is found in approximately 40% of all patients. F119L is frequent in the Northern European countries owing to a founder effect (54, 57), whereas V231M and P113L are frequent all over Europe (Table 3) (13, 58). Most patients are compound heterozygous for two different mutations, and homozygosity is not observed for R141H or other mutations that severely impair the protein (52, 53, 59–61).
Mutations in the MPI gene have been found in most patients with an MPI deficiency. The gene is localized on chromosome 15p22 and again, mutations are generally of the missense type. Because a very limited number of patients are known, the spectrum of mutations is rather limited for the moment (Table 3) (14, 16, 18, 62).
Genetic studies in CDG-Ic patients revealed a prevalence of the A333V mutation in ALG 6, probably related to a founder effect in the Dutch population. Only a handful of other mutations has been described to date (Table 3) (21, 22, 63, 64).
Prenatal diagnosis.
Prenatal diagnosis has become reliable for CDG-Ia (54, 65) since the mapping of the disease locus on chromosome 16p13 (12), and the subsequent identification of the enzymatic defect (11) and the cloning of the PMM2 gene (13). Early attempts of prenatal diagnosis on the basis of transferrin IEF in fetal blood have failed and thus revealed that this method is not reliable (66, 67). Enzymatic measurements of PMM activities in cultured amniocytes or trophoblasts are useful but may give inconclusive data (65). As a result, preference is given to the direct mutation analysis in the fetus.
Prenatal diagnosis is possible in all other types of CDG for which the molecular defect is known, on condition that the diagnosis has been confirmed in the index patient or the mutations have been detected in the parents.
LLO analysis.
The N-glycosylation pathway in the ER is highly conserved in eukaryotic cells (19), and yeast genetic techniques have been instrumental in the identification of new N-glycosylation disorders in human. By comparing LLO structures of patients with those of yeast mutant strains, the defects of CDG-Ic, -Id, and -Ie have been elucidated (19, 20, 23, 25, 26). The power of the LLO analysis is of course limited to defects in the cytosol and ER.
Glycan structure analysis.
Structural studies of the complex asparagine-linked glycans have mainly been performed on serum transferrin, i.e. IEF, chromatofocusing, Western blotting, capillary zone electrophoresis, and anion exchange chromatography (68–74). Electrospray mass spectrometry of protein-bound oligosaccharide chains (glycoproteins) can separate the different glycoforms (75), and nuclear magnetic resonance spectroscopy determines the glycan structures and molecular mass of the glycovariants (76). Such glycan structure analysis will be instrumental for the elucidation of CDG-x cases, by pinpointing candidate enzymes and genes responsible for the abnormal N-glycan synthesis.
TREATMENT
CDG-Ia.
Unfortunately, an efficient treatment is still not available for the CDG-Ia patients. Although it was reported that incubation with mannose resulted in an increased incorporation of mannose in patients' fibroblasts (77), mannose administration to CDG-Ia patients did not improve the clinical or biochemical features (78, 79). Also fucose supplementation, with the aim of enhancing the GDP-mannose pool, was not successful (T. Marquardt, personal communication). Studies with a ketogenic diet in CDG-Ia are ongoing. The rationale for this treatment is the observation that glucose starvation improves N-glycosylation in fibroblasts from CDG-Ia patients (80).
As to symptomatic treatment, we have obtained efficient prevention of stroke-like events by using 0.5 mg/kg per day acetylsalicylic acid (C. Van Geet and J. Jaeken, in preparation). In patients with recurrent fractures, biphosphonates should be considered.
CDG-Ib.
In CDG-Ib patients, treatment with oral mannose (4–6 doses per day × 100–150 mg/kg per day) has shown significant improvement (81–83). Serum mannose levels should be greater than 200 μM. Because mannose supplementation is probably a lifelong therapy for MPI-deficient patients, side effects have to be monitored carefully. High mannose intake can cause osmotic diarrhea (81). Also a slight increase of the glycosylated Hb (HbA1c) has been observed (82).
CDG-IIc.
In one patient with a GDP-fucose transporter defect, fucose supplementation (25 mg/kg per day in three doses) was reported to improve the fucosylation of glycoproteins and to control the recurring infections (36).
CONCLUSION AND OUTLOOK
In the last 2 years, the field of CDG has been rapidly expanding, thanks to the increasing awareness of the clinical variability of CDG and the use of yeast genetic technology. The extremely wide clinical spectrum of CDG makes a broad screening for these disorders in children as well as in adults mandatory by using serum transferrin IEF. It is quite certain that many new glycosylation disorders are awaiting identification inasmuch as it is estimated that some 500 genes (approximately 0.5 to 1% of the translated human genome) participate in oligosaccharide synthesis and function (84). The development of animal models offers the potential to test therapeutic approaches and will be helpful in understanding the pathogenesis of these disorders.
ADDENDUM
Very recently two new CDG have been identified: CDG-If (an ER defect) and CDG-IId (a Golgi defect). CDG-If has been reported in four patients with a severe encephalopathy and, in three of them, a dry, scaling skin with erythroderma. Mutations were found in the Lec35/MPDU1 gene involved in the use of dolichylphosphomannose and dolichylphosphoglucose (85, 86). CDG-IId was described in a patient with moderate psychomotor retardation and macrocephaly as well as myopathy. A homozygous mutation (insertion of a single nucleotide) was found in the gene for UDP-Gal: N-acetyl-glucosamine β-1,4-galactosyltransferase I (87, 88).
Abbreviations
- CDG:
-
congenital disorder(s) of glycosylation, formerly carbohydrate-deficient glycoprotein syndrome(s)
- PMM:
-
phosphomannomutase
- MPI:
-
phosphomannose isomerase
- ER:
-
endoplasmic reticulum
- IEF:
-
isoelectrofocusing
- LLO:
-
lipid-linked oligosaccharide
- Glc:
-
glucose
- Man:
-
mannose
- GlcNAc:
-
N-acetylglucosamine
- ALG 6:
-
α-1,3-glucosyltransferase
- ALG 3:
-
α-1,3-mannosyltransferase
- ASN:
-
asparagine
- GDP:
-
guanosine diphosphate
References
Kornfeld R, Kornfeld S 1985 Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem 54: 631–664
Varki A 1993 Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 3: 97–130
Participants, “First International Workshop on CDGS,” Leuven, Belgium, 1999 Carbohydrate-deficient glycoprotein syndromes become congenital disorders of glycosylation: an updated nomenclature for CDG. Glycobiology 10: 3–6
Participants, “First International Workshop on CDGS,” Leuven, Belgium, 1999 Carbohydrate-deficient glycoprotein syndromes become congenital disorders of glycosylation: an updated nomenclature for CDG. Glycoconj J 16: 669–671
Jaeken J, Vanderschueren-Lodeweyckx M, Casaer P, Snoeck L, Corbeel L, Eggermont E, Eeckels R 1980 Familial psychomotor retardation with markedly fluctuating serum proteins, FSH GH levels, partial TBG-deficiency, increased serum arylsulphatase A increased CSF protein: a new syndrome?. Pediatr Res 14: 179
Imtiaz F, Worthington V, Champion M, Besley C, Charlwood J, Clayton P, Keir G, Mian N, Winchester B 2000 Genotypes phenotypes of patients in the UK with carbohydrate-deficient glycoprotein syndrome type 1. J Inherit Metab Dis 23: 162–174
Jaeken J, Stibler H, Hagberg B 1991 The carbohydrate-deficient glycoprotein syndrome: a new inherited multisystemic disease with severe nervous system involvement. Acta Paediatr Scand Suppl 375: 1–71
Jaeken J, Matthijs G 2001 Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet 2: 139–152
de Zegher F, Jaeken J 1995 Endocrinology of the carbohydrate-deficient glycoprotein syndrome type 1 from birth through adolescence. Pediatr Res 37: 395–401
Stibler H, Blennow G, Kristiansson B, Lindehammer H, Hagberg B 1994 Carbohydrate-deficient glycoprotein syndrome: clinical expression in adults with a new metabolic disease. J Neurol Neurosurg Psychiatry 57: 552–556
Van Schaftingen E, Jaeken J 1995 Phosphomannomutase deficiency is a cause of carbohydrate-deficient glycoprotein syndrome type I. FEBS Lett 377: 318–320
Martinsson T, Bjursell C, Stibler H, Kristiansson B, Skovby F, Jaeken J, Blennow G, Strömme P, Hanefeld F, Wahlström J 1994 Linkage of a locus for carbohydrate-deficient glycoprotein syndrome type I (CDG1) to chromosome 16p, linkage disequilibrium to microsatellite marker D16S406. Hum Mol Genet 3: 2037–2042
Matthijs G, Schollen E, Pardon E, Veiga-Da-Cunha M, Jaeken J, Cassiman JJ, Van Schaftingen E 1997 Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome) [published erratum appears in Nat Genet 1997;16:316]. Nat Genet 16: 88–92
Niehues R, Hasilik M, Alton G, Körner C, Schiebe-Sukumar M, Koch HG, Zimmer KP, Wu R, Harms E, Reiter K, von Figura K, Freeze HH, Harms HK, Marquardt T 1998 Carbohydrate-deficient glycoprotein syndrome type Ib: phosphomannose isomerase deficiency mannose therapy. J Clin Invest 101: 1414–1420
de Koning TJ, Dorland L, van Diggelen OP, Boonman AM, de Jong GJ, van Noort WL, De Schryver J, Duran M, van den Berg IE, Gerwig GJ, Berger R, Poll-The BT 1998 A novel disorder of N-glycosylation due to phosphomannose isomerase deficiency. Biochem Biophys Res Commun 245: 38–42
Jaeken J, Matthijs G, Saudubray JM, Dionisi-Vici C, Bertini E, de Lonlay P, Henri H, Carchon H, Schollen E, Van Schaftingen E 1998 Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet 62: 1535–1539
de Lonlay P, Cuer M, Vuillaumier-Barrot S, Beaune G, Castelnau P, Kretz M, Durand G, Saudubray JM, Seta N 1999 Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency: a new manifestation of carbohydrate-deficient glycoprotein syndrome treatable with mannose. J Pediatr 135: 379–383
Babovic-Vuksanovic D, Patterson MC, Schwenk WF, O'Brien JF, Vockley J, Freeze HH, Mehta DP, Michels VV 1999 Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. J Pediatr 135: 775–781
Burda P, Borsig L, de Rijk-van Andel J, Wevers R, Jaeken J, Carchon H, Berger EG, Aebi M 1998 A novel carbohydrate-deficient glycoprotein syndrome characterized by a deficiency in glucosylation of the dolichol-linked oligosaccharide. J Clin Invest 102: 647–652
Körner C, Knauer R, Holzbach U, Hanefeld F, Lehle L, von Figura K 1998 Carbohydrate-deficient glycoprotein syndrome type V: deficiency of dolichyl-P-Glc:Man9GlcNAc2-PP-dolichyl glucosyltransferase. Proc Natl Acad Sci USA 95: 13200–13205
Grünewald S, Imbach T, Huijben K, Rubio-Gozalbo ME, Verrips A, de Klerk JB, Stroink H, de Rijk-van Andel JF, Van Hove JL, Wendel U, Matthijs G, Hennet T, Jaeken J, Wevers RA 2000 Clinical biochemical characteristics of congenital disorder of glycosylation type Ic, the first recognized endoplasmic reticulum defect in N-glycan synthesis. Ann Neurol 47: 776–781
Hanefeld F, Körner C, Holzbach-Eberle U, von Figura K 2000 Congenital disorder of glycosylation-Ic: case report genetic defect. Neuropediatrics 31: 60–62
Körner C, Knauer R, Stephani U, Marquardt T, Lehle L, von Figura K 1999 Carbohydrate deficient glycoprotein syndrome type IV: deficiency of dolichyl-P-Man:Man5GlcNAc2-PP-dolichyl mannosyltransferase. EMBO J 18: 6816–6822
Stibler H, Stephani U, Kutsch U 1995 Carbohydrate-deficient glycoprotein syndrome—a fourth subtype. Neuropediatrics 26: 235–237
Imbach T, Schenk B, Schollen E, Burda P, Stutz A, Grünewald S, Bailie NM, King MD, Jaeken J, Matthijs G, Berger EG, Aebi M, Hennet T 2000 Deficiency of dolichol-phosphate-mannose synthase-1 causes congenital disorder of glycosylation type Ie. J Clin Invest 105: 233–239
Kim S, Westphal V, Srikrishna G, Mehta DP, Peterson S, Filiano J, Karnes PS, Patterson MC, Freeze HH 2000 Dolichol phosphate mannose synthase (DPM1) mutations define congenital disorder of glycosylation Ie (CDG-Ie). J Clin Invest 105: 191–198
Ramaekers VT, Stibler H, Kint J, Jaeken J 1991 A new variant of the carbohydrate deficient glycoproteins syndrome. J Inherit Metab Dis 14: 385–388
Jaeken J, De Cock P, Stibler H, Van Geet C, Kint J, Ramaekers V, Carchon H 1993 Carbohydrate-deficient glycoprotein syndrome type II. J Inherit Metab Dis 16: 1041
Fukuda MN, Dell A, Scartezzini P 1987 Primary defect of congenital dyserythropoietic anemia type II: failure in glycosylation of erythrocyte lactosaminoglycan-proteins caused by lowered N-acetylglucosaminyltransferase II. J Biol Chem 262: 7195–7206
Schachter H, Jaeken J 1999 Carbohydrate-deficient glycoprotein syndrome type II. Biochim Biophys Acta 1455: 179–192
Jaeken J, Schachter H, Carchon H, De Cock P, Coddeville B, Spik G 1994 Carbohydrate deficient glycoprotein syndrome type II: a deficiency in Golgi localised N-acetyl-glucosaminyltransferase II. Arch Dis Child 71: 123–127
Charuk JH, Tan J, Bernardini M, Haddad S, Reithmeier RA, Jaeken J, Schachter H 1995 Carbohydrate-deficient glycoprotein syndrome type II: an autosomal recessive N-acetylglucosaminyltransferase II deficiency different from typical hereditary erythroblastic multinuclearity, with a positive acidified-serum lysis test (HEMPAS). Eur J Biochem 230: 797–805
Tan J, Dunn J, Jaeken J, Schachter H 1996 Mutations in the MGAT2 gene controlling complex N-glycan synthesis cause carbohydrate-deficient glycoprotein syndrome type II, an autosomal recessive disease with defective brain development. Am J Hum Genet 59: 810–817
De Praeter CM, Gerwig GJ, Bause E, Nuytinck LK, Vliegenthart JF, Breuer W, Kamerling JP, Espeel MF, Martin JJ, De Paepe AM, Chan NW, Dacremont GA, Van Coster RN 2000 A novel disorder caused by defective biosynthesis of N-linked oligosaccharides due to glucosidase I deficiency. Am J Hum Genet 66: 1744–1756
Etzioni A, Frydman M, Pollack S, Avidor I, Phillips ML, Paulson JC, Gershoni-Baruch R 1992 Recurrent severe infections caused by a novel leukocyte adhesion deficiency. N Engl J Med 327: 1789–1792
Marquardt T, Brune T, Luhn K, Zimmer KP, Körner C, Fabritz L, van der Werft N, Vormoor J, Freeze HH, Louwen F, Biermann B, Harms E, von Figura K, Vestweber D, Koch HG 1999 Leukocyte adhesion deficiency II syndrome, a generalized defect in fucose metabolism. J Pediatr 134: 681–688
Lühn K, Wild MK, Eckhardt M, Gerardy-Schahn R, Vestweber D 2001 The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nat Genet 28: 69–72
Lübke T, Marquardt T, Etzioni A, Hartmann E, von Figura K, Körner C 2001 Complementation cloning identifies CDG-IIc, a new type of congenital disorders of glycosylation, as a GDP-fucose transporter deficiency. Nat Genet 28: 73–76
Dorland L, de Koning TJ, Toet M, de Vries LS, van den Berg IET, Poll-Thé BT 1997 Recurrent non-immune hydrops fetalis associated with carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 20: 88( abstr)
Eyskens F, Ceuterick C, Martin JJ, Janssens G, Jaeken J 1994 Carbohydrate-deficient glycoprotein syndrome with previously unreported features. Acta Paediatr 83: 892–896
Acarregui MJ, George TN, Rhead WJ 1998 Carbohydrate-deficient glycoprotein syndrome type 1 with profound thrombocytopenia normal phosphomannomutase phosphomannose isomerase activities. J Pediatr 133: 697–700
Charlwood J, Clayton P, Johnson A, Keir G, Mian N, Winchester B 1997 A case of the carbohydrate-deficient glycoprotein syndrome type 1 (CDGS type 1) with normal phosphomannomutase activity. J Inherit Metab Dis 20: 817–826
Skladal D, Sperl W, Henry H, Bachmann C 1996 Congenital cataract familial brachydactyly in carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 19: 251–252
Knopf C, Rod R, Jaeken J, Berant M, Van Schaftingen E, Fryns JP, Brill-Zamir R, Gershoni-Baruch R, Lischinsky S, Mandel H 2000 Transferrin protein variant mimicking carbohydrate-deficient glycoprotein syndrome in trisomy 7 mosaicism. J Inherit Metab Dis 23: 399–403
Fletcher M, Matthijs G, Jaeken J, Van Schaftingen E, Nelson PV 2000 Carbohydrate-deficient glycoprotein syndrome: beyond the screen. J Inherit Metab Dis 23: 396–398
Stibler H, von Dobeln U, Kristiansson B, Guthenberg C 1997 Carbohydrate-deficient transferrin in galactosaemia. Acta Paediatr 86: 1377–1378
Jaeken J, Pirard M, Adamowicz M, Pronicka E, Van Schaftingen E 1996 Inhibition of phosphomannose isomerase by fructose 1-phosphate: an explanation for defective N-glycosylation in hereditary fructose intolerance. Pediatr Res 40: 764–766
Adamowicz M, Pronicka E 1996 Carbohydrate deficient glycoprotein syndrome-like transferrin isoelectric focusing pattern in untreated fructosaemia. Eur J Pediatr 155: 347–348
Charlwood J, Clayton P, Keir G, Mian N, Winchester B 1998 Defective galactosylation of serum transferrin in galactosemia. Glycobiology 8: 351–357
Landberg E, Pahlsson P, Lundblad A, Arnetorp A, Jeppsson JO 1995 Carbohydrate composition of serum transferrin isoforms from patients with high alcohol consumption. Biochem Biophys Res Commun 210: 267–274
Grünewald S, Schollen E, Van Schaftingen E, Jaeken J, Matthijs G 2001 High residual activity of PMM2 in patients' fibroblasts: possible pitfall in the diagnosis of CDG-Ia (phosphomannomutase deficiency). Am J Hum Genet 68: 347–354
Kjaergaard S, Skovby F, Schwartz M 1998 Absence of homozygosity for predominant mutations in PMM2 in Danish patients with carbohydrate-deficient glycoprotein syndrome type 1. Eur J Hum Genet 6: 331–336
Kjaergaard S, Skovby F, Schwartz M 1999 Carbohydrate-deficient glycoprotein syndrome type 1A: expression characterisation of wild type mutant PMM2 in E. coli. Eur J Hum Genet 7: 884–888
Bjursell C, Wahlström J, Berg K, Stibler H, Kristiansson B, Matthijs G, Martinsson T 1998 Detailed mapping of the phosphomannomutase 2 (PMM2) gene mutation detection enable improved analysis for Scandinavian CDG type I families. Eur J Hum Genet 6: 603–611
Vuillaumier-Barrot S, Barnier A, Cuer M, Durand G, Grandchamp B, Seta N 1999 Characterization of the 415G>A (E139K) PMM2 mutation in carbohydrate-deficient glycoprotein syndrome type Ia disrupting a splicing enhancer resulting in exon 5 skipping. Hum Mutat 14: 543–544
Kondo I, Mizugishi K, Yoneda Y, Hashimoto T, Kuwajima K, Yuasa I, Shigemoto K, Kuroda Y 1999 Missense mutations in phosphomannomutase 2 gene in two Japanese families with carbohydrate-deficient glycoprotein syndrome type 1. Clin Genet 55: 50–54
Crosby A, Jeffery S, Homfray T, Taylor R, Patton M 1999 Prenatal diagnosis the subsequent mutation analysis in a family with carbohydrate-deficient glycoprotein type I syndrome: growing evidence to support founder effects within CDG1 populations. Genet Test 3: 305–307
Matthijs G, Schollen E, Heykants L, Grünewald S 1999 Phosphomannomutase deficiency: the molecular basis of the classical Jaeken syndrome (CDGS type Ia). Mol Genet Metab 68: 220–226
Matthijs G, Schollen E, Van Schaftingen E, Cassiman JJ, Jaeken J 1998 Lack of homozygotes for the most frequent disease allele in carbohydrate-deficient glycoprotein syndrome type 1A. Am J Hum Genet 62: 542–550
Pirard M, Matthijs G, Heykants L, Schollen E, Grünewald S, Jaeken J, Van Schaftingen E 1999 Effect of mutations found in carbohydrate-deficient glycoprotein syndrome type IA on the activity of phosphomannomutase 2. FEBS Lett 452: 319–322
Schollen E, Kjaergaard S, Legius E, Schwartz M, Matthijs G 2000 Lack of Hardy- Weinberg equilibrium for the most prevalent PMM2 mutation in CDG-Ia (congenital disorder of glycosylation type Ia). Eur J Hum Genet 8: 367–371
Schollen E, Dorland L, de Koning TJ, van Diggelen OP, Huijmans JG, T. M, Babovic-Vuksanovic D, Patterson MC, Imtiaz F, Winchester B, Adamowicz M, Pronicka E, Freeze H, Matthijs G 2000 Genomic organization of the human phosphomannose isomerase (MPI) gene mutation analysis in patients with congenital disorders of glycosylation type Ib (CDG-Ib). Hum Mut 16: 247–252
Imbach T, Burda P, Kuhnert P, Wevers RA, Aebi M, Berger EG, Hennet T 1999 A mutation in the human ortholog of the Saccharomyces cerevisiae ALG6 gene causes carbohydrate-deficient glycoprotein syndrome type-Ic. Proc Natl Acad Sci USA 96: 6982–6987
Imbach T, Grünewald S, Schenk B, Burda P, Schollen E, Wevers R, Jaeken J, de Klerk JB, Berger EG, Matthijs G, Aebi M, Hennet T 2000 Multi-allelic origin of congenital disorder of glycosylation (CDG)-Ic. Hum Genet 106: 538–545
Matthijs G, Schollen E, Cassiman JJ, Cormier-Daire V, Jaeken J, Van Schaftingen E 1998 Prenatal diagnosis in CDG1 families: beware of heterogeneity. Eur J Hum Genet 6: 99–104
Clayton P, Winchester B, Di Tomaso E, Young E, Keir G, Rodeck C 1993 Carbohydrate-deficient glycoprotein syndrome: normal glycosylation in the fetus. Lancet 341: 956
Stibler H, Skovby F 1994 Failure to diagnose carbohydrate-deficient glycoprotein syndrome prenatally. Pediatr Neurol 11: 71
Jaeken J, van Eijk HG, van der Heul C, Corbeel L, Eeckels R, Eggermont E 1984 Sialic acid-deficient serum cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta 144: 245–247
Stibler H, Jaeken J 1990 Carbohydrate deficient serum transferrin in a new systemic hereditary syndrome. Arch Dis Child 65: 107–111
Foo Y, Rosalki S 1998 Carbohydrate deficient transferrin measurement. Ann Clin Biochem 35: 345–350
Iourin O, Mattu TS, Mian N, Keir G, Winchester B, Dwek RA, Rudd PM 1996 The identification of abnormal glycoforms of serum transferrin in carbohydrate deficient glycoprotein syndrome type I by capillary zone electrophoresis. Glycoconj J 13: 1031–1042
Seta N, Barnier A, Hochedez F, Besnard MA, Durand G 1996 Diagnostic value of Western blotting in carbohydrate-deficient glycoprotein syndrome. Clin Chim Acta 254: 131–140
Oda RP, Prasad R, Stout RL, Coffin D, Patton WP, Kraft DL, O'Brien JF, Landers JP 1997 Capillary electrophoresis-based separation of transferrin sialoforms in patients with carbohydrate-deficient glycoprotein syndrome. Electrophoresis 18: 1819–1826
Yamashita K, Ideo H, Ohkura T, Fukushima K, Yuasa I, Ohno K, Takeshita K 1993 Sugar chains of serum transferrin from patients with carbohydrate deficient glycoprotein syndrome: evidence of asparagine-N-linked oligosaccharide transfer deficiency. J Biol Chem 268: 5783–5789
Yamashita K, Ohkura T, Ideo H, Ohno K, Kanai M 1993 Electrospray ionization-mass spectrometric analysis of serum transferrin isoforms in patients with carbohydrate-deficient glycoprotein syndrome. J Biochem 114: 766–769
Coddeville B, Carchon H, Jaeken J, Briand G, Spik G 1998 Determination of glycan structures molecular masses of the glycovariants of serum transferrin from a patient with carbohydrate deficient syndrome type II. Glycoconj J 15: 265–273
Panneerselvam K, Etchison JR, Skovby F, Freeze HH 1997 Abnormal metabolism of mannose in families with carbohydrate-deficient glycoprotein syndrome type 1. Biochem Mol Med 61: 161–167
Mayatepek E, Schröder M, Kohlmüller D, Bieger WP, Nützenadel W 1997 Continuous mannose infusion in carbohydrate-deficient glycoprotein syndrome type I. Acta Paediatr 86: 1138–1140
Mayatepek E, Kohlmüller D 1998 Mannose supplementation in carbohydrate-deficient glycoprotein syndrome type I phosphomannomutase deficiency. Eur J Pediatr 157: 605–606
Körner C, Lehle L, von Figura K 1998 Carbohydrate-deficient glycoprotein syndrome type 1: correction of the glycosylation defect by deprivation of glucose or supplementation of mannose. Glycoconj J 15: 499–505
Alton G, Kjaergaard S, Etchison JR, Skovby F, Freeze HH 1997 Oral ingestion of mannose elevates blood mannose levels: a first step toward a potential therapy for carbohydrate-deficient glycoprotein syndrome type I. Biochem Mol Med 60: 127–133
Freeze HH, Aebi M 1999 Molecular basis of carbohydrate-deficient glycoprotein syndromes type I with normal phosphomannomutase activity. Biochim Biophys Acta 1455: 167–178
Niehues R, Hasilik M 2000 Carbohydrate-deficient glycoprotein syndrome (CDGS) type Ib: a hereditary metabolic disease its therapy. MMW Fortschr Med 142: 171–172
Varki A, Marth JD 1995 Oligosaccharides in vertebrate development. Semin Dev Biol 6: 127–138
Kranz C, Denecke J, Lehrman MA, Ray S, Kienz P, Kreissel G, Sagi D, Peter-Katalinic J, Freeze HH, Schmid T, Jackowski-Dohrmann, Harms E, Marquardt T 2001 A mutation in the human MPDU1 gene causes congenital disorder of glycosylation type If (CDG-If). J Clin Invest 108: 1613–1619
Schenk B, Imbach T, Frank CG, Grubenmann CE, Raymond GV, Hurritz H, Raas-Rotschild A, Luder AS, Jaeken J, Berger EG, Matthijs G, Hennet T, Aebi M 2001 MPDU1 mutations underlie a novel human congenital disorder of glycosylation, designated type If. J Clin Invest 108: 1687–1695
Hansske B, Thiel C, Lübke T, Hasilik M, Hönig S, Peters V, Heidemann PH, Hoffmann GF, Berger EG, von Figura K, Körner C 2002 Deficiency of UDP-galactose: N-acetylglucosamine β-1,4-galactosyltransferase I causes the congenital disorder of glycosylation type IId. J Clin Invest 109: 725–733
Peters V, Penzien JM, Reiter G, Körner C, Hackler R, Assmann B, Fang J, Schaefer JR, Hoffmann GF, Heidemann PH 2002 Congenital disorder of glycosylation IId (CDG-IId)—a new entity: clinical presentation with Dandy-Walker malformation myopathy. Neuropediatrics 33: 27–32
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Grünewald, S., Matthijs, G. & Jaeken, J. Congenital Disorders of Glycosylation: A Review. Pediatr Res 52, 618–624 (2002). https://doi.org/10.1203/00006450-200211000-00003
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200211000-00003
This article is cited by
-
Untangling adaptive functioning of PMM2-CDG across age and its impact on parental stress: a cross-sectional study
Scientific Reports (2023)
-
Clinical staging and genetic profiling of Korean patients with primary lymphedema using targeted gene sequencing
Scientific Reports (2022)
-
A Double-Blind, Placebo-Controlled, Crossover Study of Magnesium Supplementation in Patients with XMEN Disease
Journal of Clinical Immunology (2022)
-
AP1S1 missense mutations cause a congenital enteropathy via an epithelial barrier defect
Human Genetics (2020)
-
An Update on XMEN Disease
Journal of Clinical Immunology (2020)
