
Abstract
L-Alloisoleucine (2 S, 3 R), a diastereomer of L-isoleucine (2 S, 3 S), is a normal constituent of human plasma. Considerable amounts accumulate in maple syrup urine disease, in which the branched-chain 2-oxo acid dehydrogenase step is impaired. The mechanism of L-alloisoleucine formation, however, is unclear. We addressed this issue by performing oral L-[1-13C]isoleucine loading (38 μmol/kg body wt, 50% 1-13C) in overnight-fasted healthy subjects (n = 4) and measuring the 3-h kinetics of 13C-label incorporation into L-isoleucine plasma metabolites. Compared with L-isoleucine, the time course of 13C-enrichment in the related 2-oxo acid, S-3-methyl-2-oxopentanoate, was only slightly delayed. Peak values, amounting to 18 ± 4 and 17 ± 3 mol percent excess, respectively, were reached within 35 and 45 min, respectively. The kinetics of 13C-enrichment in S- and R-3-methyl-2-oxopentanoate enantiomorphs were similar and linearly correlated (p ≪ 0.001). In L-alloisoleucine, however, 13C-label accumulated only gradually and in minor amounts. Our results indicate that R-3-methyl-2-oxopentanoate is an immediate and inevitable byproduct of L-isoleucine transamination and further suggest that alloisoleucine is primarily formed via retransamination of 3-methyl-2-oxopenanoate in vivo.
Similar content being viewed by others


Main
The branched-chain L-amino acids, L-leucine, L-valine, and L-isoleucine (2 S, 3 S), and the corresponding 2-oxo acids, 4-methyl-2-oxopentanoate, 3-methyl-2-oxobutyrate, and S-3-methyl-2-oxopentanoate, are normal constituents of human plasma. Additionally, L-alloisoleucine (2 S, 3 R) and derived R-3-methyl-2-oxopentanoate are generally present, although at barely detectable concentrations. Significant amounts of the latter compounds are only found in MSUD [McKusik 248600 (1a)], in which an inherited impairment of the activity of the mitochondrial branched-chain 2-oxo acid dehydrogenase complex (EC 1.2.4.4) causes accumulation of branched-chain compounds in blood and other body fluids [see Chuang and Shih (1) for a comprehensive review].
In fact, occurrence of L-alloisoleucine in humans was first detected in MSUD plasma. Initially mistaken as methionine (2), the entity of alloisoleucine in MSUD plasma was identified by Norton et al. in 1962 (3), and the L-chirality was finally established by Halpern and Pollock in 1970 (4). As to the related R-2-oxo acid, two gas chromatographic peaks originally assigned to the oxim derivatives of R- and S-3-methyl-2-oxopentanoate (5) later turned out to be actually caused by E- and Z-isomerism (6). Recently, data on R-3-methyl-2-oxopentanoate in human plasma have become available (7).
It is well documented that the catabolism of the diastereomers L-isoleucine and L-alloisoleucine is initiated by reversible transamination catalyzed by branched-chain L-amino acid aminotransferase (EC 2.6.1.42). S- and R-3-methyl-2-oxopentanoate thus formed are oxidatively decarboxylated by the branched-chain 2-oxo acid dehydrogenase complex, and the ensuing S- and R-branched-chain acyl-CoA derivatives are further degraded via the so-called S- and R-pathways (8). An important difference between the two amino acids is that L-alloisoleucine, in contrast to L-isoleucine, is not used for protein synthesis (9).
With respect to the mechanism of L-alloisoleucine formation, Meister and White suggested in 1951 (10) an S- to R-3-methyl-2-oxo acid racemization via oxo enol tautomerization as the crucial link. This proposal was later adapted (11) toward an explanation of L-alloisoleucine production in MSUD (Fig. 1). Matthews and coworkers (13) took a somewhat delayed transfer of 15N-label from L-[15N]leucine into L-alloisoleucine versus L-isoleucine in two MSUD patients as evidence that L-isoleucine and L-alloisoleucine equilibrate via the corresponding 2-oxo acids in vivo. The validity of this conclusion is dubious, however, because of the multiplicity of α-amino group cycling occurring in vivo (12, 14). Furthermore, 2-oxo acid racemization is not observed at pH < 8.4 in vitro (15), and a recently conducted 1H-nuclear magnetic resonance spectroscopy study rigorously proved that spontaneous racemization does not occur under physiologic conditions (12).

The two mechanisms proposed for the formation of L-alloisoleucine from L-isoleucine in vivo. The interconversion of these amino acids has been suggested to occur via keto-enol-tautomerism of the 2-oxo acids and subsequent retransamination (10, 11), and, alternatively, via C3-epimerisation of the L-amino acids when bound to the pyridoxyl moiety of the branched-chain L-amino acid aminotransferase and subsequent release from the enzyme by hydrolysis (12).
The obvious discrepancy between the virtually absent 2-oxo acids racemization in vitro and an apparent relatively rapid S- to R-3-methyl-2-oxopentanoate conversion in vivo (16) prompted the notion that “the interconversion is evidently catalyzed by a racemase somewhere in the body” (17). However, this notion has never been substantiated experimentally. Mamer and Reimer (12), in critically evaluating the available evidence, finally refused the 2-oxo acid racemization hypothesis of L-alloisoleucine formation and concluded, by default, L-alloisoleucine to be formed directly from L-isoleucine at the aminotransferase step (Fig. 1).
In principle, application of appropriately 13C-labeled substrates provides an excellent (and safe) means to assess whether L-alloisoleucine may be formed via 2-oxo acids or rather directly from L-isoleucine in humans. In the past, such studies were impeded, however, by the lack of a method for differential 13C-label enrichment analysis in the S- and R-3-methyl-2-oxopentanoate enantiomorphs (12, 18). Having recently established a reliable method (7), we now directly addressed the issue of L-alloisoleucine formation in vivo by performing oral L-[1-13C]isoleucine loading studies in healthy volunteers and measuring comparatively the kinetics of 13C-label transfer into the four L-isoleucine metabolites in plasma: L-isoleucine, S-3-methyl-2-oxopentanoate, L-alloisoleucine, and R-3-methyl-2-oxopentanoate.
METHODS
Chemicals.
Unless otherwise noted, all chemicals and reagents were purchased in the highest available purity from Merck (Darmstadt, Germany), Sigma Chemical Co. Chemie (Munich, Germany), or Boehringer-Mannheim (Mannheim, Germany). L-[1-13C]isoleucine (99% 1-13C) was obtained from Promochem (Wesel, Germany) and checked for isotope content by gas chromatography–mass spectrometry analysis as detailed below. Naturally enriched L-isoleucine and L-alloisoleucine preparations were from Merck and Bachem Biochemica (Heidelberg, Germany), respectively. As checked by automatic amino acid analysis, the 13C- and naturally labeled L-isoleucine contained <0.2% and <0.3% alloisoleucine, respectively. Alloisoleucine contained <0.1% isoleucine. As examined by the D,L-amino acid HPLC-analysis described by Lam (19), the L-isoleucine and L-alloisoleucine preparations showed an enantiomeric purity of >99.8%.
Subjects.
Five healthy volunteers participated in the study (four men, one woman; mean (±SD) age, 32 ± 8 y; height, 179 ± 6 cm; weight, 76 ± 8 kg). The results of their physical examination were normal. None of them received medication or had an acute or chronic illness. Four of the subjects received L-[13C]isoleucine only. One of the subjects, having normal L-isoleucine and L-alloisoleucine metabolism on previous occasions (18, 20), simultaneously ingested L-alloisoleucine (38 μmol/kg body wt). Written informed consent was obtained from all subjects before they entered the study. The protocol used in this study had been approved by the Ethics Committee of the Heinrich-Heine Universität of Düsseldorf.
L-[1-13C]isoleucine loading tests.
Starting at 0800–0830 h, the overnight-fasted subjects were kept in the supine position throughout the experiment. As detailed previously (21), an indwelling cannula was inserted into the basilic vein and kept patent by infusion of sterile saline. After a 45-min resting period, samples of blood were withdrawn into EDTA-containing tubes just before the load as control values. Likewise, expired air was collected using breathing bags (1.5 L Tecobag; Tessaurex Container, Bürstadt, Germany). The subjects then ingested 1-13C-labeled L-isoleucine (50% 1-13C, 38 μmol/kg) dissolved in 150 mL of diluted citric acid (5 mM). Thereafter, further samples of venous blood and, in parallel, exhaled air were collected during a 3-h experimental period according to the following time schedule: 0–45 min, every 5 min; 45–60 min, every 7.5 min; 60–180 min, every 15 min. Total CO2 output was measured by indirect calorimetry (Deltatrac Metabolic Monitor, Datex Instruments, Helsinki, Finland).
Determination of metabolites.
L-Isoleucine and L-alloisoleucine—as well as their corresponding 2-oxo acids after enzymatic conversion to the L-amino acids (7)—were quantitated by automatic amino acid analysis using ninhydrin detection (18). For analyses, plasma samples were treated as follows.
Plasma (1.9 mL) was mixed with SSA (60 g/100 mL, 0.1 mL) fortified with sodium 2-oxohexanoate (2 mM) and DL-norleucine (2 mM) as internal standards and deproteinized by centrifugation (10 min, 4°C, 10 000 ×g). For separation of branched-chain L-amino and 2-oxo acids, the supernatant (1.5 mL) was applied onto a Dowex 50 WX8 column (200–400 mesh, H+-form, 0.5 mL) equilibrated with SSA (3 g/100 mL) and washed with 0.5 mL SSA (3 g/100 mL). The first 0.5 mL of the eluate was discarded. The remaining eluate contained the 2-oxo acids. It was collected, neutralized by addition of NH4OH (2.5 M, 0.15 mL), and mixed with NH4Cl-NH4OH buffer (5 M, pH 8.3, 0.3 mL). For preparation of the corresponding L-amino acids, NADH (100 mM, 0.1 mL) and L-leucine dehydrogenase solution (12.5 mM/min, 0.02 mL) were added, and the mixture was incubated at 30°C for 2 h. The reaction was stopped by addition of SSA (20 g/100 mL, 3 mL), and the mixture was applied onto a 1.8-mL column of Dowex 50 WX8 (200–400 mesh, H+-form). After four washes with water (2 mL), the enzymatically formed amino acids were eluted from the column using NH4OH (2.5 M, 4 mL). The eluate was evaporated to dryness. The residue was dissolved in water (0.5 mL) and centrifuged (10 000 ×g, 2 min). Aliquots of the supernatant were evaporated to dryness, and the residues were used for quantitation by automatic amino acid analysis and determination of 13C-label enrichment by gas chromatography–mass spectrometry.
Plasma branched-chain L-amino acids were recovered from the initial Dowex 50 WX8 column after elution of the 2-oxo acids. The column was washed with four volumes of H2O (2 mL) to remove SSA, and the L-amino acids were then eluted with NH4OH (2.5 M, 1.2 mL) as described above. The eluate was evaporated to dryness, and the residue was further used for analysis of concentration and 13C-enrichments as above.
13C-label enrichment analysis.
The residues, containing the preformed or 2-oxo acid-derived branched-chain L-amino acids, were dissolved in 50 μL of N-methyl-N-(t-butyldimethylsilyl)-trifluoroacetamide (Pierce Chemicals, Rockford, IL) in CH3CN (1:1, vol/vol), and the mixture was incubated at 70°C for 1 h to yield the N,O-t-butyldimethylsilyl amino acid derivatives. For analysis by gas chromatography–mass spectrometry, an HP 6890 gas chromatograph equipped with an HP-5 mass spectrometry capillary column (5% phenylmethylpolysiloxane; 30 m, inner diameter 0.25 mm, 0.25 μm film thickness) and directly connected to an HP mass selective detector was used (Hewlett-Packard, Waldbronn, Germany). Helium was the carrier gas (0.35 mL/min). The injector and the transfer line to the spectrometer were held at 250°C. The column temperature was 100°C initially. After 1.5 min, the temperature was increased to 176°C at 4°C/min and then raised to 320°C for 5 min. Electron impact-mode was used. The ion source was operated at 140°C and an electron energy of 40 eV. For measurement of label enrichment, the ratio (R) of the ion intensities at m/z 303 and m/z 302 for labeled and unlabeled isoleucine metabolites, respectively, were measured using selected ion monitoring. The standard deviation of 13C-enrichment measurements in L-isoleucine and S-3-methyl-2-oxopentanoate was typically ±0.1 MPE. Because of the low plasma concentration of L-alloisoleucine and R-3-methyl-2-oxopentanoate, the standard deviation of label enrichment measurement in these compounds was about ±0.2 MPE.
Determination of 13CO2 in expired air was performed using isotope ratio–mass spectrometry as detailed previously (21).
Calculations.
The 13C-label MPE in metabolites was calculated from the natural abundance ratio as measured in the samples withdrawn before (R0) and the values after (R) oral administration of 13C-labeled L-isoleucine according to Wolfe (22) as MATH

Increase in 13C-label (APE) in expired CO2 was calculated as the difference of 13CO2 in breath samples withdrawn before (control value) and after the oral loads. Rates of extra 13CO2 exhalation per min were calculated from the APE on the basis of the total CO2 exhalation rate. Cumulative extra 13CO2 exhalation was calculated using trapezoidal methods.
Unless otherwise noted, results are presented as means ± SEM with the number of determinations in parentheses. Correlations were checked by linear regression analysis (least square method; RGP function of MS Excel 5.0; Microsoft, Redmond, WA) yielding the slope and constant of the regression line ± SE.
RESULTS
Time course of plasma concentrations.
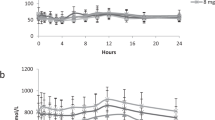
When healthy subjects ingested L-[13C]isoleucine (50% 1-13C; 38 μmol/kg body wt), a distinct, although interindividually variable, time course of isoleucine metabolites in plasma was observed (Fig. 2). The mean initial L-isoleucine plasma concentrations amounted to 62.5 ± 8.5 μM. After load, peak values were reached within 10 to 35 min with mean peak increases amounting to 80.0 ± 16.7 μM. Interestingly, the height of the peak appeared to be inversely correlated to the peak time. Thereafter, plasma concentrations gradually declined but remained still elevated at the end of the 3-h study period (80.9 ± 4.1 μM). The kinetics of the corresponding 2-oxo acids were slightly delayed and attenuated. The initial levels of S- and R-3-methyl-2-oxopentanoate were 27.6 ± 2.9 and 0.78 ± 0.11 μM, respectively. Peak increases in the enantiomeric 2-oxo acids were reached in parallel within 15 to 45 min and amounted to 17.4 ± 2.9 and 0.43 ± 0.12 μM, respectively. After 3 h, the plasma concentrations had declined to 36.4 ± 1.6 and 0.94 ± 0.05 μM, respectively.

Time course of the concentration of L-isoleucine metabolites in plasma of overnight-fasted healthy volunteers after ingestion of L-[13C]isoleucine (50% 1-13C, 38 μmol/kg body wt). Ile, L-isoleucine;Allo, L-alloisoleucine. Results are means ± SEM (n = 4).
With L-alloisoleucine, the kinetic was different. This metabolite exhibited no distinct peak increase but rather appeared to increase more gradually by a mean of approximately 10%, from 2.23 ± 0.40 μM at the beginning to 2.46 ± 0.39 μM at the end of the study.
Time course of 13C-label enrichment.
The load-induced increase in the 13C-label enrichment is depicted in Figure 3. The data show that the time course of the labeling pattern in the isoleucine metabolites largely paralleled that of the plasma concentrations, i.e. fast appearance of 13C-label in L-isoleucine directly followed by an increase in 13C-label in the enantiomeric 2-oxo acids and significantly delayed and attenuated 13C-enrichment in L-alloisoleucine.

Time course of (A) 13C-label enrichment in L-isoleucine metabolites in plasma of overnight-fasted healthy volunteers after ingestion of L-[13C]isoleucine (50% 1-13C, 38 μmol/kg body wt) and (B) relationship of 13C-label enrichment (MPE) in Ile, S- and R-KMV, and Allo. Results are means ± SEM (n = 4). In (B), data are expressed as the ratio of metabolite label enrichment and MPE in Ile (≡ 1 at each time point).
A somewhat unexpected finding was that both time course and amount of 13C-label enrichment of S- and R-3-methyl-2-oxopentanoate were very similar in each subject and were linearly correlated (p ≪ 0.001;Fig. 4), but were quite different from those of L-alloisoleucine. The latter is accentuated in Figure 3 (inset), in which the 13C-label enrichments in the 2-oxo acids and L-alloisoleucine are related to the enrichment in the common metabolic precursor, L-isoleucine. It is evident that label introduction into the two enantiomeric 2-oxo acid pools lagged behind that of the L-isoleucine pool, but finally a rather constant and nearly equal 13C-enrichment was reached within 1 h after administration of L-[13C]isoleucine. In contrast, with the L-alloisoleucine pool, incorporation of label was gradual, and 13C-enrichments comparable to those found in plasma L-isoleucine were not reached before the end of the experimental period. In these experiments, the cumulative mean 13CO2 output within 3 h amounted to 2.85 ± 0.40 μmol/kg body wt, equivalent to 15.0 ± 2.1% of the 13C-dose applied.

Linear relationship of 13C-label enrichment (MPE) in S- and R-KMV in plasma after oral loading with L-[1-13C]isoleucine. Individual data of the four healthy subjects who underwent the experiments depicted in Figure 3 are shown. Dotted lines are regression lines (least squares method; subject GD, y = 0.99 (±0.02) x − 0.48 (±0.18); subject CH, y = 0.99 ± (0.02) x + 0.63 (±0.18); subject MP, y = 0.71 (±0.03) x + 0.03 (±0.18); subject DH, y = 1.08 (±0.04) x − 0.45 (±0.28); each, n = 21, r > 0.989, p ≪ 0.001).
The results presented so far suggested that, in vivo, the L-alloisoleucine pool primarily served as a final trap for the 13C-label, via 2-oxo acid retransamination, which had been originally introduced into the 3-methyl-2-oxopentanoate pools from L-[13C]isoleucine.
Effect of simultaneous L-alloisoleucine loading.
To further substantiate the traplike function of L-alloisoleucine, we performed an L-[13C]isoleucine loading experiment in which the L-alloisoleucine pool was simultaneously enlarged by oral application of naturally labeled L-alloisoleucine (38 μmol/kg body wt, each). As shown in Figure 5, the L-isoleucine plasma concentration increased about 2-fold within 20 min as above, from a control value of 47.7 μM to a peak value of 123.3 μM. In parallel, plasma L-alloisoleucine was enhanced about 30-fold, from 2.4 to 80.8 μM. S- and R-3-methyl-2-oxopentanoate showed an increase from 16.4 and 0.88 μM initially, respectively, to a delayed maximum of 31.3 (at 30 min) and 8.2 μM (at 45 min), respectively.

Time course of concentration and 13C-label enrichment of Ile metabolites in plasma of an overnight-fasted subject after combined oral loading with 50% enriched L-[1-13C]isoleucine plus naturally labeled Allo (38 μmol/kg body wt, each).
The 13C-labeling pattern of plasma L-isoleucine and the corresponding 2-oxo acid was similar to the previous experiments. It appeared not to be significantly influenced by the simultaneous presence of excess L-alloisoleucine. Obviously, this was also valid for the rate of whole body L-[13C]isoleucine oxidation. In agreement with the exhalation data described above, 15.0% (i.e. 2.86 μmol/kg body wt) of the 13C-dose applied was recovered as 13CO2 in expired air within 3 h.
However, the very high dilution caused by the administered L-alloisoleucine now prevented any detection of an increase in 13C-label in plasma L-alloisoleucine. In contrast, label enrichments in R-3-methyl-2-oxopentanoate remained reliably measurable in the initial phase of the experiment but were at variance with the concentration curve, e.g. the peak enrichment at 20 min clearly preceded the concentration maximum and amounted to 4.3 MPE compared with 19.4 and 20.8 MPE in S-2-methyl-2-oxopentanoate and L-isoleucine, respectively. Thus, 13C- label enrichment in R-3-methyl-2-oxopentanoate was about 5-fold lower than in the S-2-oxo acid, and this appears to be in excellent agreement with the approximately 5-fold L-alloisoleucine-induced increase in R-3-methyl-2-oxopentanoate concentration (5.8 μM) at this time point.
DISCUSSION
It is generally held that L-alloisoleucine is derived from L-isoleucine in vivo (1). However, no strict direct proof for this assumption has been presented. So far, the accumulated experimental evidence is indirect: in patients with MSUD, the kinetics of plasma L-alloisoleucine approach those of L-isoleucine (11, 23). Administration of L-isoleucine preparations to patients with MSUD (11, 24) and healthy subjects (18) gave rise to a more or less marked and persistent increase in L-alloisoleucine concentrations in vivo, and similar observations have been made in L-isoleucine loading studies with normal and MSUD skin fibroblasts in vitro (18). Furthermore, besides L-isoleucine, some L-alloisoleucine consistently accumulated when (huge) amounts of S-3-methyl-3-oxopentanoate preparations were administered to patients (17) or laboratory animals (16, 25). There are some indications that the reverse pathway may also be operative in vivo to some extent: in animals on an L-isoleucine-deficient diet, supplementation with R- or racemic R,S-3-methyl-2-oxopentanoate, but apparently not L-alloisoleucine (26), promoted growth, although at lower rates than when the S-2-oxo acid isomer was supplemented (10, 25). The present findings on the 13C-labeling now unequivocally show that the carbon skeleton of L-isoleucine is the physiologic precursor of L-alloisoleucine in man.
The physiologic enteral route was used for application of L-[1-13C]isoleucine and L-alloisoleucine in the present study. The plasma kinetics of L-isoleucine and L-alloisoleucine in the combined loading test indicate that there was no discrimination in the gut with respect to the rates of carrier-mediated absorption and release of the two diastereomeric branched-chain L-amino acids into the circulation. The time course of 13C-label enrichment in the derived plasma L-isoleucine metabolites, including L-alloisoleucine, distinctly and consistently lagged behind that of L-isoleucine. Therefore, it is highly improbable that appreciable first-pass conversion of L-isoleucine to L-alloisoleucine took place in the gastrointestinal tract. The present 13C-labeling kinetics rather suggest that L-alloisoleucine formation is ubiquitous and, as will be discussed below in detail, may primarily proceed in organs exhibiting high capacities for L-amino acid transamination, i.e. essentially in muscle tissue (27, 28).
In our experiments, the timely order of 13C-incorporation into plasma metabolite pools was L-isoleucine >S-3-methyl-2-oxopentanoate ≅R-3-methyl-2-oxopentanoate ≫ L-alloisoleucine, strongly suggesting that, starting with L-isoleucine, the two enantiomeric branched-chain 2-oxo acids had been formed rather quickly and simultaneously, although at quite different amounts, followed by a gradual increase in L-alloisoleucine. Because interconversion of L-isoleucine metabolites into each other by spontaneous nonenzymatic reactions is firmly excluded under physiologic conditions, the branched-chain L-amino acid aminotransferase reaction remains the only known catalytic step that can account for the present 13C-labeling kinetics.
We infer from our data that R-3-methyl-2-oxopentanoate is an immediate and probably inevitable byproduct of enzyme-catalyzed L-isoleucine transamination. Thus, the aminotransferase may also function as an apparent S-3-methyl-2-oxopentanoate racemase. The existence of such an enzyme activity has long been postulated (17) to explain the difference between the relatively rapid S- to R-2-oxo acid racemization observed in dogs in vivo and the virtual absence of any racemization in buffered solutions in vitro (10, 12, 16).
With respect to the mechanism, Mamer and Reimer (12) recently pointed out that L-isoleucine, once bound to the pyridoxyl-5-phosphate moiety of the aminotransferase in the ketimine form, may, theoretically, undergo tautomerization, via the enamine, at the β-carbon. Thus, during the course of the enzymatic reaction, S- and R-forms of the bound ketimine would be formed. On hydrolysis, R-3-methyl-2-oxopentanoate would be liberated from the R-ketimine. Alternatively, after rearrangement to the R-aldimine, L-alloisoleucine would be released directly from the enzyme. Mamer and Reimer (12) strongly favored a direct formation of L-alloisoleucine from L-isoleucine at the aminotransferase step without, however, providing experimental evidence.
If L-alloisoleucine had been formed in our experiments directly from ingested 13C-labeled L-isoleucine, the 13C-label would have first appeared in L-alloisoleucine and the label transfer into R-3-methyl-2-oxopentanoate would have been time delayed. Furthermore, when, in the combined loading with stable isotope-labeled L-isoleucine and unlabeled L-alloisoleucine, label transfer from L-isoleucine into R-3-methyl-2-oxopentanoate had occurred via L-alloisoleucine, it would have remained undetectable because of the dilution by the amount of administered unlabeled L-alloisoleucine. Actually, however, quite the contrary was observed in our study. In addition, if L-isoleucine is the immediate precursor of L-alloisoleucine, the rate of L-alloisoleucine formation should be dependent on the concentration of L-isoleucine, and plasma L-alloisoleucine kinetics can be expected to mirror those of L-isoleucine. This was not observed, however, either in the present experiments or in our previous high dose loading studies (1.5 mmol of L-isoleucine/kg body wt, p.o). In the latter experiments, plasma L-isoleucine concentrations peaked within 1–2 h, whereas plasma L-alloisoleucine peaked with a considerable delay after 6–8 h (18). In contrast, under either loading condition, plasma 3-methyl-2-oxopentanoate peaked largely in parallel with the precursor amino acid, thus indicating the concentration dependency of transamination. These striking differences in the plasma kinetics of 3-methyl-2-oxopentanoate versus L-alloisoleucine after an L-isoleucine challenge appear to be further accentuated under in vitro conditions (18). Finally, β-epimerization at the amino acid level during transamination would not be compatible with the delayed increases of plasma L-alloisoleucine in patients with MSUD as discussed below. Taken together, the available experimental evidence is rather consistent with an L-alloisoleucine formation in vivo occurring primarily via 3-methyl-2-oxopentanoate intermediates and subsequent retransamination than with the L-amino acid β-epimerization hypothesis.
The R-2-oxo acid enantiomorph produced during L-isoleucine transamination, however, may not represent the sole metabolic precursor of L-alloisoleucine in vivo. Because of the reversibility of the aminotransferase reaction, some L-alloisoleucine is presumably formed as a byproduct of natural S-3-methyl-2-oxopentanoate retransamination. The present data and the previous findings in S-3-methyl-2-oxopentanoate loading studies (16, 17, 25) suggest that the rate of byproduct formation during transamination is comparatively low. Therefore, the rate constant for L-alloisoleucine formation from the R-2-oxo acid is likely to be considerably higher than that for L-alloisoleucine production via retransamination of the S-2-oxo acid. Nevertheless, because of the obviously quite different pool sizes of R- and S-3-methyl-2-oxopentanoate in vivo (relationship in human plasma, approximately 1:30), it cannot be excluded that transamination of either enantiomorph contributes substantially to net L-alloisoleucine production. The relationship of L-alloisoleucine formation from R- and S-3-methyl-2-oxopentanoate, however, cannot be deduced from the present experiments and remains to be established, e.g. in stable isotope studies with simultaneous application of differentially labeled 2-oxo acid enantiomorphs.
In any case, when L-alloisoleucine formation in the body primarily proceeds gradually, via transamination, from the 3-methyl-2-oxopentanoate pool, one faces the question of what the underlying mechanisms are. A possible explanation may be based on differences in sizes and interactions of the intracellular and extracellular L-amino and 2-oxo acid pools as follows. Reported data suggest that intracellular concentrations of branched-chain 2-oxo acids in mammalian tissues are far lower than in the extracellular space (29, 30), quite in contrast to the branched-chain L-amino acids, which are concentrated within the cells to some extent (31). Therefore, 3-methyl-2-oxopentanoate in the extracellular compartment may represent the main 2-oxo acid pool of the body. Obviously, this pool is fed by a rather effective extrusion of the 2-oxo acid that escaped oxidative degradation (Fig. 5). On the other hand, the considerable changes of 13C-labeling in plasma 3-methyl-2-oxopentanoate pools as opposed to the relatively minor changes of concentrations, together with the rather similar labeling kinetics in the L-amino acid precursor, point to a considerable cycling of carbon skeletons between the intracellular L-isoleucine pool and the extracellular 2-oxo acid pool, which is catalyzed by combined action of the branched-chain L-amino acid aminotransferase and cellular transport mechanisms. In vivo (13, 32) and in vitro studies (33, 34) indicate that extraneous 2-oxo acids (re)entering the intracellular compartment are (re)transaminated to yield the L-amino acid rather than degraded directly by oxidative decarboxylation. Thus, it can be envisaged that 3-methyl-2-oxopentanoate taken up from the extracellular compartment may function as the primary precursor for the feeding of the L-alloisoleucine pool by intracellular 2-oxo acid transamination. Although transient, storage of the 2-oxo acid precursor in the metabolically largely inactive extracellular compartment, whether occurring within tissues or in the overall extracellular space of the body, might finally be causative of the delayed increase of L-alloisoleucine versus L-isoleucine in plasma of patients with MSUD and in our healthy subjects.
In the present experiments, some of the R-3-methyl-2-oxopentanoate once formed intracellularly presumably underwent degradation via the R-pathway of L-isoleucine catabolism (Fig. 5). Reportedly, degradation in the R-pathway is incomplete. 2-Ethylhydracrylic and ethyl malonic acid are the main products to be finally cleared by the kidney (8, 35). In humans, up to approximately 0.05 μmol·h−1·kg−1 of the latter compounds may be excreted in the urine [as estimated from the data presented in Mamer et al. (8) and Stalder (35)]. The rate of whole body L-isoleucine oxidation amounts to about 10 μmol·h−1·kg−1 in postabsorptive healthy subjects (21). By comparison, these data indicate that, under normal conditions, metabolic flux through the R-pathway is well below 1% of the total S-pathway flux.
Physiologically, the R-pathway may function as a relief mechanism against unavoidable appearance of the R-2-oxo acid (and L-alloisoleucine) in the aminotransferase reaction. Obviously, however, some of the R-2-oxo acid temporarily escaped degradation and was transported into the extracellular space or was diverted and gradually accumulated in the L-alloisoleucine pool. Being formed by chance because of the reversibility of the transamination reaction, L-alloisoleucine appears to have no use other than representing a comparatively inert metabolic trap: it is not required for protein synthesis (9), has a low renal clearance rate (36), and is a comparatively poor substrate for the branched-chain L-amino acid aminotransferase (7). The slower rates of formation and degradation of L-alloisoleucine in vivo compared with its precursor, L-isoleucine, are most evident in patients with MSUD in whom the plasma kinetics of L-alloisoleucine have consistently been found to lag behind those of L-isoleucine (11, 23).
In conclusion, the present 13C-labeling patterns in plasma metabolites of healthy subjects undergoing oral L-[1-13C]isoleucine loads for the first time directly show that the carbon skeleton of L-alloisoleucine is derived from L-isoleucine in vivo. The tracer kinetics further show that R-3-methyl-2-oxopentanoate is rapidly formed as a byproduct of L-isoleucine transamination, yielding L-alloisoleucine in a subsequent retransamination step. The role of byproduct formation during retransamination of S-3-methyl-2-oxopentanoate in total L-alloisoleucine formation in vivo, however, remains to be established.
Abbreviations
- APE:
-
atom percent excess
- KMV:
-
3-methyl-2-oxopentanoate (2-keto-3-methylvalerate)
- MPE:
-
mol percent excess
- MSUD:
-
maple syrup urine disease
- SSA:
-
5-sulfosalicylic acid
References
McKusick VA 1994 Mendelian Inheritance in Man, 11th Ed. John Hopkins University Press, Baltimore
Chuang DT, Shih VE 1995 Disorders of branched chain amino acid and keto acid metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Metabolic and Molecular Bases of Inherited Disease. McGraw Hill, New York, pp 1239–1277
Dancis J, Levitz M, Westall RG 1960 Maple syrup urine disease: branched-chain keto-aciduria. Pediatrics 25: 72–79
Norton PM, Roitman E, Snyderman SE, Holt LE Jr 1962 A new finding in maple syrup urine disease. Lancet 1: 26–27
Halpern B, Pollock GE 1970 The configuration of the alloisoleucine present in maple syrup urine disease plasma. Biochem Med 4: 352–356
Jakobs C, Solem E, Ek J, Halvorsen K, Jellum E 1977 Investigation of the metabolic pattern in maple syrup urine disease by means of glass capillary gas chromatography and mass spectrometry. J Chromatogr 143: 31–38
Mamer OA, Montgomery JA, Taguchi VY 1980 Origin of the two peaks for 2-keto-3-methylvaleric acid produced by the oxidation of the keto acids occurring in maple syrup urine disease. J Chromatogr 182: 221–225
Schadewaldt P, Wendel U, Hammen H-W 1996 Determination of R- and S-3-methyl-2-oxopentanoate enantiomers in human plasma: suitable method for label enrichment analysis. J Chromatogr B 682: 209–218
Mamer OA, Tjoa SS, Scriver CR, Klassen GA 1976 Demonstration of a new mammalian isoleucine catabolic pathway yielding an R series of metabolites. Biochem J 160: 417–426
Batshaw ML, Brusilow S, Walser M 1976 Long-term management of a case of carbamyl phosphate synthetase deficiency using ketoanalogues and hydroxyanalogues of essential amino acids. Pediatrics 58: 227–235
Meister A, White J 1951 Growth response of the rat to the keto analogues of leucine and isoleucine. J Biol Chem 191: 211–216
Snyderman SE, Norton PM, Roitman E, Holt LE Jr 1964 Maple syrup urine disease with particular reference to dietotherapy. Pediatrics 34: 454–462
Mamer OA, Reimer MLJ 1992 On the mechanism of the formation of L- alloisoleucine and the 2-hydroxy-3-methylvaleric acid stereoisomers from L- isoleucine in maple syrup urine disease patients and in normal humans. J Biol Chem 267: 22141–22147
Matthews DE, Ben-Galim E, Haymond MW, Bier DM 1980 Alloisoleucine formation in maple syrup urine disease: isotopic evidence for the mechanism. Pediatr Res 14: 854–857
Schadewaldt P, Wendel U, Hammen H-W 1995 Human branched-chain L- amino acid aminotransferase: activity and subcellular localization in cultured skin fibroblasts. Amino Acids 9: 147–160
Meister A 1951 Studies on D - and L -α-keto-β-methylvaleric acids. J Biol Chem 190: 269–276
Weinberg RB, Walser M 1977 Racemization and amination of the keto-analog of isoleucine in the intact dog. Biochem Med 17: 164–172
Walser M, Sapir DG, Mitch WE, Chan W 1981 Effects of branched-chain ketoacids in normal subjects and patients. In: Walser M, Williamson JR (eds) Metabolism and Clinical Implication of Branched Chain Amino and Ketoacids. Elsevier, North Holland, pp 291–299
Schadewaldt P, Hammen H-W, Dalle-Feste C, Wendel U 1990 On the mechanism of L- alloisoleucine formation: studies on a healthy subject and in fibroblasts from normals and patients with maple syrup urine disease. J Inher Metab Dis 13: 137–150
Lam S 1986 Resolution of D - and L- amino acids after precolumn derivatization with o-phthalaldehyde by mixed chelation with Cu(II)- L- proline. J Chromatogr 355: 157–164
Schadewaldt P, Dalle-Feste C, Langenbeck U, Wendel U 1991 Oral L- alloisoleucine loading studies in healthy subjects and in patients with maple syrup urine disease. Pediatr Res 30: 430–434
Bodner A, Hammen H-W, Renn W, Wendel U, Schadewaldt P 1997 Whole body branched-chain L- amino acid oxidation in overnight fasted human subjects. Isotopes Environ Health Stud 33: 189–196
Wolfe RR 1992 Radioactive and Stable Isotope Tracers in Biomedicine: Principles and Practice of Kinetic Analysis. Wiley-Liss, New York, pp 230–232
Wendel U, Langenbeck U, Seakins JWT 1989 Interrelation between the metabolism of L- isoleucine and L- allo-isoleucine in patients with maple syrup urine disease. Pediatr Res 25: 11–14
Dent CE, Westall RG 1961 Studies in maple syrup urine disease. Arch Dis Child 36: 259–268
Funk MA, Lowry KR, Baker DH 1987 Utilization of the L- and DL- isomers of α-keto-β-methylvaleric acid by rats and comparative efficacy of the keto analogs of branched-chain amino acids provided as ornithine, lysine and histidine salts. J Nutr 117: 1550–1555
Greenstein JP, Levintow L, Baker CG, White J 1951 Preparation of the four stereoisomers of isoleucine. J Biol Chem 188: 647–663
Goto M, Shinno H, Ichihara A 1977 Isoenzyme patterns of branched-chain amino acid transaminase in human tissues and tumors. Gann 68: 663–667
Suryawan A, Hawes JW, Harris RA, Shimomura Y, Jenkins AE, Hutson SM 1998 A molecular model of human branched-chain amino acid metabolism. Am J Clin Nutr 68: 72–81
Crowell PL, Miller RH, Harper AE 1988 Measurement of plasma and tissue levels of branched-chain α-keto acids by gas-liquid chromatography. Methods Enzymol 166: 39–46
Matuso Y, Yagi M, Walser M 1993 Arteriovenous differences and tissue concentrations of branched-chain ketoacids. J Lab Clin Med 121: 779–784
Askanazi J, Carpentier YA, Michelsen CB, Elwyn DH, Furst P, Kantrowitz LR, Gump FE, Kinney JM 1980 Muscle and plasma amino acids following injury: influence of intercurrent infections. Ann Surg 192: 78–85
Chinkes D, Klein S, Zhang X-J, Wolfe RR 1996 Infusion of labeled KIC is more accurate than labeled leucine to determine human muscle protein synthesis. Am J Physiol 270: E67–E71
Schadewaldt P, Radeck W, Hammen H-W, Wendel U 1988 Transamination and oxidative decarboxylation rates of branched-chain 2-oxo acids in cultured human skin fibroblasts. Pediatr Res 23: 40–44
Schadewaldt P, Radeck W, Hammen H-W, Staib W 1989 Transamination and oxidative decarboxylation of L- isoleucine, L- alloisoleucine and related 2-oxo acids in perfused rat hind limb muscle. Biochim Biophys Acta 992: 115–123
Stalder K 1959 Über das Vorkommen von Äthylmalonsäure im Harn. Hoppe-Seyler's Z Physiol Chem 314: 205–210
Schadewaldt P, Hammen H-W, Ott A-C, Wendel U 1999 Renal clearance of branched-chain L- amino and 2-oxo acids in maple syrup urine disease. J Inherit Metab Dis 22: 706–722
Acknowledgements
The authors thank Dr. H. Brösicke and Mrs. A. Pfundstein (Berlin) for IRMS measurements.
Author information
Authors and Affiliations
Additional information
Supported, in part, by Grant We 614/9–2 from the Deutsche Forschungsgemeinschaft.This communication contains parts of the Thesis of A.B.-L.
Rights and permissions
About this article
Cite this article
Schadewaldt, P., Bodner-Leidecker, A., Hammen, HW. et al. Formation of L-Alloisoleucine In Vivo : An L-[13C]Isoleucine Study in Man. Pediatr Res 47, 271 (2000). https://doi.org/10.1203/00006450-200002000-00020
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200002000-00020
