
Abstract
Barbiturates are known to inhibit glucose transport mediated by the facilitative sugar transporter GLUT1. We have studied such inhibition in children with GLUT1-deficiency. Zero-trans influx of 14C-labeled 3-O-methyl glucose (3OMG) into erythrocytes of patients (n= 3) was 35% of controls (n= 6). Preincubation with 10 mM phenobarbital or pentobarbital reduced patients' 3OMG influx to 17%. In patients and controls, preincubation with barbiturates significantly decreased Vmax in a dose-dependent manner (for pentobarbital, IC50 = 0.84 mM, patient 2). The apparent Km in individuals remained largely unchanged. Three-OMG influx without preincubation resulted in a stronger inhibition at lower barbiturate concentrations. The patients' data are discussed in the light of individual missense mutations (patient 1: R126L and K256V; patient 2: T310I; patient 3: S66F) in the GLUT1 gene. In conclusion, in controls and patients with GLUT1-deficiency barbiturates interact with GLUT1, lowering its intrinsic activity. The use of barbiturates in this condition for anesthesia or as anticonvulsants could therefore potentially aggravate the existing glucose transport defect and may put these patients at increased risk.
Similar content being viewed by others
Main
Transport of D-Glucose across the blood-brain barrier is mediated by a sodium-independent, facilitative transport protein, GLUT1 (1–4). A GLUT1-defect in brain has been identified as the pathogenetic mechanism of GLUT1-deficiency. Immunoblotting and cytochalasin-B binding studies suggest that in this condition about 50% of the normal complement of GLUT1 transporters are expressed; uptake studies into erythrocytes and fibroblasts reveal a reduced GLUT1-intrinsic activity (5–8). The defect has recently been described at the molecular level (5). The failure to meet the glucose demand by the brain leads to hypoglycorrhachia, infantile seizures, acquired microcephaly, and developmental delay (6–8), a state of haploinsufficiency.
Additional inhibition of GLUT1 activity can be expected to aggravate the already impaired intracerebral glucose homeostasis in these patients. In this context, barbiturates have been reported to inhibit GLUT1-mediated glucose transport both in vitro (9) and in vivo (10–13). Barbiturates are known to limit cerebral blood flow and metabolism (11, 14–16). They are also known to interact directly with the purified GLUT1 protein and to inhibit the intrinsic activity of GLUT1 (10, 17). Importantly, recent studies suggest that the maximal velocity for glucose transfer (Vmax) across the blood-brain barrier is lowered by barbiturates due to a direct effect on GLUT1 in the endothelial cells of cerebral microvessels (11, 18). They suggest that the mechanism responsible is either a decrease in intrinsic activity or a decrease in the number of functional GLUT1 transporters—mechanisms that have been identified in GLUT1-deficiency (5–8). Hence, treatment of GLUT1 deficient patients with barbiturates could potentially result in a significant inhibition of glucose transport and thus put them at increased risk.
In the present study we investigated the effect of barbiturates on GLUT1-mediated glucose transport in erythrocytes of three patients with GLUT1-deficiency and parental controls. Our results confirm the hypothetical risk that these drugs pose for these patients.
METHODS
Clinical material.
Blood samples were obtained from three GLUT1-deficient patients as described (7). Blood samples from parents (n= 6) were used as intrassay controls.
Patient 1 (male, 5 y) was born after an uncomplicated full-term pregnancy. Postnatal development was normal. At age 6 wk he developed his first seizure and phenobarbital treatment was started. Multiple mixed seizures increased throughout infancy and early childhood despite anticonvulsive treatment; phenobarbital was discontinued eventually. By the age of 6 mo, motor and language delay and acquired microcephaly were prominent. At age 3 y hypoglycorrhachia was noted and a ketogenic diet was introduced. The patient became seizure-free and all anticonvulsant medication was withdrawn.
Patient 2 (female, 6 y) presented with seizures at age 3 mo after an uneventful pregnancy, delivery, and neonatal period. She was treated with phenytoin and phenobarbital without effect. Hypoglycorrhachia was noted at 3.5 mo and a ketogenic diet was initiated. Seizures were controlled, but she continued to develop clinical features of GLUT1-deficiency.
Patient 3 (male, 13 y) was born after an uncomplicated full-term pregnancy, delivery, and newborn period. Seizures started at 31/2 mo of age and continued throughout infancy. Phenobarbital treatment from age 6 to 21 mo was ineffective in controlling seizures. The child continued to develop microcephaly and significant motor and language delay. GLUT1-deficiency was diagnosed at age 4 y and a ketogenic diet was introduced. This resulted in gradual clinical improvement with cessation of seizures and improvement of motor skills.
Controls (two males, four females, mean age 30 y)—six parents of patients with GLUT1-deficiency were used as intraassay controls for the kinetic studies as described previously, including the mother of patient 1 and both parents of patient 2. All were healthy individuals with no clinical or laboratory features of GLUT1-deficiency present. The six parents of the three patients served as controls for the GLUT-1 genomic analyses.
RBC uptake studies.
Techniques are as in Lowe and Walmsley, with the modifications we previously described (19). Blood samples were mixed with citrate phosphate dextrose anticoagulant solution (5:1, vol/vol) and immediately stored at 4°C. Samples had to be processed within 10 d (8). Because samples could only be obtained at limited times and quantities, not every patient could be tested for barbiturate inhibition of 3OMG influx. Samples were excluded if RBCs were hemolyzed, and none of the subjects had been transfused in recent months. Blood samples from both parents served as intrassay controls.
Solutions.
To keep assay conditions close to the clinical setting, 3OMG glucose concentrations were in the range of physiologic blood glucose concentrations (∼5 mmol/L), and the barbiturates were added using aliquots of the unmodified solution for i.v. use below. Solutions were:1) Citrate phosphate dextrose solution: 2.45 g dextrose, 2.63 g sodium citrate, 298 mg citric acid, all into 100 mL;2) PBS (PBS), in mmol/L: 137 NaCl, 2.7 KCl, 4.3 Na2HPO4·H2O, 1.4 KH2PO4 (pH 7.4). Radiolabeled solution. 14C-labeled 3OMG (stock: 56.4 mCi/mmol) was from DuPont NEN (Boston, MA). Final radiolabel concentration in the uptake samples was 2.5 μCi/mL. The stop solution was (in μmol/L): 100 mercuric chloride and 50 phloretin in ice-cold PBS, and was prepared immediately before use.
Reagents.
3OMG, phloretin, and mercuric chloride were from Sigma Chemical Co. (St. Louis, MO). Barbiturate solutions were prepared using i.v. solutions from Wyeth (Philadelphia, PE). Barbiturates were phenobarbital-sodium (215 mmol/L stock) and pentobarbital-sodium (560 mmol/L stock). Vehicle for barbiturates was (vol/vol) 10% EtOH, 75% 1,2-propanediol, and 15% distilled H2O.
Preparation of erythrocytes.
Procedures were performed at 4°C. Blood specimens (2–5 mL) were centrifuged at 1000 × g for 10 min. The supernatant was discarded, and the RBC pellet (∼1–2 mL) was washed three times with 50 mL PBS. RBC was then resuspended in PBS at approximately the original blood sample volume. Fifty microliter aliquots were taken for immediate assay, and 300 μL were used for a cell count.
3OMG uptake.
All data points were obtained in quadruplicate. One hundred microliters of 14C-labeled 3OMG solution at the specified concentrations was added to the 50 μL aliquot of the RBC suspension; the assay was terminated at 15 s by rapid addition of 1 mL of ice cold stop solution. After centrifugation at 2000 × g for 5 min, the RBC pellet was washed twice with 1 mL stop solution. After final centrifugation, pellets were digested in 0.5 mL of quaternary ammonium hydroxide solubilizing agent (Soluene, Packard, Meriden, CT) for 2 h at 60°C. After cooling to room temperature, samples were bleached with 0.25 mL 30% hydrogen peroxide. The mixture was swirled until all foaming subsided and was subsequently incubated for 30 min at 60°C (20). Five milliliters/sample of Hionic Fluor™ scintillation fluid (Packard) were added, and aliquots were counted with a Packard TR 2300 Scintillation Counter.
Use of barbiturates.
Unless specified otherwise, 50 μL aliquots of PBS containing barbiturate were added to 50 μL aliquots of erythrocyte suspension. The mixture was preincubated for 15 min at 4°C. As an internal control experiment, aliquots of erythrocyte suspension were preincubated with the solvent vehicle used to dissolve barbiturates. These control samples were at vehicle final concentrations corresponding to those reached at each barbiturate concentration tested. Influx was started by adding 100 μL of the radiolabeled 3OMG solution at the specified 3OMG concentrations to the erythrocyte/barbiturate suspension. Barbiturate and unlabeled 3OMG concentrations were adjusted appropriately so as to obtain the final concentrations of interest (0.6 to 10 mmol/L). Influx was terminated after 15 s by the rapid addition of 1 mL ice-cold stop solution. The assay was then continued as described above.
In another group of experiments, in patient 3 and his control, influx was initiated without barbiturate preincubation. For this purpose, 50 μL of PBS containing barbiturate were mixed with 100 μL of radiolabeled 3OMG solution, and at time zero they were added to 50 μL aliquots of erythrocyte suspension to initiate influx. Uptake was terminated after 15 s and the assay continued as described above.
GLUT1 genomic analyses.
DNA analyses were performed in the three patients and their parents as described previously (5, 8). A detailed analysis of molecular techniques and results will be discussed elsewhere (Wang et al. in preparation).
Statistical analyses.
Data are expressed as the mean ± SD. Data analyses and best fits were generated using Origin 228 software (Microcal 228 Software Inc. Northampton, MA).
RESULTS
Zero-trans influx of 3OMG into erythrocytes of those three patients and six parental controls was measured as a function of substrate concentration in the absence of barbiturates. In both patients and controls, the expected saturation kinetics was apparent (insert, Fig. 1). These data are given in a double reciprocal plot of 3OMG concentration versus uptake in Fig. 1. From the linear fit mean Vmax values were, in fmol s−1 (106RBC)−1: controls, 1500 ± 297; patients, 529 ± 21 (35% ± 4% of controls), and apparent Km values were, in mmol/L: controls, 3.2 ± 0.2; patients, 2.3 ± 0.05 (72% of controls).

Zero-trans influx of 3OMG into erythrocytes of GLUT1-deficiency patients (n= 3) and controls (n= 6). Each point represents the mean of patients or controls; individual experiments were run in quadruplicate. The data are presented as a double-reciprocal plot of 3OMG transport velocity (v) in fmol s−1 106RBC−1vs substrate concentration (S) in mmol/L. Insert shows saturation kinetics of transport as velocity (v) vs[S] (0.6–10 mmol/L). Kinetic parameters for transport were determined as linear fits in the double-reciprocal plot.
Time course of 3OMG influx in the presence and absence ofbarbiturates.
To decide on a suitable uptake interval, we followed the time course of 3OMG uptake over a period of 30 s (Fig. 2). For technical reasons, a 15-s time interval was used for all uptake assays. Barbiturates clearly impaired the uptake values in both controls and patients.

Initial time course (0–30 s) of zero-trans influx of 3OMG (0.3 mmol/L) at 4°C into erythrocytes in the presence or absence of barbiturates at a concentration of 10 mmol/L. The results represent the mean of individual experiments (two determinations/datapoint).
Phenobarbital or pentobarbital (10 mmol/L) inhibition of zero-trans3OMG influx.
Erythrocytes were preincubated with barbiturates at a concentration of 10 mmol/L, and 3OMG influx was measured at 15 s in the presence of either phenobarbital (control 1, patients 1 and 2, Fig. 3A) or pentobarbital (control 2, patient 2, Fig. 3B). The pooled data shown in these two figures correspond to the data of Fig. 1, and serve as a graphic baseline to appreciate the effects of barbiturates. Both barbiturates inhibited 3OMG transport; in patients the mean 3OMG influx was reduced to 17% ± 5% SD. The individual data for Vmax and apparent Km values are summarized in Figs. 3C and 3 D; once more the results of the pooled data of Fig. 1 are shown for comparison. Fig. 3C shows the barbiturate-induced decrease of Vmax in both controls and patients, and the fact that pentobarbital is a stronger inhibitor than phenobarbital. With the exception of patient 2, barbiturates did not seem to affect the Km values in either controls or patients.

(A-D) Zero-trans influx of 3OMG ([3OMG]: 0.6–10 mmol/L; 4°C; 15-s interval) into erythrocytes of patients and controls in the presence of phenobarbital (A) or pentobarbital (B) both inhibitors at a concentration of 10 mmol/L. The pooled data of 3OMG influx in the absence of barbiturates shown in Fig. 1 are repeated here for comparison. The data (four determinations/datapoint) is presented as a double-reciprocal plot of 3OMG transport velocity (v) in fmol s−1 106RBC−1, vs substrate concentration [S] in mmol/L. The kinetic parameters determined are shown in (C) (vmax) and (D) (Km).
Influence of barbiturate concentration (0–10 mmol/L) on zero-trans3OMG influx.
Erythrocytes of control 2 and patient 2 were preincubated with phenobarbital or pentobarbital at various concentrations, and 3OMG influx was assayed. The concentration of 3OMG was 5 mmol/L, which approximates the concentration in blood of the physiologic D-glucose substrate. Figure 4A shows that, in control 2, no significant inhibitory effect was observed at concentrations < 1 mmol/L barbiturate. At higher concentrations of barbiturate, 3OMG influx was increasingly inhibited, resulting in a 50% inhibition by phenobarbital and a 70% inhibition by pentobarbital at a concentration of 10 mmol/L. In the patient, even low concentrations of barbiturates inhibited 3OMG influx (note that 100% influx in the patient equals 46% influx of the control, which results in a more shallow slope for patient influx). Influx of 3OMG into the patient's erythrocytes, already impaired to 46% of the control in the absence of barbiturate, was inhibited to 20% of the control value after preincubation with 10 mmol/L phenobarbital. Preincubation of the patient's erythrocytes with pentobarbital 10 mmol/L resulted in total loss of transport. Preincubation with the barbiturate vehicle solution in control 2 did not affect 3OMG influx (Fig. 4A). As in Fig. 3C, the results illustrated in Fig. 4A indicate a stronger inhibitory effect of pentobarbital compared with phenobarbital. Pentobarbital at 500 μmol/L, caused a 46% reduction in uptake activity in the patients' erythrocytes (Fig. 4A). This concentration approximates high doses of pentobarbital in clinical setting (see Table 1).

(A) Barbiturate inhibition of 3OMG influx (5 mmol/L). Isolated erythrocytes of patient 2 (ptnt,○) and control 2 (ctrl,•) were preincubated for 15 min at 4°C containing the indicated concentrations of phenobarbital (full lines), pentobarbital (dotted lines) or vehicle (dashed line). The means of individual experiments (4 determinations/datapoint) are presented and results are expressed as % of control 2 in the absence of barbiturates. (B) Pentobarbital inhibition of 3OMG influx (5 mmol/L) with and without preincubation (patient 3 and control 3).
As mentioned above, Fig. 4A shows that at concentrations of barbiturate lower than 1 mmol/L, inhibition was negligible or absent in the control. This suggested that the 15 min preincubation period with barbiturates might be having a modifying effect on 3OMG uptake. Preincubation was used by Honkanen and colleagues (9), and we adopted their procedure (except that they used 2-deoxyglucose as a substrate). Therefore in patient 3 and his control, 3OMG influx at a concentration of 5 mmol/L was determined with and without preincubation with pentobarbital (Fig. 4B). In the control, preincubation with pentobarbital at lower concentrations (0.3–2 mmol/L) resulted in higher uptake than when substrate and inhibitor were added simultaneously. As the pentobarbital concentration increased further, this difference disappeared at 5 mmol/L and was reversed at 10 mmol/L. A similar trend was observed in the data of patient 3 (Fig. 4B).
Kinetic characteristics of pentobarbital inhibition in a patientwith GLUT1-deficiency.
The inhibition of GLUT1-mediated hexose transport by barbiturates has been reported to be noncompetitive (17) in normal human erythrocytes. However, that study also found that barbiturates bound preferentially to the unoccupied form of the carrier. To analyze the character of pentobarbital inhibition in the setting of GLUT1-deficiency, we analyzed the uptake of 3OMG in patient 2 in the presence of pentobarbital (1, 3, and 10 mmol/L) and found a very pronounced inhibitory effect of pentobarbital on the already impaired GLUT1 transport (data of patient 2, Fig. 5). Vmax was inhibited in a hyperbolic dose-dependent fashion; extrapolation yielded an IC50 of 0.84 mmol/L (insert, Fig. 5).

[S]/ v vs[S] plot of zero-trans influx of 3OMG in patient 2 at the indicated substrate concentrations in the presence of 1, 3, and 10 mmol/L pentobarbital. Note that in this type of plot {[S]/ v=Km/ Vmax + [S]/ Vmax}, the linear fit (y=A+Bx) yields Vmax = 1/ B, Km =A/ B. The insert clarifies the Vmax and Km values vs[inhibitor] (four determinations/datapoint).
The apparent affinity revealed a more complicated pattern. The Km values at 1 and 3 mmol/L of inhibitor concentration (∼1 mmol/L) were comparable to the apparent Km without inhibitor described in the pooled patient data of Fig. 3D. However, at 10 mmol/L pentobarbital, the Km increased to ∼5.6 mmol/L (insert, Fig. 5).
Mutations in the GLUT1 gene.
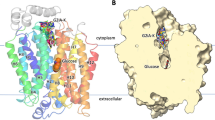
All three patients carried missense mutations in the GLUT1 gene. In Fig. 6, mutations are shown using the 12-helix model for GLUT1 (21). In patient 1, two isolated mutations were identified. Each was on a separate allele. Within the extracellular loop between helices 3 and 4, arginine-126 was replaced with leucine (R126L); and in the cytoplasmic loop connecting helices 6 and 7, lysine-256 was substituted by valine (K256V). This second mutation also was found in the mother's GLUT-1 gene. She is asymptomatic with normal glucose uptake in her isolated erythrocytes. Thus, we believe that this mutation represents a nonpathogenic polymorphism. Further mutagenesis studies in Xenopus Oocyte preparation are underway to confirm this assumption. In patient 2, threonine 310 in helix 8 was replaced by isoleucine (T310I). Lastly, in patient 3, a change from serine-66 to phenylalanine was identified within the extracellular loop connecting helices 1 and 2. Molecular screening of the parents was negative in all cases except the mother of patient 1 as mentioned earlier (vide supra).

Model for the orientation of GLUT1 in the membrane (21). The missense mutations identified in patients 1–3 are indicated as circles.
DISCUSSION
The human brain is entirely dependent upon glucose as an energy source (22–24). GLUT1-deficiency causes impaired glucose transport across the blood-brain barrier resulting in hypoglycorrhachia, seizures and developmental delay (5–8). Identifying potential inhibitors of GLUT1 is essential in preventing further impairment of glucose transport in these children. Barbiturates have been shown to inhibit glucose transport both in vivo and in vitro (9–14). Our present study represents the first in vitro analysis of barbiturate inhibition in the diseased state, and provides evidence for a possible inhibitory effect of both phenobarbital and pentobarbital in patients with this syndrome.
Kinetic data obtained in human erythrocytes are likely to reflect transendothelial cell transport across the blood-brain-barrier, as the human erythrocyte glucose transporter is immunochemically identical to the human brain GLUT1 (25). The effect of barbiturates on glucose transport into human erythrocytes of normal individuals has been studied previously (9, 17, 26). Zero-trans 2-deoxy-glucose and 3OMG influx after preincubation with barbiturates at a concentration of 10 mmol/L was inhibited to ∼50% by phenobarbital, and to ∼80% by pentobarbital (9, 17). Both reports observed a significant reduction of Vmax with no change in affinity. These findings are in agreement with our control data (Fig. 4A). Mechanisms thought responsible for the barbiturate effect are a decrease in the number of functional transporters (e.g. by translocation to the intracellular compartment) and/or a decrease in the intrinsic activity by interacting with the occupied or unoccupied GLUT-1 transporter.
In GLUT-1 patients, transport function is about half the normal, as shown by immunoblotting, cytochalasin B binding and glucose influx studies in the past (5–8). Most of these studies have been done (8) or are under way for the three patients studied here (Fig. 1), and show similar results. At this point, it is unclear whether the functional transporters are derived from the unmutated allele only, or represent a combination of transcription of both the unmutated and the mutated allele. The molecular aspects of these and other GLUT1 mutations are the subject of a separate communication (Dong et al. in preparation); the mutation in patient 2 has been discussed previously (8).
Our data show that the patients' transporters maintain sensitivity to barbiturates (Figs. 3–5). The effect of barbiturates is modulated by the presence of the specific GLUT1 substrate-3OMG (Figs. 3 and 5), which suggests that barbiturates interact directly with GLUT1, an observation shared with other investigators (17). In the absence and the presence of barbiturates, the apparent Km value in most patients and controls was 2–4 mmol/L (Fig. 3D, and De Vivo et al. unpublished data). This range resembles Km values reported in the literature for zero-trans influx conditions similar to those used here (27). There was an interesting exception in the case of patient 2; in the presence of 0, 1, and 3 mmol/L pentobarbital, Km values were as described immediately above, but at 10 mmol/L of pentobarbital, Km increased to ∼5.6 mmol/L. This observation cannot be explained fully at the moment; one might speculate that the mutation in this patient (T310I in helix 8, Fig. 6) might affect the pattern of pentobarbital-GLUT1 interaction at higher pentobarbital concentrations, assuming that the mutant transporter is functional. To be noted, this increased Km was not seen with phenobarbital (Fig. 3D). Overall, the affinity of the transporter for its substrate appears unchanged by barbiturates in both the normal and the GLUT1 deficient state.
Our data indicate that a 15-min preincubation period with barbiturates alters 3OMG influx (Fig. 4B). As shown, preincubation with relatively low pentobarbital concentrations (0.3–2 mmol/L) results in higher uptake values. Because without preincubation, inhibition is evident during a 15-s uptake interval (Fig. 4B, dotted lines), we hypothesize that the inhibition is due to pentobarbital interacting with the exofacial side of GLUT1. To account for the decreased inhibition after preincubation (Fig. 4B, solid lines), we presume that the relatively long preincubation period would allow the lipid-soluble pentobarbital to penetrate the plasma membrane and reach the intracellular compartment. Once there, pentobarbital could either 1) induce a conformational change in GLUT1 that decreases extracellular pentobarbital binding, or 2) recruit or “lock” an increased proportion of GLUT1 into the outward-facing conformation, thus increasing the numbers of available transporters for zero-trans influx. At 5 mmol/L and higher pentobarbital concentrations, this effect disappears and reverses, as if the increased extracellular inhibitor might be blocking larger numbers of outward facing conformers. Lastly, the site(s) of interaction of barbiturate with GLUT1 are not known. From Fig. 4B, this effect appears less marked in the patient than in its control; in other words, preincubation has less inhibition in the patient at low pentobarbital concentrations. It is therefore conceivable that the S66F mutation might have increased the affinity of GLUT1 for pentobarbital. This speculation assumes that the mutant transporter is functional. In contrast to serine, phenylalanine is more hydrophobic and hence presumably better able to interact with pentobarbital; moreover, the position of the mutation at residue 66 would place it at the end of the first predicted extracellular loop (Fig. 6), where it could interact with extracellular pentobarbital.
In the case of patient 1, both mutations (R126L and K256V) turn positively charged residues into hydrophobic ones in predicted exofacial and endofacial loops of GLUT1 (Fig. 6). Again, assuming that the mutant transporter is functional, this might have resulted in increased interactions with barbiturates. The profound inhibition of 3OMG uptake seen in the presence of phenobarbital in this case (∼60% inhibition versus∼40% in its control, Fig. 3C) is consistent with this notion.
Several in vivo studies of glucose transport across the blood-brain barrier via GLUT1 in animals and humans have been performed under barbiturate anesthesia (for reviews, see (24), (28), and (29). The rates of transport they reported might be lower than normal because of barbiturate inhibition of intracerebral glucose transport under general barbiturate anesthesia. In fact, there is a report of a 40% decreased maximal glucose transport capacity into rat brain under pentobarbital anesthesia when compared with normal (10) that was attributed to decreased cerebral blood flow under anesthesia and other factors unknown at the time. Lastly, there is a comprehensive study of in vivo transfer of glucose across the blood-brain barrier of rats (11) that suggested that pentobarbital (50 mg/kg i.p.) decreases both the Vmax and the Km of glucose transport. These reports together with our current evidence strongly support the notion that barbiturates can aggravate the clinical consequences of an already compromised glucose transport system in GLUT1-deficiency.
In brain, GLUT1 mediates transport 1) across the brain capillary endothelium, and 2) from the interstitial fluid into astrocytes, oligodendrocytes, and neurons (27). The mode of transport differs. In the first case, since the glucose concentration is sizable on both sides of the endothelium, transport is close to equilibrium exchange conditions. In the second case, intracellular glucose concentration is very low and transport is practically zero-trans. In GLUT1-deficiency, given that the glucose concentration in CSF (and presumably brain interstitium) is about half the normal level, conditions will drift toward zero-trans for the endothelium as well. Our data have all been obtained under zero-trans conditions, which makes them representative of the expected clinical conditions. In addition, a previous report (17) suggests that in normal human erythrocytes, inhibition is greater at zero-trans than at equilibrium exchange conditions. This implies that even when no barbiturate effect is seen in normal individuals, cellular glucose uptake in the brain of these patients could still be greatly impaired by these drugs (cf. Fig. 4A).
Barbiturates are the treatment of choice for infantile seizures (30), the dominant clinical feature of GLUT1-deficiency. Furthermore, clinical use of barbiturates includes sedation, sleep induction, treatment of hyperbilirubinemia in neonates, pentobarbital coma in status epilepticus (31), and anesthesia for neurosurgery to lower elevated intracranial pressure (32–34). In vivo concentrations of barbiturates (Table 1) (35) are at the lower end of the concentrations [0–10 mmol/L] used in our assays for reproducibility of previous studies (9). Inhibition in vivo is probably partial at therapeutical or even toxic concentrations, but even that could potentiate the clinical effect of GLUT1-deficiency. In addition, barbiturate levels in brain, due to their lipophilic nature, are supposedly higher than in plasma. The concentration at which partial inhibition is observed in our study is consistent with the estimated brain levels of barbiturates in both animal and clinical studies (36, 37). The observation of barbiturate inhibition in GLUT1-deficiency at concentrations that do not yet affect normal individuals (Fig. 4A) is further supported by clinical data. Eighteen of 20 patients diagnosed with GLUT1-deficiency received phenobarbital for seizure control (De Vivo et al. in preparation). In 17 patients (including the 3 in our current study), no benefit was observed and seizure activity increased; only one patient has been apparently reported to have benefited from phenobarbital (De Vivo, unpublished observations). These clinical data therefore are quite consistent with our present in vitro findings. No data about the response of these patients to barbiturate anesthesia is available.
We conclude that barbiturates inhibit the intrinsic activity of the GLUT1 carrier in normal individuals and in GLUT1-deficiency. By implication, these results suggest that impaired glucose transport across the blood-brain barrier in these patients is further aggravated by the inhibitory effect of barbiturates. The use of barbiturates in general anesthesia or for seizure control might put these patients at further risk. A GLUT1-deficient mouse model is currently being developed to investigate this concern in vivo.
Abbreviations
- GLUT1:
-
facilitated glucose transport protein isoform 1
- 3OMG:
-
3-O-Methyl-D–Glucose
- RBC:
-
red blood cells, erythrocytes
- Vmax:
-
maximal transport velocity
- Km:
-
substrate affinity
References
Pardridge WM, Boado RJ, Farrell CR 1990 Brain-type glucose transporter (Glut-1) is selectively localized to the blood-brain-barrier. J Biol Chem 265: 18035–18040
Baldwin SA 1993 Mammalian passive glucose transporters: members of active and passive transport proteins [review]. Biochim Biophys Acta 1154: 17–49
Bell IG, Burant CF, Takeda J, Gould GW 1993 Structure and function of mammalian facilitative sugar transporters [review]. J Biol Chem 268: 19161–19164
Gould GW, Holman GD 1993 The glucose transporter family: structure, function and tissue-specific expression [review]. Biochem J 295: 329–341
Seidner G, Garcia-Alvarez M, Yeh JI, O'Driscoll KR, Klepper J, Stump TS, Wang D, Spinner NB, Birnbaum MJ, De Vivo DC 1998 Glut-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet 18: 1–4
De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI 1991 Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med 325: 703–709
De Vivo DC, Garcia-Alvarez M, Ronen G, Trifiletti R 1995 Glucose transport protein deficiency: an emerging syndrome with therapeutic implications. Int Pediatr 10: 51–56
Klepper J, Wang D, Fischbarg J, Vera JC, Jarjour IT, O'Driscoll KR, De Vivo DC 1999 Defective glucose transport across brain tissue barriers: a newly recognized neurological syndrome. Neurochem Res 24: 587–594
Honkanen RA, McBath H, Kushmerick C, Callender GE, Scarlata SF, Fenstermacher JD, Haspel HC 1995 Barbiturates inhibit hexose transport in cultured mammalian cells and human erythrocytes and interact directly with purified GLUT-1. Biochemistry 34: 535–544
Gjedde A, Rasmussen M 1980 Pentobarbital anesthesia reduces blood-brain glucose transfer in the rat. J Neurochem 35: 1382–1387
Ingwar M, Abdul-Rahman A, Siesjo BK 1980 Local cerebral glucose consumption in the artificially ventilated rat: influence of nitrous oxide analgesia and phenobarbital anesthesia. Acta Physiol Scand 109: 177–185
Otsuka T, Wei L, Acuff VR, Shimizu A, Pettigrew KD, Patlak CS, Fenstermacher J 1991 Variation in local cerebral blood flow response to high-dose pentobarbital sodium in the rat. Am J Physiol 261: H110–H120
Otsuka T, Wei L, Bereczki D, Acuff VR, Patlak CS, Fenstermacher J 1991 Pentobarbital produces dissimilar changes in glucose influx and utilization in brain. Am J Physiol 261: R265–R275
Nilsson L, Seisjo BK 1975 The effect of phenobarbitone anesthesia on blood flow and oxygen consumption in the rat brain. Acta Anaesthesiol Scand 57: 18–24
Saija A, Princi P, De Pasquale R, Costa G 1989 Modifications of the permeability of the blood-brain barrier and local cerebral metabolism in pentobarbital- and ketamine-anaesthetized rats. Neuropharmacology 28: 997–1002
Todd MM, Warner DS 1992 A comfortable hypothesis reevaluated: cerebral metabolic depression and brain protection during ischemia. Anesthesiology 76: 161–164
el-Barbary A, Fenstermacher JD, Haspel HC 1996 Barbiturate inhibition of GLUT-1 mediated hexose transport in human erythrocytes exhibits substrate dependence for equilibrium exchange but not unidirectional sugar flux. Biochemistry 35: 15222–15227
Wei L, Otsuka T, Acuff V, Bereczki D, Pettigrew K, Patlak C, Fenstermacher JD 1993 The velocities of red cell and plasma flows through parenchymal microvessels of rat brain are decreased by pentobarbital. J Cereb Blood Flow Metab 13: 487–497
Lowe AG, Walmsley AD 1986 The kinetics of glucose transport in human red blood cells. Biochim Biophys Acta 857: 146–154
Naftalin RJ, Holman GD 1977 Transport of sugars in human red cells. In: Ellory JC, Lew VL (eds) Membrane Transport in Red Cells. Academic, New York, pp 257–300
Mueckler M, Caruso C, Baldwin SA, Panico M, Blench I, Morris HR, Allard WJ, Lienhard GE, Lodish HF 1985 Sequence and structure of a human glucose transporter. Science 228: 941–945
Chugani HT, Phelps ME, Mazziotta JC 1978 Positron emission tomography study of human brain functional development. Ann Neurol 22: 487–497
Clarke DD, Sokoloff L 1994 Circulation and energy metabolism of the brain. In: Siegel GJ (ed) Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 5th Ed. Raven Press, New York, pp 645–748
Cremer JE 1982 Substrate utilization and brain development (review). J Cereb Blood Flow Metab 2: 394–407
Dick APK, Harik SI, Klip A, Walker DM 1984 Identification and characterization of the glucose transporter of the blood/brain barrier by cytochalasin B binding and immunological reactivity. Proc Natl Acad Sci USA 81: 7233–7237
Salah-Abu KM, Hampton KK, Findlay JBC 1982 The effects of general anesthetics on glucose and phosphate transport across the membrane of the human erythrocyte. Biochim Biophys Acta 688: 163–168
Vannucci SJ, Maher F, Simpson IA 1997 Glucose transporter proteins in brain: delivery of glucose to neurons and glia [review]. Glia 21: 2–21
Ito M, Miyaoka M, Ishii S 1984 Alterations in local cerebral glucose utilization during various anesthesia - the effect of urethane and a review. No To Shinkei 36: 1191–1199
Sokoloff L, Reivich M, Kennedy CM, Des Fosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M 1977 The [14C]deoxy-glucose method of the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized rat. J Neurochem 29: 897–916
Volpe JJ 1995 Neurology of the Newborn. WB Saunders Company, Philadelphia, pp 195
Haslam RHA 1996 The nervous system. In: Behrman RE, Kliegman RM, Arvin AM (eds). Nelson Textbook of Pediatrics. Saunders, Philadelphia, pp 1699
Anonymous 1996 The use of barbiturates in the control of intracranial hypertension. Brain Trauma Foundation review. J Neurotrauma 13: 711–714
Shapiro HM 1975 Intracranial hypertension: therapeutic and anesthetic considerations. Anesthesiology 43: 445–471
Willow M, Johnston GAR 1983 Pharmacology of barbiturates: electrophysiological and neurochemical studies. Int Rev Neurobiol 24: 15–49
Nelson WE 1996 Nelson textbook of pediatrics. WB Saunders Company, Philadelphia, pp 2057
Richter JA, Waller MB 1975 Effects of pentobarbital on acetylcholine content and release in different regions of rat brain. Biochem Pharmacol 26: 609–615
Richter JA, Holtman JR Jr 1982 Barbiturates: their in vivo effects and potential biochemical mechanisms. Prog Neurobiol 18: 275–319
Acknowledgements
We are grateful for the skillful assistance of Pamela Kranz-Eble in the laboratory, helpful discussions with Kevin O'Driscoll, Ph.D. and Anton Zellner, M.D., and the generous support of David Diuguid, M.D., Hematology, Columbia Presbyterian Hospital for blood cell counts. CPD-anticoagulant solution was generously provided by Baxter Healthcare Corporation, Fenwal Division, Deerfield, IL 60014.
Author information
Authors and Affiliations
Additional information
This work was supported in part by the Colleen Giblin Charitable Foundation for Pediatric Neurology Research (D.C.D.), the Will Foundation (D.C.D.), USPHS grants NS37949–01 (D.C.D.), EY08918 (J.F.) and CA30388 (J.C.V.), Research to prevent blindness, Inc. (J.F.), Memorial Sloan-Kettering Cancer Center institutional funds (J.C.V.), and the Deutsche Forschungsgemeinschaft (J.K.).
Rights and permissions
About this article
Cite this article
Klepper, J., Fischbarg, J., Vera, J. et al. GLUT1-Deficiency: Barbiturates Potentiate Haploinsufficiency in Vitro. Pediatr Res 46, 677 (1999). https://doi.org/10.1203/00006450-199912000-00006
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199912000-00006
This article is cited by
-
Experience in the Use of a Ketogenic Diet in a Patient with Type I Glucose Transporter Deficiency Syndrome (clinical observations)
Neuroscience and Behavioral Physiology (2016)
-
Dietary Treatments and New Therapeutic Perspective in GLUT1 Deficiency Syndrome
Current Treatment Options in Neurology (2014)
-
Phenotypic Spectrum of Glucose Transporter Type 1 Deficiency Syndrome (Glut1 DS)
Current Neurology and Neuroscience Reports (2013)
-
Effects of single-nucleotide polymorphisms in the human holocarboxylase synthetase gene on enzyme catalysis
European Journal of Human Genetics (2012)
