
Abstract
The conversion of labeled formate to methionine and serine, as a measure of remethylation of homocysteine to methionine and folate coenzyme cycling, has been studied in control and mutant human fibroblasts. Fibroblasts in monolayer culture were incubated with [14C]formate, and labeled methionine sulfone and serine were determined in hydrolysates of oxidized cell proteins. In control cells, methionine and serine were clearly measurable (n = 21, 1.7-5.5 and 2.4-9.7 nmol/mg protein/16 h, respectively). In contrast, methionine formation was reduced in cells from patients with methylenetetrahydrofolate reductase (MR) deficiency (MR mutant, n = 11, 0.05-0.44), combined methylmalonic aciduria/homocystinuria[cobalamin(cbl)C/D mutant, n = 12, 0.014-0.13), and methionine synthase deficiency (MS mutant, n = 3, 0.04-0.23). Furthermore, serine formation was low in cblC/D mutant (0.08-0.98) and MS mutant(0.17-0.94) cells, but normal or high in MR mutant cells (5.2-11.4). Growth of cblC/D mutant cells in medium supplemented with high concentrations of hydroxo-cbl resulted in significant increases of both methionine and serine formation. Taken together these findings provide clear evidence for the existence of the formate to serine pathway described by W. B. Strong and V. Schirch in cultured fibroblasts and indicate that disturbed MS function due to a specific genetic disorder is associated with reduced serine formation in vitro, which reflects availability of reduced folate coenzymes. The correction of this defect by vitamin B12 alone, in cblC/D mutant cell lines, correlates well with the clinical response in the patients and fits in well with the idea that reduced availability of folate coenzymes occurs in functional MS deficiency, in agreement with the methyl trap hypothesis.
Similar content being viewed by others


Main
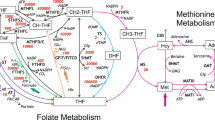
Regeneration of methionine from homocysteine plays a vital role in humans in maintaining adequate methionine levels, in the recycling of one carbon units for S-adenosylmethionine-dependent methylation reactions, as well as in the removal of potentially toxic homocysteine(2). This regeneration depends on the function of B12 requiring MeTHF:homocysteine methyltransferase (MS, EC 2.1.1.13,Fig. 1, reaction 1). MeTHF is produced from tetrahydrofolate by the action of a series of enzymes, the last of which is MR(EC 1.1.1.68, Fig. 1, reaction 5). Once formed, MeTHF must be demethylated to allow recycling of intracellular reduced folates(3).

Interaction of folate and methionine metabolism. The numbers represent the following enzymes or sequences: 1, MeTHF:homocysteine methyltransferase; 2, 10-formyltetrahydrofolate synthase; 3, 5,10-methenyltetrahydrofolate cyclohydrolase;4, 5,10-methylenetetrahydrofolate dehydrogenase; 5, MR;6, serine hydroxymethyltransferase; 7, Protein synthesis;MeCbl, methyl-cbl; *radioactively marked compounds in the assay of methionine and serine formation. The shaded areas denote the substrates and products in this assay.
Disturbance of MS activity leads to a variety of disease states. Inherited enzyme deficiencies that affect the function of MS lead to homocystinuria and low to normal plasma methionine levels. MS deficiency is associated with methylmalonyl CoA mutase (EC 5.4.99.2) deficiency in combined homocystinuria and methylmalonic aciduria due to the cblC, D, or F defect in which there is defective synthesis of both vitamin B12 coenzymes, deoxy-adenosyl-cbl and methyl-cbl(4). Isolated MS deficiency, associated with reduced synthesis of methylcbl, occurs in the cblE and cblG defects(5). These disorders usually result in neurologic abnormalities together with megaloblastic anemia, and all patients have shown some degree of clinical and biochemical improvement with vitamin B12(cbl), although the long-term outcome is poor(6).
MR deficiency which results in low levels of MeTHF is usually associated with severe neurologic abnormalities without hematologic abnormalities, and most patients have not responded to vitamin therapy(3). This disorder results in an abnormal distribution of folates in cultured fibroblasts(7) and liver and kidney(8) with normal intracellular folate concentrations but a much reduced proportion of the 5-methyl form. In contrast there was a reduced total folate concentration with a high proportion of the 5-methyl form in liver and kidney of a patient with MS deficiency due to the cblC defect(8).
Elevated plasma homocyst(e)ine has been well documented in both vitamin B12(9) and folate deficiency(10), reflecting disturbed MS function. In addition MS is inactivated dramatically in rats exposed to nitrous oxide(11). Administration of this gas has led to megaloblastic anemia in humans and to subacute combined degeneration of the spinal cord in monkeys, both mimicking clinical signs of MS deficiency states(12). In one related study, reduced purine synthesis was shown to accompany lowered MS activity in cultured lymphoblasts exposed to nitrous oxide(13). In addition, 14C-labeled formate was shown to be converted to serine, and this was lower in nitrous oxide-treated cells. It is likely that serine formation is due to the activity of serine hydroxymethyltransferase (EC 2.1.2.1). This enzyme has been characterized from rabbit liver(1) and is the final step in the conversion of formate to serine, a process dependent on the availability of reduced folate coenzymes. Previous studies have shown reduced synthesis of methionine from [14C]formate(14) and [35S]homocysteine(15) in cultured fibroblasts from patients with MR deficiency.
We have studied the formation of methionine and serine from radioactively labeled formate as a measure of both MS function and availability of reduced folates in intact fibroblasts in monolayer culture. These activities have been compared in fibroblasts from patients with genetic deficiencies of either MS or MR. This system provides an additional model to nitrous oxide-treated rats or lymphoblasts for the study of interactions of folate and cbl and also provides a valuable diagnostic tool.
METHODS
Cell culture. Cultured skin fibroblasts were established from pinch biopsies obtained with the informed consent of patients, or when appropriate their parents, and grown in Eagle's minimum essential medium containing undialyzed FCS (10% vol/vol), nonessential amino acids (1% wt/vol), and kanamycin (100 mg/L). This medium contains 1 mg/L folic acid and approximately 50 ng/L vitamin B12, which originates from the FCS. Cultures were maintained at 37°C in a 5% CO2/95% air atmosphere and periodically tested to exclude mycoplasma contamination. Cells were subcultured up to a maximum of 25 times or harvested for assay using trypsin(0.25% wt/vol)(16). Fibroblasts were set up for intact cell assay or harvested for specific enzyme assay (stored at -70°C) when just confluent, except for assay of MR, in which case cells were harvested 3 d after confluence. In some experiments the medium was changed after 4 d and replaced by medium supplemented with various vitamins or cofactors.
Specific enzyme assays. Cell pellets were disrupted either by sonication in a 10-fold dilution of the buffer used in the enzyme assay or by standing in the same buffer containing Lubrol (0.15%, wt/vol) for 30 min at 4°C. The supernatant obtained by centrifugation at 10,000 ×g for 10 min at 4°C was used for assays.
MR activity was determined by the method described by Rosenblatt and Erbe(17) except that the radioactive substrate was preextracted with dimedone and toluene to reduce radioactivity in the blank. The total assay volume was 100 μL, and extraction of the product formed in the reaction was achieved with 1 mL of toluene.
MS was assayed essentially as described previously(18) except that the separation of radioactive methionine produced in the assay was achieved batchwise with Dowex-1 anion exchange resin. Serine hydroxymethyltransferase activity was performed by measuring the conversion of [14C]serine and tetrahydrofolate to 5,10-methylenetetrahydrofolate as described by Taylor and Weissbach(19) except that DTT was used instead ofβ-mercaptoethanol as a reducing agent. Enzyme activities were related to protein determined by the Lowry method(20).
Intact cell assay of methionine and serine formation. The previously described method(15) for the determination of methionine formation from homocysteine in fibroblast monolayer cultures was modified by using [14C]formate as the labeled precursor instead of[35S]homocysteine. Duplicate 75-cm2 flasks of confluent cells were washed with 5 mL of Earle's basic salt solution, then incubated for 16 h at 37°C with 5 mL of Earle's basic salt solution that contained[14C]formate (2 μCi/mL), sodium formate (250 μmol/L), and L-homocysteine (200 μmol/L) prepared freshly from its thiolactone as previously described(16). Cells were harvested with 0.25% wt/vol trypsin solution, and proteins were precipitated with 5% trichloroacetic acid and oxidized with performic acid, then hydrolyzed with 6 M hydrochloric acid. Labeled amino acids were separated by high voltage paper electrophoresis, and radioactivity in the area of the electrophoretogram corresponding to oxidized methionine and serine standards, was quantitated by liquid scintillation counting(15). In some experiments the identity of the radioactively labeled amino acids was confirmed by analysis of hydrolysates, which were desalted on cation exchange resin using two-dimensional thin layer chromatography(21). After drying, chromatograms were stained with ninhydrin, and the areas corresponding to the individual amino acids were cut out and counted by liquid scintillation counting. Incorporation of label from [14C]propionate into protein was assayed in intact fibroblast monolayer cultures(22).
Cell lines. Control skin fibroblasts were obtained from healthy controls, from patients with a disease not related to homocysteine metabolism, or from their parents. Eleven cell lines from patients with clinical and biochemical evidence of MR deficiency were studied (designated MR mutant). Twelve cell lines were studied from patients with increased urinary or plasma homocystine and methylmalonic acid and accompanying clinical features(classified as cblC/D mutants). Three cell lines were studied from patients with biochemical and clinical features consistent with a functional deficiency of MS without methylmalonic aciduria. The cell lines were sent from various centers as follows: Dr. A. Burlina, Padua Italy, three lines; Dr. R. Cerone, Genoa Italy, five lines; Dr. A. Green, Birmingham UK, three lines; Dr. Knapp, Braunschweig, Germany; Dr. A. Kohlschutter, Hamburg Germany; Dr. E. Kvittingen, Oslo Norway; Prof. J. Leonard, London UK, three lines; Dr. H. Mandel, Haifa Israel; Dr. J. R. Moore, Leicester UK; Dr. I. B. Sardharwalla, Manchester UK; Dr. R. Schutgens, Amsterdam Netherlands, two lines; Dr. Shawinsky, Johannesburg Republic of South Africa Mr. R. Smith, Bradford UK, two lines; Dr. G. Zeman, Prague, Czech Republic.
Materials and chemicals. Medium, reagents, and disposable plastic ware for tissue culture were obtained from GIBCO Europe.[14C]Formate and 5-[14C]methyltetrahydrofolic acid were purchased from Amersham Corp. Hydroxo-cbl was obtained from Fluka Chemicals. All other chemicals were of the highest purity commercially available from Sigma Chemical Co. or Merck.
RESULTS
Cell line characteristics. MR activities in the 11 cell lines from patients with biochemical and clinical features of this enzyme deficiency and controls are shown in Table 1. Activities were greatly reduced in each patient cell line both with and without FAD, although activities tended to be higher in the presence of added coenzyme as also seen in controls. In mutant cells the level of activity assayed without FAD was 0-2.3% of the mean control value, confirming their classification as MR mutants, whereas activity was less than 6% of control when assayed with added FAD in all but two cell lines, which exhibited appreciable residual activity of 8 and 14%, respectively, of control values.
Incorporation of label from [14C]propionate into trichloroacetic acid-precipitable proteins in cells grown in normal and hydroxo-cbl-supplemented medium in the group of cell lines from patients with clinical and biochemical features of combined methylmalonic aciduria and homocystinuria is shown in Table 2. This activity was reduced in all cell lines when grown in normal medium ranging from 9 to 32% of the mean control value. Importantly this activity increased considerably in each cell line when grown in hydroxo-cbl-supplemented medium, reaching between 68 and 562% higher values in supplemented compared with values in basal medium. These findings together with homocystinuria in the patients confirm the combined defect and allow classification of these cell lines as cblC/D mutants.
Three cell lines were studied from patients with clinical features suggestive of a disorder of MS function with hypomethioninemia but without methylmalonic aciduria. In cells from these three patients MS activity was virtually absent in one cell line, which was shown by complementation studies to belong to the cblG class. The two other cell lines exhibited normal activity in the presence of excess, but reduced activity in the presence of low, concentrations of reducing agent and were confirmed as cblE by complementation studies.
Analysis of radioactively labeled amino acids. This is illustrated in hydrolysates of cell proteins by high voltage electrophoresis in Figure 2. In the control, clear peaks of radioactivity with migration which corresponds exactly with that of standards of methionine sulfone and serine are evident. In contrast, both peaks are much smaller in the cells from a cblC patient, whereas in the MR-deficient cells, low counts in the methionine position are accompanied by a large peak in the position of serine. In representative control cell lines two-dimensional thin layer chromatography of cell protein hydrolysates revealed a high number of counts in the positions of serine and methionine sulfone, confirming the identity of these two labeled amino acids. In contrast, only a few counts above background were found in the positions of threonine, aspartic acid, glycine, and histidine, whereas no radioactivity was detected in the remaining protein amino acids.

Analysis of labeled amino acids in cell proteins. Fibroblast monolayers were incubated with [14C]formate, and cell proteins were precipitated, oxidized, hydrolyzed, and analyzed by high voltage electrophoresis as described in “Methods.” The positions of marker methionine sulfone (oxidation product of methionine, MetO2) and serine (Ser) are indicated by broken lines: A, control cell line; B, cblC mutant cell line; C, MR mutant cell.
Formation of both methionine and serine was shown to be linear with time up to 24 h in duplicate flasks of confluent control fibroblasts. The coefficients of variation (R2) were 0.983 and 0.986 for methionine and serine, respectively.
Formation of methionine and serine in control and mutant fibroblasts. The formation of methionine and serine, in cells grown in normal medium from patients with the three types of defect and controls, is shown in Table 3 and Figure 3.Table 3 summarizes the range, mean, and SD of values obtained in control and mutant cells, whereas the individual values are shown in Figure 3.

Formation of methionine and serine in individual control and mutant cells grown in normal medium. Cells were incubated with labeled formate and harvested, and proteins were precipitated, oxidized, hydrolyzed, and analyzed by high voltage electrophoresis as described in“Methods.” Note the logarithmic scale.
The control cells show clearly measurable formation of both methionine and serine with clearly, similarly reduced methionine formation in each type of remethylation defect with a mean value of 7.5, 3, and 3.5% of the mean control value for MR, cblC/D, and MS mutant cells, respectively. In both cblC/D and MS mutant cells, there is a simultaneous reduction of labeled serine formation of 9.5 and 10% of the mean control value, respectively. In contrast, MR mutant cells show on average 175% higher serine formation than control cells, although most values fall within the upper limit of the control range. Inspection of the values in the individual cell lines (Fig. 3) reveals that methionine formation is clearly lower than the lowest control value in all mutant cells (at the most 25%), whereas serine formation is also reduced in every cblC/D and MS mutant cell line (at the most 41%) without overlap.
The formation of methionine in 10 and serine in 9 of the cbl mutant cells, grown for 3 d in various concentrations of hydroxo-cbl before assay, is shown in Figure 4. All cell lines showed a significant increase of the reduced methionine and serine formation in the higher B12-containing media, sometimes reaching the control range values. Most required the highest concentration of 1000 μg/L to elicit the maximum response, whereas exceptional cell lines did this even at 100 μg/L.

Formation of methionine and serine in cells grown with varying amounts of cbl in the medium. Cells were grown for 4 d in normal medium then for 3 d in medium supplemented with hydroxo-cbl at the depicted concentrations. Cells were incubated with labeled formate and harvested, and proteins were precipitated, oxidized, hydrolyzed, and analyzed by high voltage electrophoresis as described in “Methods.”
Serine hydroxymethyltransferase activity in control cultured fibroblasts. Clearly measurable serine hydroxymethyltransferase activity was found in 10 control cell lines with a slight stimulation of activity in the presence of added pyridoxal 5′-phosphate (Table 4). This activity was dependent on time with linearity up to 60 min and on the amount of cell extract protein included in the assay with linearity up to 230 μg of protein (Fig. 5). The relation of activity to varying concentrations of the two substrates is shown in Lineweaver-Burke linear plots in Figure 6. These kinetic studies revealed Km values of 0.47 mmol/L for serine and 0.38 mmol/L for tetrahydrofolate.

Effect of varying incubation time (A) and varying amount of cell extract (B) on serine hydroxymethyltransferase activity in cultured fibroblasts. The activity was measured as described under “Methods.” In experiment A, the protein content was 113 μg and in experiment B the incubation time was 60 min.

Effect of varying substrate concentrations on serine hydroxymethyltransferase activity in control cultured fibroblasts.(A) Varied serine concentrations with tetrahydrofolate(THF) maintained at 2 mmol/L. (B) Varied THF concentration with serine maintained at 1.25 mmol/L.
DISCUSSION
These studies using cultured fibroblasts from patients with clinical, biochemical, and enzymologic evidence of three different types of remethylation defect provide evidence for the presence of the involved pathways and information on their disturbance in this in vitro model system. Incorporation of label from [14C]formate into methionine and serine, but not to any significant extent in other amino acids, reflects the existence in cultured fibroblasts of the pathway from formate to MeTHF illustrated in Figure 1. This probably involves the coupled cyclic system described by Strong and Schirch(1) based on studies in rabbit liver. In this system formate is converted to 5,10-methylenetetrahydrofolate by the action of 10-formyltetrahydrofolate synthase, 5,10-methenyltetrahydrofolate cyclohydrolase, and 5,10-methylenetetrahydrofolate dehydrogenase. In the next step, 5,10-methylenetetrahydrofolate is reduced by MR to MeTHF, whose methyl group, derived from formate, is transferred to homocysteine by MS. Finally, 5,10-methylenetetrahydrofolate donates its C1 unit to glycine, catalyzed by serine hydroxymethyltransferase, yielding serine and releasing tetrahydrofolate, which can react once more with formate, thereby completing this cycle. That serine formation reflects activity of serine hydroxymethyltransferase is supported by the demonstration, in this study, of the presence of this enzyme activity in cultured fibroblasts by specific assay.
The clearly reduced formation of labeled methionine, using labeled formate as a precursor, in cells with each of the different remethylation defects indicates the critical role of both MR and MS in remethylation of homocysteine. This extends previous observations of defective methionine formation in MR mutants using labeled homocysteine(15), or labeled formate(14).
An important additional finding in the present study is the clearly reduced formation of serine in cells with deficient MS activity, so far shown only in nitrous oxide-treated lymphoblasts(13). This is explained by a lack of availability of reduced folate coenzymes, especially 5,10-methylenetetrahydrofolate, as shown in the liver of such patients(8) due to lack of release of C1 units from MeTHF,i.e. methyl group trapping. This idea is supported by the opposite finding of elevated serine formation in MR-deficient cells. Presumably this is due to 5,10-methylenetetrahydrofolate accumulation, which would be expected because non-methyl forms of folate accumulate in tissues and fibroblasts in this defect, leading to increased serine hydroxymethyltransferase activity.
Taken together these findings in control and different types of mutant cells provide clear evidence for the existence of the formate to serine pathway of Strong and Schirch(1) in cultured fibroblasts. They also provide further evidence that reduced availability of reduced folate coenzymes is associated with disturbed MS function in cell lines with a specific genetic disorder.
There is much evidence that reduced availability of folate coenzymes occurs in functional MS deficiency due to the so-called methyl trap hypothesis(12). According to this theory, once MeTHF is formed it cannot be reoxidized by MR, because this reaction is irreversible under physiologic conditions. The ensuing functional folate deficiency leads to low levels of folate coenzymes, thus impairing other folate-dependent reactions. However, this theory has been reputed by some workers who provided evidence that disturbed folate metabolism in cbl deficiency is best explained by formate starvation rather than methyl group trapping(23, 24). Also the reduction of quinonoid dihydrobiopterin by MeTHF, catalyzed by MR, has been proposed as an alternative mechanism for recycling of reduced folate coenzymes(25).
This genetic model of MS deficiency, in a system plentiful in other related nutrients and with normal activities of other enzymes, clearly demonstrates the consequences of a single enzyme block on folate coenzyme availability. Importantly, the correction of MS activity, in the cells with the cbl defect, by hydroxo-cbl was also associated with improvement of serine formation and presumably correction of the deficiency of reduced folates. Thus in this study vitamin B12 corrected methyl trapping, independent of changes in other constituents of the medium, for example folate. The idea that formate deprivation is the cause of decreased availability of reduced folates seems less likely, because methionine and formate levels in this culture system and assay, respectively, are relatively high and no different in those experiments where hydroxo-cbl improved both MS activity and serine formation. This idea is supported by other studies, for example the observation by Hoffbrand and Jackson(26) that the DNA synthesis defect in vitamin B12 deficiency can be corrected by tetrahydrofolate administration.
The increase of the abnormally low serine formation in response to supplementation with hydroxo-cbl in the cblC/D mutant cell lines correlates well with the clinical response in the patients. For example, the megaloblastic anemia, which is a key feature of the cblC/D defect(3, 27), probably results from low availability of reduced folates and consequent scarcity of purine and pyrimidine precursors of DNA synthesis. Correction of this defect in vivo and in vitro by vitamin B12 provides strong evidence that the low serine formation in fibroblasts reflects low reduced folates in these mutant cells with a functional deficiency of MS and therefore fits in well with the methyl trap hypothesis.
Finally these studies demonstrate that normal and mutant cultured skin fibroblasts provide an appropriate model for the study of the interaction of methionine and folate metabolism and cofactor and vitamin influences. Also the measurement of methionine and serine from labeled formate in cultured fibroblasts is invaluable in the enzymatic confirmation of remethylation defects and can also be used for prenatal diagnosis(28).
Abbreviations
- cbl:
-
cobalamin
- FAD:
-
flavin adenine dinucleotide
- MR:
-
5,10-methylenetetrahydrofolate reductase
- MeTHF:
-
5-methyltetrahydrofolate
- MS:
-
methionine synthase
References
Strong WB, Schirch V 1989 In vitro conversion of formate to serine: effect of tetrahydropteroylglutamates and serine hydroxymethyltransferase on the rate of 10-formyltetrahydrofolate synthetase. Biochemistry 28: 9430–9439.
Finkelstein J.D 1990 Methionine metabolism in mammals. J Nutr Biochem 1: 228–237.
Rosenblatt DS 1995 Inherited disorders of folate transport and metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Metabolic and Molecular Basis of Inherited Disease, 7th Ed. McGraw-Hill, New York, pp 3111–3128.
Fenton WA, Rosenberg LE 1995 Inherited disorders of cobalamin transport and metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Metabolic and Molecular Basis of Inherited Disease, 7th Ed. McGraw-Hill, New York, pp 3129–3150.
Watkins D, Rosenblatt DS 1989 Functional methionine synthase deficiency (cblE and cblG): clinical and biochemical heterogeneity. Am J Med Genet 34: 427–434.
Seashore MR, Rosenblatt DS, Aspler AL, Shevell MI, Pletcher BA, Fenton WA 1994 Clinical heterogeneity and prognosis in combined MMA and homocystinuria (CBLC). 32nd Symposium of the Society for the Study of Inborn Errors of Metabolism, Edinburgh, September 6–9, Abstract no. O-28
Rosenblatt DS, Cooper BA, Lue-shing S, Wong PWK, Berlow S, Narisawa K, Baumgartner R 1979 Folate distribution in cultured human cells: studies on 5,10-CH2-H4PTEGLU reductase deficiency. J Clin Invest 63: 1019–1025.
Baumgartner ER, Stokstad ELR, Wick H, Watson JE, Kusano G 1985 Comparison of folic acid coenzyme distribution patterns in patients with methylenetetrahydrofolate reductase and methionine synthetase deficiencies. Pediatr Res 19: 1288–1292.
Higginbottom MC, Sweetman L, Nyhan WL 1978 A syndrome of methylmalonic aciduria, homocystinuria, megaloblastic anemia and neurologic abnormalities in a vitamin B12 deficient breastfed infant of a strict vegetarian. N Engl J Med 299: 317–323.
Kang SS, Wong PWK 1987 Homocysteinemia due to folate deficiency. Metabolism 36: 458–462.
Horne DW, Patterson D, Cook RJ 1989 Effect of nitrous oxide inactivation of vitamin B12-dependent methionine synthetase on the subcellular distribution of folate coenzymes in rat liver. Arch Biochem Biophys 270: 729–733.
Shane B, Stokstad ELR 1985 Vitamin B12 folate interrelationships. Ann Rev Nutr 5: 115–141.
Boss GR 1985 Cobalamin inactivation decreases purine and methionine synthesis in cultured lymphocytes. J Clin Invest 76: 213–218.
Boss GR, Erbe RW 1981 Decreased rates of methionine synthesis by methylene tetrahydrofolate reductase-deficient fibroblasts and lymphoblasts. J Clin Invest 67: 1659–1664.
Fowler B 1982 Transsulphuration and methylation of homocysteine in control and mutant human fibroblasts. Biochim Biophys Acta 721: 201–207.
Fowler B, Kraus J, Packman S, Rosenberg LE 1978 Homocystinuria: Evidence for three distinct classes of cystathionine β synthase mutants in cultured fibroblasts. J Clin Invest 61: 645–653.
Rosenblatt DS, Erbe RW 1977 Methylenetetrahydrofolate reductase in cultured human cells. 1. Growth and metabolic studies. Paediatr Res 11: 1137–1141.
Mellman I, Willard HF, Rosenberg LE 1978 Cobalamin binding and cobalamin-dependent enzyme activity in normal and mutant human fibroblasts. J Clin Invest 62: 952–96019.
Taylor RT, Weissbach H 1965 Radioactive assay for serine transhydroxymethylase. Anal Biochem 13: 80–84.
Lowry OH, Rosebrough NHJ, Farr AL, Randall RJ 1951 Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275.
Fowler B 1988 Amino acids. In: Gowenlock AH (ed) Practical Clinical Biochemistry, 6th Ed. William Heinmann, London, pp 368–400.
Willard HF, Ambani LM, Hart AC, Mahoney MJ, Rosenberg LE 1976 Rapid prenatal and postnatal detection of inborn errors of propionate, methylmalonate and cobalamin metabolism: a sensitive assay using cultured cells. Hum Genet 34: 277–283.
Chanarin I, Deacon R, Lumb M, Perry J 1989 Cobalamin-folate Interrelations. Blood Rev 3: 211–215.
Deacon R, Perry J, Lumb M, Chanarin I 1990 Formate metabolism in the cobalamin-inactivated rat. Br J Haematol 74: 354–359.
Kaufman S 1991 Some metabolic relationships between biopterin and folate: Implications for the methyl trap hypothesis. Neurochem Res 16: 1031–1036.
Hoffbrand AV, Jackson BFA 1993 Correction of the DNA synthesis defect in vitamin-B12 deficiency by tetrahydrofolate: evidence in favour of the methyl-folate trap hypothesis as the cause of megaloblastic anaemia in vitamin-B12 deficiency. Br J Haematol 83: 643–647.
Bellini C, Cerone R, Bonacci W, Caruso U, Magliano CP, Serra G, Fowler B, Romano C 1992 Biochemical diagnosis and outcome of 2 years treatment in a patient with combined methylmalonic aciduria and homocystinuria. Eur J Pediatr 151: 818–820.
Berant M, Mandel H, Aizin A, Schwartz Y, Behar J, Fowler B 1990 Methylenetetrahydrofolate reductase deficiency. 1. Prenatal diagnosis. 2. Lysinuric protein intolerance in the same family. 5th International Congress of Inborn Errors of Metabolism, Asilomar, June 1–5, Abstract no. W8-7
Acknowledgements
The authors thank those colleagues who sent cell lines for investigation.
Author information
Authors and Affiliations
Additional information
Supported by a grant from the Research Trust for Metabolic Diseases in Children, UK.
Rights and permissions
About this article
Cite this article
Fowler, B., Whitehouse, C., Wenzel, F. et al. Methionine and Serine Formation in Control and Mutant Human Cultured Fibroblasts: Evidence for Methyl Trapping and Characterization of Remethylation Defects. Pediatr Res 41, 145–151 (1997). https://doi.org/10.1203/00006450-199701000-00023
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199701000-00023
This article is cited by
-
Characterization and review of MTHFD1 deficiency: four new patients, cellular delineation and response to folic and folinic acid treatment
Journal of Inherited Metabolic Disease (2015)
-
Three new cases of late-onset cblC defect and review of the literature illustrating when to consider inborn errors of metabolism beyond infancy
Orphanet Journal of Rare Diseases (2014)
-
Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism
Nature Genetics (2012)
-
Causes of and diagnostic approach to methylmalonic acidurias
Journal of Inherited Metabolic Disease (2008)
-
Folate restriction and methylenetetrahydrofolate reductase 677T polymorphism decreases adoMet synthesis via folate-dependent remethylation in human-transformed lymphoblasts
Leukemia (2007)
