
Abstract
Extract: Metabolism of 14C-propionate and methylmalonate was severely curtailed in fibro-blasts cultured from an infant with massive transient hyperammonemia (1370 $mUg/100 ml), severe metabolic acidosis, and excretion of large amounts of methylmalonic acid (580 mg at 24°). Metabolism of succinate was normal.
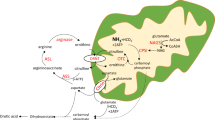
Metabolism of methylmalonate was not enhanced by the addition of excessive amounts of the cofactor, 5′-deoxyadenosylcobalamin (DBCC). The DBCG content of the liver was within normal limits.
Homogenates of liver and fibroblasts metabolized methylmalonate approximately one-half as well as control samples when tritiated racemic methylmalonyl coenzyme A (CoA) was added. Inasmuch as L-methylmalonyl-CoA and not D-methylmalonyl-CoA is the substrate for the enzyme, methylmalonyl-CoA mutase, which converts L-methylmalonyl-CoA to succinyl-CoA, this indicates that the mutase was intact.
Mitochondrial homogenate from liver, in contrast to normal samples, did not incorporate tritium during the metabolism of synthetic methylmalonyl-CoA, which indicates that activity of racemase was deficient.
Activities of the urea cycle enzymes were low but not rate limiting.
Speculation: A diet low in isoleucine, threonine, methionine, and valine may offer a rational therapeutic approach to other affected patients. The hyperammonemia observed resembles that reported in propionyl-CoA carboxylase deficiency and Reye's syndrome, which raises the possibility of a common denominator in these several disorders.
Similar content being viewed by others


Article PDF
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Kang, E., Snodgrass, P. & Gerald, P. Methylmalonyl Coenzyme A Racemase Defect: Another Cause of Methylmalonic Aciduria. Pediatr Res 6, 875–879 (1972). https://doi.org/10.1203/00006450-197212000-00004
Issue Date:
DOI: https://doi.org/10.1203/00006450-197212000-00004
Keywords
This article is cited by
-
Methylmalonacidurie
Klinische Wochenschrift (1977)
