
Abstract
As more and more links between cancer and metabolism are discovered, new approaches to treat cancer using these mechanisms are considered. Dietary restriction of either calories or macronutrients has shown great potential in animal studies to both reduce the incidence and growth of cancer, and to act synergistically with other treatment strategies. These studies have also shown that dietary restriction simultaneously targets many of the molecular pathways that are targeted individually by anticancer drugs. The insulin/insulin-like growth factor-1 (IGF-1) system has thereby emerged as a key regulator of cancer growth pathways. Although lowering of insulin levels with diet or drugs such as metformin and diazoxide seems generally beneficial, some practitioners also utilize strategic elevations of insulin levels in combination with chemotherapeutic drugs. This indicates a broad spectrum of possibilities for modulating the insulin/IGF-1 system in cancer treatment. With a specific focus on dietary restriction, insulin administration and the insulin-lowering drug diazoxide, such modifications of the insulin/IGF-1 system are the topic of this review. Although preclinical data are promising, we point out that insulin regulation and the metabolic response to a certain diet often differ between mice and humans. Thus, the need for collecting more human data has to be emphasized.
Similar content being viewed by others
Introduction
It is increasingly recognized that not only age but also the denaturalization of our food, lifestyle and environment are partly responsible for the current rise in non-communicable diseases such as obesity, type II diabetes mellitus (T2D) and related types of cancer.1, 2, 3, 4 This has motivated research into lifestyle interventions and drugs for prevention and treatment of these diseases. One of the most promising interventions is dietary restriction (DR) of either calories in general or specific macronutrients, as it consistently has been shown to prolong life- and healthspan in a broad range of model organisms and possibly humans, too, when compared with unrestricted food intake.5, 6 DR targets whole-body metabolism, impacting hormones, metabolic substrates and molecular signaling pathways that have a role in metabolic disorders such as obesity and T2D. Intriguingly, the same pathways are increasingly implicated in the development and growth of cancer, as more and more associations and parallels between obesity, T2D and an abnormal metabolism of cancer patients become evident. An important example is insulin resistance: reduced glucose uptake in cancer patients compared with healthy controls during an euglycemic hyperinsulinemic glucose clamp has commonly been observed not only during7, 8 but also before weight loss or malnutrition.8, 9, 10 This has been linked to chronic low-level inflammation induced by pro-inflammatory cytokines released by the tumor and tumor-associated macrophages (see review on cachexia in the same issue of this topical issue). Along these lines, insulin resistance in T2D and obesity seems connected to low-grade chronic inflammation induced by an increased release of pro-inflammatory cytokines from predominantly visceral adipose tissue and its associated immune cells, combined with a decreased release of the insulin-sensitizing hormone adiponectin.11 Furthermore, hyperglycemia itself, resulting from insulin resistance, induces a pro-inflammatory environment through its effect on immune cells.12, 13, 14 Together, the pathological features of T2D—notably elevated serum concentrations of inflammatory cytokines, glucose, insulin and free insulin-like growth factor-1 (IGF-1)—provide a pro-tumorigenic environment that may account for the increased risk of diabetic and obese patients for the development of cancer at various sites11 as well as the worse prognosis of patients with cancer that display one or more of these abnormalities.15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25
Although inflammation is a powerful driver of tumor growth,26 it is the aim of this review to focus on the connection between insulin/IGF-1 signaling and cancer, and discuss possibilities to modulate these interactions through DR and pharmaceutical interventions to improve cancer outcomes. We here refer to DR as any intervention that either restricts the total amount of energy consumed without changing the macronutrient ratio (calorie restriction; CR) or restricts a particular macronutrient without necessarily lowering the energy content of the diet. Usually, CR involves a 20–50% reduction in energy intake while maintaining sufficient intake of essential vitamins and minerals.27 It can be achieved via chronic energy restriction, the most extreme form of which is short-term starvation (STS, corresponding to water-only fasting) or intermittend fasting (IF) regimes such as only eating every other day.
Insulin, IGF-1 and cancer
Molecular pathways
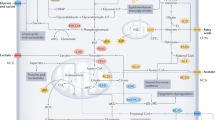
Insulin and the IGFs, IGF-1 and IGF-2, are structurally similar peptides with important roles in controlling metabolism and growth in response to nutrient signals and nutritional status. IGF-1-1 and IGF-2 are primarily produced in the liver and to a lesser extent locally in target tissues where they exert autocrine and paracrine actions. Both have similar biological effects that are caused by binding to the IGF-1 receptor (IGF-1R), whereas the IGF-2R is specific for IGF-2 and thought to have no physiological role except serving to degrade IGF-228 (Figure 1). Serum concentrations of IGF-2 increase during childhood and then level off at ~500 ng/ml,29 although abnormal IGF-2 concentrations can occur in certain conditions such as IGF-2-producing tumors.30 IGF-1 is low at birth, increases throughout puberty and declines with older age, its concentration being roughly three times lower than that of IGF-2.29 Bioavailability of IGFs is regulated by a class of six IGF-binding proteins (IGFBP-1–6), which are also produced in the liver. Production of IGF-1 and the most abundant binding protein in plasma, IGFBP-3, occurs via growth hormone-mediated signaling, and insulin influences IGF-1 bioavailability by controlling the transcription of IGFBP-1.31

Insulin/IGF receptor binding. As tyrosine kinase receptors, the IR and the IGF receptors, consist of an extracellular ligand-binding domain and a cytosolic tyrosine kinase domain that autophosphorylates upon ligand binding and transphosphorylates several substrates that initiate downstream signaling. The IR shares ~50 and 80% homology with the ligand-binding and tyrosine kinase domain, respectively, of the IGF-1 receptor (IGF-1R).30 It exists in two isoforms, IR-A and IR-B, which promote either mainly mitogenic or metabolic effects, depending on the ligand and the cellular context, allowing cells flexibility in responding to mainly one or the other stimulus. In general, IR-A is preferentially associated with mitogenic and anti-apoptotic signaling, whereas IR-B is associated with cell differentiation and metabolic effects.30 A predominant expression of IR-A has correspondingly been found in fetal tissue and tumors with autocrine production of IGF-2, which binds this receptor with 30–40% affinity compared with insulin.210 In this way, these tumors promote cell proliferation in an autocrine manner.30, 211 IGF-2 also binds to the IGF-1R, whereas IGF-1 binds to its own IGF-1R and to hybrid receptors of IGF-1R and IR-A as well as IGF-1R and IR-B.30, 212 Physiological concentrations of insulin show no measurable binding to the IGF-1R both in vitro30 and in vivo.213 Nevertheless, in mammals, insulin may be the major controller of insulin/IGF-1 action due to its effect on the bioavailability of IGF-1.43
Insulin is a key hormone for coordinating nutrient intake with energy production and storage through both excitatory and inhibitory actions. Insulin is secreted from pancreatic β-cells with blood glucose being the main secretagogue in humans. As an anabolic hormone, insulin accelerates glucose uptake in various tissues and promotes lipid synthesis in the liver while simultaneously inhibiting lipolysis, proteolysis, glycogenolysis, ketogenesis and gluconeogenesis. The latter function is particularly important for insulin’s well-known ability to lower and regulate blood glucose levels within a narrow physiological range during the postprandial phase.32
Insulin is also well established as a growth-stimulating hormone.33 In this respect, its effects parallel those of IGF-1 and IGF-2. This is due not only to the structural similarity between insulin and the IGFs, but also to the high degree of homology between the insulin receptor (IR) and the IGF receptors (Figure 1). The IR, IGF-1R and their hybrid receptors are expressed by most human tumors, whereby predominant expression of the IR-A isoform correlates with a poor differentiation grade.30 Among the pathways activated by IR and IGF-1R signaling are the RAS−RAF−MEK1/2−extracellular signal-regulated kinase (ERK)-1/2 pathway34 and the phosphatidylinositol-3-kinase (PI3K)−AKT−mammalian target of rapamycin (mTOR) pathway, two fundamental pathways for tumor cell proliferation and survival (Figure 2). The serine/threonine protein kinase mTOR is part of a protein complex called mTORC1, which has important roles in tumor cell growth and metabolism. Reduced glucose, insulin and IGF-1 levels or—more generally—DR activate an energy-sensing network consisting of AMPK, SIRT1, PPARα and PGC-1α with the potential to counteract tumor cell proliferation.35, 36 AMPK activation by anti-diabetic drugs such as metformin is currently considered a beneficial adjunct to standard cancer therapy.37, 38

Insulin/IGF-1 signaling network and its modulation by dietary restriction. Dietary restriction in the form of overall calorie restriction or specific restriction of carbohydrates or protein has specific effects on the insulin/IGF-1 system that transduces cellular signals through its insulin and IGF-1 tyrosine kinase receptors. This picture can only provide a partial overview of the complexity of this signaling network. The classical action of activated extracellular signal-regulated kinase (ERK)-1 and ERK-2 is their translocation into the nucleus where they activate mitogenic transcription factors. Similarly, mTORC1 targets transcription factors that increase proliferation and counteract apoptosis. Activation of mTORC1 via IR/IGF-1R−PI3K−AKT converges with its activation by amino acids at the lysosomal membrane. There, the guanosine triphosphatase (GTPase) Rheb (Ras homolog enriched in brain) stimulates mTOR activity, whereas a lack of growth signals activates the tumor suppressor tuberin (TSC2), which translocates to the lysosomal membrane and inhibits Rheb-stimulated mTORC1 activation.214 High insulin levels activate AKT that phosphorylates and inactivates TSC2, whereas CR or glucose withdrawal induce energy stress, decrease the intracellular ATP/AMP ratio and activate TSC2 through liver kinase B1 (LKB1)—adenosine monophosphate-activated protein kinase (AMPK) signaling. AMPK can also directly inhibit mTORC1 by phosphorylating the regulatory-associated protein of mTOR (Raptor). AMPK has similar actions to the class III histone deacetylase SIRT1, which is a NAD+-dependent enzyme that is also activated under DR-induced energy stress through an increase in the NAD+/NADH ratio.36 AMPK and SIRT1 amplify each other and both activate the peroxisome proliferator-activated receptor gamma 1α coactivator (PGC-1α) protein that cooperates with peroxisome proliferator-activated receptor α (PPARα) to induce major metabolic shifts under DR such as an upregulation of lipid oxidation and downregulation of glycolysis.35 mTORC1 inhibits these actions, providing another link to insulin/IGF-1 signaling.
The insulin/IGF-1 system and tumor cell metabolism
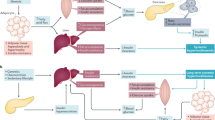
Apart from a few exceptions, glucose has a key role in tumor cell metabolism and its connection to proliferation and cell protection. As known since the seminal studies of Otto Warburg et al.39, 40, 41 most tumors ferment glucose to lactate even under sufficient oxygen supply, which in normal cells of the same tissue would shuffle pyruvate, the end product of glycolysis, into the mitochondria for further oxidative utilization. This peculiar feature of tumor cell metabolism is now known as the ‘Warburg effect’ or ‘aerobic glycolysis’. It is the basis of molecular 2-(18 F)fluoro-2-deoxy-d-glucose (FDG)-positron emission tomography (PET) imaging with the radioactively labeled glucose analog FDG (Figure 3). A high uptake of glucose in tumor cells not only serves for energy production but also for protection from endogenous and exogenous reactive oxygen species because NADPH, a by-product of the oxidative pentose phosphate pathway, is used to regenerate glutathione, an important cellular antioxidant.

Glycolytic pathways in tumor cells. Sketch of the most important glucose-degrading metabolic pathways in a tumor cell. Glucose uptake into the cytoplasm is accomplished via specific transpcorters (GLUTs) that are often overexpressed in tumor cells. Here the enzyme hexokinase (HK) phosphorylates glucose to glucose-6-phosphate (glucose-6-P). This metabolite either gets degraded to pyruvate via several intermediate steps of glycolysis or serves as the precursor for conversion into ribulose-5-phosphate in the oxidative part of the pentose phosphate pathway (PPP). In the PPP, CO2 gets released and the reducing equivalent NADPH is produced. The generated ribulose-5-phosphate either serves as the basis for de novo synthesis of nucleotides or is converted to various C3–C7 sugars through the transketolase/transaldolase reaction in the non-oxidative part of the PPP. Pyruvate, the end product of glycolysis, usually gets transported into the mitochondria, converted to acetyl-CoA and channeled into the TCA cycle for oxidative degradation. In case of insufficient oxygenation, dysfunctional mitochondria or metabolic reprogramming through hyperactivation of AKT–mTOR signaling, pyruvate is increasingly converted to lactate via the enzyme lactate dehydrogenase (LDH). Lactate gets transported out of the cell by monocarboxylate transporters (MCTs). The PI3K–AKT pathways increases glycolytic rate by the mechanisms depicted and described in the main text. Dashed arrows indicate several intermediate steps.
On a molecular basis, the PI3K–AKT–mTORC1 pathway has been found to significantly contribute to the high glycolytic activity of many tumor cells.42 AKT directly and indirectly—by the activation of mTOR, which promotes the stabilization of the transcription factor hypoxia-inducible factor-1α—stimulates the expression of glucose transporters and key glycolytic enzymes (Figure 3). AKT also phosphorylates the pro-apoptotic and anti-proliferative transcription factor FOXO1, which leads to its exclusion from the nucleus and cytosolic degradation, thereby connecting tumor cell metabolism with cell cycle progression and survival.
High IGF-1 and insulin levels in the microenvironment therefore provide a plausible mechanism of carcinogenesis and early tumor growth through anti-apoptotic signaling and metabolic reprogramming mediated by the PI3K–AKT–mTORC1 pathway. This is consistent with the finding that diabetes and obesity mainly raise the risk for those cancers that exhibit a Warburg phenotype.43 The relevance of this pathway for tumorigenesis is demonstrated by the fact that humans with the Laron syndrome, a recessively inherited defect in the growth hormone receptor, display extremely low IGF-1 and reduced insulin concentrations and usually do not develop cancer despite high prevalence of obesity and dyslipidemia.44, 45
Human tumors stimulated by insulin in vitro include breast cancer,46, 47 colon cancer,48 various leukemia cells lines49, 50, 51 or melanomas.52 In addition, most cancer cells are extremely vulnerable to glucose withdrawal,53, 54, 55, 56, 57, 58, 59 a feature they owe to metabolic reprogramming, leading to constitutively active proliferation pathways and ‘glucose addiction’. In fact, hyperglycemia itself stimulates tumor growth through distinct mechanisms and often amplifies the growth-promoting action of insulin.60 Thus, 100 ng/ml of insulin increased proliferation rates of human breast, colon, prostate and bladder cancer cell lines in a glucose-dependent manner, achieving 7–44% higher proliferation when combined with diabetogenic glucose concentrations of 11 mm compared with glucose concentrations of 5.5 mm without added insulin.61 Furthermore, high glucose and insulin altered the activity of several cell adhesion and migration genes, increasing migratory ability and the duration of locomotion. This coincided with an upregulation of the PI3K pathway,61 and a 29 and 66% increase of Akt expression in MDA-MB-468 breast and SW480 colon cancer cell lines, respectively.62
Despite these indications of tumor sensitivity to insulin, it is not clear to which extent the modulation of insulin and IGF-1 levels is able to influence the proliferation of progressive cancers that have become self-sufficient in growth and metabolic signals as illustrated for example by: (i) expression of insulin-independent glucose transporter isoforms; (ii) concurrent overexpression of IR-A and IGF-2; (iii) gain of function mutations in the PIK3CA gene, encoding the catalytic subunit of human PI3K; and (iv) loss of function mutations in PTEN, encoding the phosphatase PTEN which inhibits PI3K. This may not only pose a resistance mechanism against specific IR and IGF-1R inhibitors but also against dietary and pharmacological insulin and IGF-1 modulation. Kalaany and Sabatini had shown in NOD-SCID (non-obese diabetic, severe combined immunodeficient) mice that xenografted tumors with constitutive activation of AKT by either gains in PIK3CA or loss of PTEN are resistant against CR, and insensitive to insulin and IGF-1 treatment in vitro. At the same time, tumors without such a constitutive activation were stimulated by insulin and IGF-1, and responded to CR with increased rates of apoptosis mediated through FOXO1.63 This is reminiscent of the classical studies of the 7,12-dimethylbenz(a)anthracene (DMBA)-induced mammary carcinoma of the rat by Heuson and Legros. In rats bearing this tumor, induction of type-1-like diabetes with alloxan was followed in up to 90% of cases by rapid onset of remission, whereas treatment with insulin and especially combined treatment with insulin and glucose stimulated tumor growth considerably.64 These insulin-sensitive tumors also regressed in response to 60% CR, and insulin treatment in vitro induced a parallel rise of DNA synthesis and DNA polymerase activity.65 Some tumors however had apparently reached an autonomous growth without responding to insulin withdrawal or CR, similar to the tumors with constitutively activated AKT studied by Kalaany and Sabatini.
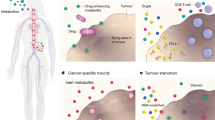
Some inconsistencies, however, remain. For example, the U87-MG glioma that was found resistant to CR when grown as a subcutaneous xenograft in NOD-SCID mice63 was responsive to CR when grown orthotopically in mice of a different genetic background.66 The CT-2A malignant mouse astrocytoma responded to CR when grown orthotopically or subcutaneously in C57BL/6J mice despite its PTEN and TSC2 deficiency, and constitutive AKT activation.67 Notably, growth retardation was accompanied by decreased phosphorylation of AKT and IGF-1R/IR tyrosine kinase domains, decreased production of IGF-1 and downregulation of IGF-1R protein expression in the tumors (Figure 4).

Effects of CR on IGF-1 and IGF-1R mRNA expression and growth of the CT-2A astrocytoma. For conditions a and b, tumors were implanted into the brains of C57BL/6J mice. At 10 days post tumor implantation, mice were randomly switched to either an unrestricted (UR; n=9) or CR (n=9) diet that aimed at reducing body weight by ~30%. In condition c, tumors were implanted subcutaneously and CR started at day 14; the Kaplan–Meier survival curve indicates significantly longer survival for CR compared with UR (P=0.01). Figure parts reproduced with permission from67.
It was further shown that in some cases, including a large percentage of human non-small cell lung cancers, AKT activity can be low despite loss of PTEN; in an experimental setting, such cancers were sensitive to upstream activation by insulin and IGF-1, and regressed during CR.68 Other common mutations in oncogenes such as RAS and BRAF or tumor suppressors such as TP53 did not affect the sensitivity of such tumor cells to insulin and IGF-1 in vitro or to CR in vivo.63 Together, these findings indicate that (i) the sensitivity of tumors to insulin and IGF-1 parallels their response to CR, and (ii) neither the activation status of PI3K−AKT per se nor mutation status of individual genes predicts for the sensitivity of tumors to both growth factors and CR; instead, the metabolic environment of the host (NOD-SCID mice display signs of both type 1 and type 2 diabetes69) and genetic conformity between host and tumor (xenograft/allograft/isograft) seem to have the dominant role.
There are hints that constitutive activation of PI3K–AKT signaling could be exploited therapeutically because it underlies a differential stress response between malign and benign cells, such that only the latter increase their resistance against cytotoxic insults upon reduction of growth factors induced by CR.70 In C57BL/6J mice, CR promoted stress resistance in a FOXO1-dependent manner,71 which would not occur in tumor cells with FOXO inactivation due to constitutively active AKT. We have recently argued that a similar, albeit less pronounced, differential stress response may also be induced by a ketogenic diet (KD).72 A KD is usually defined as an isocaloric diet low enough in CHO and high enough in fat to induce significant elevations (⩾0.5 mmol/l in humans) of the ketone bodies (KBs) β-hydroxybutyrate and acetoacetate (termed ‘ketosis’). It therefore can be considered a fasting mimicking diet. This would be of special value for patients undergoing several week long radiotherapy during which prolonged fasting is no option. The state of ketosis, induced by low insulin levels, may generally benefit cancer patients, as KBs have been shown to inhibit glycolysis in various tumor cell lines73, 74 and probably also patients,75, 76, 77 and tumor cells often lack the enzymes78, 79, 80, 81 or oxygenation82 to effectively use ketones for energy production. Furthermore, in glioblastoma xenografts it was shown that KBs can partly reverse the genetic alterations that occur in these tumors.83
In conclusion, beneficial effects may be achieved by lowering insulin and IGF-1 levels in patients with both insulin-sensitive and insulin-insensitive tumors.
As a final note, some experimental tumors have been described whose growth is suppressed by insulin and stimulated by induction of type 1 diabetes, with the R3230AC adenocarcinoma of the rat being the most intensively studied tumor of this type.84, 85, 86 A tumor-suppressing effect of insulin and glucagon, and especially of their combination has been found by Salter et al.87 and was reproduced in different experimental systems.88, 89 The Morris hepatoma, which was also suppressed by insulin,90 was stimulated by acute fasting,91 and it was later discovered that linoleic and arachidonic acid released from adipose tissue were the main substrates promoting tumor growth.92 Similarly, in a hamster model of pancreatic cancer, high substrate levels of glucose, linoleic acid and other fatty acids mobilized through streptozocin-induced diabetes significantly enhanced tumor growth, which was prevented by insulin treatment.93 Collectively, these experimental tumors provide evidence not for a direct tumor growth-suppressing effect of insulin but indirect effects such as an influence on metabolic growth-promoting substrates whose global abundance is controlled by insulin.
DR and its effect on the insulin/IGF-1 system
The importance of the insulin/IGF-1 system for the antitumor effects of DR is exemplified by the fact that in vivo IGF-1 administration94, 95, 96 completely rescued CR-sensitive tumors from CR-induced growth inhibition. Therefore, it is important to review the effects of different DR regimes on circulating levels of these growth factors and other substrates influenced by them. Relevant to many preclinical studies are the extensive investigations in male C57BL/6 mice that are summarized in Table 1 and compared with the data from insulin-resistant humans, because many cancer patients are also insulin resistant. From Table 1, it is already evident that mice and humans can respond differently to the same DR regime, an important fact we will elaborate on later.
Calorie restriction
Because of the simultaneous lowering of all three macronutrients and energy, CR induces a complex metabolic response that is not straightforward to attribute to one of these individual factors. Mitchell et al.97 have shown that 10–40% CR over 3 months in C57BL/6 mice (corresponding to approximately a tenth of their lifespan) decreases blood glucose, insulin and IGF-1 concentrations, the latter showing the greatest dependence upon the severity of CR.
Many of the metabolic effects of CR in model organisms are also observed in humans.5 CR reduces fasting insulin levels and improves insulin sensitivity in overweight individuals, whereby these effects may be more pronounced with regular IF compared with chronic CR.98, 99 A CR diet supplying 600 kcal/day over 8 weeks significantly decreased fasting insulin from 151 to 65 pmol/l and glucose levels from 9.2 to 5.7 mmol/l in overweight individuals with T2D, with the greatest declines already apparent after 1 week.100 Kitada et al.101 showed that 7 weeks of 25% CR significantly reduced insulin levels and inflammatory markers in healthy obese males. Interestingly, incubation of human skeletal muscle cells with serum obtained from these subjects after the intervention resulted in an increase in PGC-1α expression, AMPK and SIRT1 activity, and mitochondrial biogenesis. Mercken et al.102 investigated tissue samples from m. vastus lateralis of individuals following long-term CR (average 9.6 years) of ~30% compared with a typical Western diet. Thereby, PI3K and AKT transcription was downregulated 1.7- and 2-fold, respectively, whereas PGC-1α transcripts were increased 7-fold and AKT phosphorylation was reduced by 35–50%. This indicates that chronic CR reduces PI3K−AKT signaling in humans.
In contrast to rodents, IGF-1 levels in humans are usually not reduced with chronic CR unless protein is also restricted103 (see below). STS, on the other hand, decreases IGF-1, glucose and insulin levels, and increases KB levels comparable to 40% CR over 3 weeks in mice.104
Clinically, it is well established that weight loss and physical activity can reduce insulin levels by 10–30%, and that lowering of insulin by 25% may be associated with a 5% absolute improvement in breast cancer mortality.105 Although this is not generalizable to other cancer patients, especially those at risk for malnourishment, IF and STS could be considered as therapeutic options in such cases; recommendations for their implementation are given by Simone et al.106 or Klement and Champ.72
Carbohydrate restriction
The metabolic response of different mice strains to CHO restriction differs and is also influenced by the total energy intake and the percentage of the remaining macronutrients. A classical biomarker of CHO restriction is the amount of circulating KBs.
Insulin and glucagon are key hormones regulating ketogenesis by controlling the flux of non-esterified fatty acids to the liver for KB production.107 Paradoxically, C57BL/6 mice gain excessive body weight, display hyperlipidemia, and have concurrently elevated KB and glucose levels when fed an unrestricted KD with >10% energy (E%) from protein.108, 109 Thereby, insulin levels are only slightly decreased, but drop significantly together with blood glucose levels and body weight once calorie intake is also restricted.109 This is in contrast to healthy humans where unrestricted KDs with adequate protein intake tend to lower body weight and insulin levels,110 improve body composition by increasing fat-free mass110, 111 and decrease inflammatory markers.112 Some authors argue that CHO restriction should be the first approach in the treatment of T2D, as it improves long-term glucose control, lowers insulin levels and allows a reduction or even elimination of anti-diabetic drugs.113 Indeed, investigation of glucose kinetics and insulin secretion after a low-CHO meal (20E% CHO, 65E% fat) in five young adult baboons revealed minimal perturbations of glucose homeostasis, in stark contrast to a high-CHO (65E% CHO, 20E% fat) meal.114 Consistently, it has been shown that the metabolic effects of fasting in humans are largely mediated by the absence of CHO.115 This is the rationale for using KDs as fasting mimicking diets in the oncological setting where maintenance of fat-free mass is important.116, 117
Protein restriction
Mitchell et al.97 found that isocaloric protein restriction down to 12E% over 3 months was not able to reproduce the beneficial metabolic changes induced by CR in male C57BL/6 mice, in particular the decrease in IGF-1 concentrations. Other studies, however, have reported reduced IGF-1 levels in mice of this strain when protein was restricted to <10E% within the context of either ad libitum high-CHO118 or high-fat119 diets. In mice fed ad libitum an 8-week protein restriction to 5E%120 or methionine restriction121 have been shown to mimic several of the metabolic effects of CR, such as decreases in triglycerides, blood glucose and insulin levels. In the long run, however, such diets may impair gains in lean body mass, and five out of nine experimental diets with protein restricted to 5E% turned out to be not sustainable due to excessive weight loss (>20%), rectal prolapse or failure to thrive (Supplementary Table S1 in118).
A change in IGF-1 concentrations is a marker for acute changes in nitrogen balance that depends on protein intake, but also on total energy intake.122, 123, 124, 125 Insulin, for example, inhibits protein breakdown, facilitating the maintenance of positive nitrogen balance when protein is replaced with CHO. Accordingly, IGF-1 levels in healthy humans dropped significantly during STS122, 123 or the initiation phase of a KD126 but were unaltered after several weeks of a KD110 or long-term CR with adequate protein intake.127
Another biomarker of protein restriction is fibroblast growth factor 21 (FGF21). FGF21 has originally been described as a fasting hormone that is upregulated in the liver via PPARα and partly regulates ketogenesis during starvation or KDs;128 an important role for SIRT1 in FGF21 expression was also recently demonstrated.129 However, a study using Fgf21-knockout mice found that FGF21 was not required for ketogenesis or other adaptions to a KD.119 It was later shown that it is the de facto protein restriction during starvation or protein-deficient diets that increases FGF21 concentrations as only diets with <10E% from protein,128, 130 but not those with higher protein intake,119, 131 led to an upregulation of hepatic FGF21 expression and secretion, independent of energy or CHO intake.131 This is consistent with the data in humans showing that a low-protein diet (5E% protein) increased plasma FGF21 concentrations by 171% over 4 weeks despite caloric overfeeding.131 Other data indicate that depletion of single amino acids such as methionine is sufficient for hepatic FGF21 production.121 There are data showing that FGF21 acts as an insulin-sensitizing and glucose-normalizing hormone in diabetic states and contributes to the action of anti-diabetic drugs.132
Protein restriction also limits mTORC1 activation by mechanisms distinct from its regulation via IR/IGF-1R−PI3K−Akt. It is generally believed that activation of mTORC1 by specific amino acids, notably leucine, starts with its recruitment to the lysosomal membrane by GTPases called Rags, which are concentrated there as heterodimers consisting of RagA or RagB combined with RagC or RagD. These heterodimers are part of an amino-acid-responsive supercomplex also containing the Ragulator and vacuolar adenosine triphosphatase protein complexes. This supercomplex in turn is thought to be activated by amino acids transported from the lysosomal lumen by transmembrane proteins, which in this way act as amino-acid sensors.133 Two alternative, Rag-independent ways of mTORC1 activation were also recently described. Glutamine, but not leucine, was found to activate mTORC1 by a pathway requiring the ADP ribosylation factor-1 GTPase and vacuolar adenosine triphosphatase for mTORC1 translocation and fixation, respectively, to the lysosomal membrane.134 Thomas et al.135 described a pathway involving amino-acid- but not insulin-stimulated binding of the small GTPase Rab1A and mTORC1 with subsequent recruitment to Golgi membranes where mTORC1 gets activated by Rheb. This study identified Rab1A as an oncogene in certain human cancers whose overexpression promotes amino-acid-stimulated tumor growth but also renders these cells vulnerable to amino-acid restriction.135
In humans, AMPK activation from training in a glycogen-depleted state was not influenced by protein intake.136 Other data have shown that CHO restriction is sufficient to activate the AMPK–SIRT1–PGC-1α network in humans even under caloric overconsumption.137 These findings complement the previously mentioned hormonal and metabolic changes induced by CHO restriction115 and indicate that in humans CHOs have a more dominant role than protein in the response to fasting.
In summary, protein restriction exerts specific effects on IGF-1, FGF21 and mTOR activity that probably contribute to the life-prolonging and anticancer effects seen when rodents are placed on low-protein diets. These effects can partly be mimicked by the restriction of certain amino acids. On the other hand, severe restriction of total protein intake, that is, either very-low-protein diets (<10E%) or moderate protein intake combined with CR, with the aim of reducing IGF-1 levels bears the risk of weight and fat-free mass loss. This would have detrimental effects for cancer patients, thus precluding severely protein restricted diets from a role as supportive interventions in cancer patients.
DR and tumor growth retardation
Animal data
Two meta-analysis have evaluated the evidence for tumor growth inhibition by CR. Focusing on studies on spontaneous breast tumors in mice published between 1942 and 1995, Dirx et al.138 found that CR led to an average of 55% less tumor development in CR-fed mice than in ad libitum controls. In a recent meta-analysis, 40 out of 44 studies (90.9%) showed a tumor inhibitory effect of CR in laboratory animals with respect to tumor incidence, progression or metastasis.27 The evidence for a protective role of various IF protocols was weaker, but still mostly positive. Furthermore, eight out of nine preclinical studies evaluated in this meta-analysis showed that a KD was able to slow down tumor growth, often even as a monotherapy.27
In a study not included in these meta-analyses, Frimberger et al.139 not only achieved a retardation of tumor growth but in 36% of cases a complete remission of benzo(α)pyren-induced cutaneous squamous cell carcinomas. The mice in this study were placed on a maximally tolerable restriction of both calories and protein which was accompanied by an extreme loss of up to 50% body weight.
In C57BL/6 mice, an unrestricted KD with 13E% from protein reduced tumor growth after transplantation of Lewis lung carcinoma cells compared with a high-CHO diet (77E% CHO), and this became significant when protein was further restricted to 5E% and replaced with fat.119 In another study, a lower number of lung metastases was observed when mice of this strain bearing the B16 melanoma were fed a zero-CHO, zero-protein (100E% from polyunsaturated fatty acids) diet.140 In contrast, growth of the CT-2A Astrocytoma in these mice was not influenced by an unrestricted KD containing 17E% protein,141 but significantly reduced on a calorically restricted KD containing only 8E% protein, along with significant reductions in blood glucose and IGF-1.142 These findings might be correlated to the metabolic abnormalities that these mice develop on KDs that are not concurrently low in protein as discussed above.
In the majority of studies, antitumor effects of CHO restriction have been achieved without concurrent CR. In the studies that proofed tumor growth inhibition by a KD fed ad libitum, there was a significant increase in KB levels, but not necessarily decreases in blood glucose levels or body weight.74, 140, 143, 144, 145, 146, 147, 148 In some models, also non-ketogenic low-CHO diets (10–15E% CHO) led to significant tumor growth retardation that was correlated to low blood glucose and insulin levels.149, 150
Clinical data
The large preclinical support for CR as an antitumor therapy implies a possible role for CR in human cancer prevention, treatment and survivorship.151 However, clinical trials to test its effects in patients have only recently started, and published results of CR interventions are restricted to small pilot studies or case reports,75, 76, 77, 152, 153, 154, 155, 156, 157, 158, 159 which we summarize in Table 2. One study reported the successful treatment of a patient with end-stage ovarian cancer by a diet allowing only 300–400 kcal/day.158 Notably, bioimpedance analysis indicated that of 21 kg body weight loss within 6 months <2 kg consisted of muscle mass.
In an evaluation of 10 patients, it was found that STS before and/or after chemotherapy reduced therapy-related weakness, fatigue and gastrointestinal side effects.152 Importantly, fasting did not interfere with the cytotoxic effect of chemotherapy on tumor cells. This might indicate that the fasting-induced differential stress response between tumor and normal tissue may also be achieved in humans.
Seven case reports exist for the treatment of glioblastoma multiforme with a CR-KD. Although this diet achieved one remission of 5 years when used as a monotherapy,75 the usual progress within 12 weeks occurred in two other cases.159 The results obtained by combining the CR-KD with chemo- or radiotherapy had been more promising, with remissions between 4 months and 4 years having been achieved.75, 153, 160, 161
Ninteen more patients with recurrent glioblastoma were treated in the ERGO trial by Rieger et al.157 with a KD consumed ad libitum. Here, too, no clinical effects on tumor growth were achieved with the KD as a monotherapy, with a median progression-free survival of 5 weeks (range 3–13 weeks). However, subsequent salvage therapy with bevacizumab and continuation of the KD led to a response in six out of seven patients, and the authors confirmed in a mouse experiment that the combination of KD and bevacizumab, but not the KD alone, was superior to bevacizumab combined with standard chow with respect to survival and tumor size.157 A partial explanation for the failure of the KD as a monotherapy in the ERGO trial could be based on the fact that there was no significant drop in blood glucose or HbA1c levels, and only part of the patients reached stable ketosis.157 According to Seyfried et al.162 the ratio between blood glucose and KB levels (both measured in mmol/l) should be below ~1.5 to enable metabolic management of malignant brain cancer. As insulin inhibits ketogenesis, low insulin levels are a prerequisite to achieve such a metabolic state.
The importance of low insulin and high ketone body levels is also implicated by the pilot trial of Fine et al.76 in which five out of nine patients with previous progression responded to a 4-week KD with partial remission or stable disease as judged by FDG-PET scans; these five patients exhibited significantly higher KB levels compared with their baseline values than the four non-responders. Ketosis was thereby inversely correlated to insulin levels. Together with the other reports measuring less FDG uptake or a decline in intratumoral lactate levels as summarized in Table 2, this implies that a KD is able to influence tumor cell metabolism by lowering insulin levels and increasing ketone body concentrations.
In addition, small parenteral feeding studies obtained direct hints that a lipid-based diet (fat contributing 80% non-protein calories) retards tumor cell proliferation while a dextrose-based diet (dextrose contributing 100% non-protein calories) accelerates it.163 A mixed diet (fat contributing 45% non-protein calories) investigated in another study had no effect on tumor cell kinetics.164 Finally, some, but not all, studies found worse survival in patients who received nutritional support in the form of high glycemic supplements165 or total parenteral nutrition.166 This could be taken as a warning to monitor the caloric and glycemic load of the diet to avoid overnutrition of patients that would resemble the feeding of control animals in the preclinical DR studies.
Critically questioning the concept of DR: implications for humans
Together, it seems that humans are more sensible to the amount and quality of CHO in their diet than mice in which high-protein intake can decisively stimulate insulin output.108, 118 This is not surprising, given that the species-specific diet in mice is very different from humans who are omnivores and during much of their evolution consumed low-glycemic load diets.167 Furthermore, mice and humans differ substantially in insulin kinetics and blood glucose control.69, 168 Expression of mouse carbohydrates and lipids on xenografted human tumor cells has also been described and could potentially alter the response of such tumors to the metabolic microenvironment.169 Together, these findings question the relevance of such tumor models for humans and complicate the translation of interventions tested in mice to human subjects.170
This also applies to DR: a thorough investigation of the life-prolonging effects of DR in mice shows that weight gain in the ad libitum fed control group is the most important covariate explaining most of the variation in the response of different strains to DR.171 Making the reasonable assumption that the mechanisms behind the life-prolonging effects of DR are also responsible for its antitumor action, one would have to conclude that DR in humans is most effective if it could replace a diet leading to weight gain and metabolic disturbances when consumed ad libitum.117 The prototype of such a diet is the Western diet, which is concurrently high in refined CHOs and fat. Thus, it could be expected that CR, CHO restriction or other fasting mimicking diets172 could be effective against tumor growth in humans. It must be considered, however, that mice have a metabolic rate approximately seven times higher than humans, and their experimental tumors display faster doubling times and larger relative weights.173, 174 Contrary to mice that appear fit and viable even with very high tumor masses, humans generally develop advanced cachexia and die when tumor masses have already reached 0.1% of body weight.174 Furthermore, relative weight loss in rodents under a particular DR regime is more rapid and extreme than in humans, with up to 50% weight loss being tolerated in tumor-bearing animals.139 Thus, the effects of any DR regime are exacerbated in mice, explaining why DR as a monotherapy has worked in preclinical studies but not in humans. Studying humans is therefore urgently needed to determine the dose–effect relation of DR interventions in human cancer treatment.
Pharmaceutical interventions
The modulation of plasma insulin and IGF-1 levels by pharmaceutical interventions is a promising approach for cancer treatment. Broad interest is now focused on the biguanide metformin37, 38 (see review in the same issue of this topical issue) as well as IR and IGF-1R inhibitors.37 The α-glucosidase inhibitor acarbose, which suppresses several tumors, but simultaneously promotes kidney tumors of the Sprague Dawley rat,175 has been recently reviewed.176 As an extensive discussion of these drugs is beyond the scope of this review, we refer the reader to the referenced work and focus our discussion on two less frequently mentioned interventions for which preclinical and clinical experience has been made by one of us (MKF). These are the administration of insulin itself to potentiate the effect of chemotherapy, and administration of the insulin-lowering drug diazoxide.
Insulin administration for cancer treatment
Although somewhat counterintuitive, reports have been published suggesting that insulin can be used therapeutically to treat cancer under certain conditions. One approach is to utilize the glucose-lowering effects of insulin to withdraw this preferred metabolic substrate from cancer cells. The idea is not new, as already Otto Warburg et al.177‘…kept tumor animals in very low blood sugar content in insulin convulsions for hours’. Although these experiments had no effects on tumor viability, the concept was later picked up by Wilhelm Brünings178,179 who combined maximally tolerable doses of insulin treatment with a KD into a ‘de-glycation method’ (‘Entzuckerungsmethode’) for the treatment of head and neck cancer patients in his clinic. The results, published in 1941/1942, indicated a very high rate of partial and complete remissions after a few weeks of treatment, but tumors became refractory after 2–3 months. Although the results could not be replicated by others,180 15 years later Joseph Weiss181 was able to achieve significant tumor growth inhibition in 20 out of 90 incurable cancer patients with a similar method. Finally, two case reports exist according to which repeatedly administered high doses of insulin resulting in hypoglycemic coma (lowest blood glucose reading 22 mg/dl) were able to bring metastatic cancer in complete remission of at least 1-year duration.182
A second approach using insulin administration consists of giving low dosed chemotherapeutic drugs at onset of hypoglycemia after intravenous administration of (typically 0.3–0.4 IU/kg) insulin, followed by hypertonic glucose. This so-called insulin potentiation therapy is hypothesized to increase drug uptake into tumor cells and additionally sensitize them to the chemotherapeutic substances through insulin’s ability to accelerate cell cycle progression into S-phase.183 However, in vitro both of these mechanisms were not responsible for insulin’s drastic enhancement of cytotoxicity of the folic acid analog methotrexate to MCF-7 human breast cancer cells.184 But, in a preclinical study on the DMBA-induced rat mammary carcinoma, this enhanced effectiveness of methotrexate was far outweighted by the growth-stimulating effect of 3-day pretreatment with combined insulin/glucose infusions.185 In a clinical pilot trial on 14 advanced cancer patients, combined insulin/glucose infusions were therefore started at most 18 h before the administration of methotrexate/5FU.186 Although results were mixed regarding an increased efficacy of combined treatment, there were some indications that tumor-associated pain could be reduced, possibly due to insulin’s anti-inflammatory action.186 Currently, insulin potentiation therapy is practiced by over 400 therapists worldwide, and the few data available indicate that it could allow a reduction of chemotherapeutic doses without compromising efficacy.187
Reduction of insulin levels with diazoxide
Diazoxide is a nondiuretic benzothiadiazine that has an antihypertensive effect and produces hyperglycemia via lowering insulin levels by activation of ATP-sensitive K+-channels, which have a key function in the control of insulin release.188, 189 In addition, diazoxide stimulates insulin degradation in the lysosomal system.190 With 300 mg diazoxide per day, fasting insulin levels decreased from 177 to 123 pmol/l (P<0.01), and insulin release in response to 100 g oral glucose administration decreased from 223 to 55.6 nmol × min/l (P<0.002) in obese patients with polycystic ovary syndrome.191 This effect was less pronounced in healthy non-obese women with a decrease of insulin release from 108 to 49.3 nmol × min/l (P=0.05).192 In moderately overweight patients with polycystic ovary syndrome, 300 mg diazoxide per day also reduced IGF-1 from 314.5 to 219.5 ng/ml (P<0.01).193 Furthermore, diazoxide inhibits glucagon secretion in healthy man194 and in the dog,195 but stimulates glucagon release in rats.196
Typical oral doses of diazoxide for the treatment of patients suffering from hyperinsulinemia197, 198 and resistant hypertension199 were 400 mg per day, with maximal doses of 800199 and 1500 mg/day,197 respectively. The main recognized side effects of orally given diazoxide are fluid retention, nausa and the growth of lanugo hair.197, 198, 199
The effect of diazoxide on cancer growth was examined in DMBA- and N-methyl-N-nitrosurea (MNU)-induced mammary carcinomas of the rat.200, 201 The determination of glucose and insulin levels in the blood of DMBA-induced tumor-bearing animals showed that tumor induction itself led to significantly higher glucose and lower insulin levels than in control animals. Increasing dosages of diazoxide led to an increasing number of remissions. After cessation of diazoxide treatment due to progression, 30% rebound responses were observed in animals that had a first remission due to diazoxide. This second remission after withdrawal of the drug is characteristic of hormonal therapy. In contrast to the rapid onset of remissions observed after diabetes induction with alloxan,64 onset of remissions with diazoxide was delayed and began about 2 weeks after the start of treatment; the cause of this delay is unclear.
Treatment of the more aggressively growing MNU-induced mammary carcinoma of the rat with 300 mg/kg diazoxide given on 5 days/week induced a remission in 55% of the animals. This effect was completely abolished by additional treatment with 2 IU depot insulin/day.200 Thus, an insulin-mediated effect of diazoxide was proven.
Combined therapy with a low dose (75 mg/kg) of diazoxide and the alkylating agents melphalan or N-(2-chloroethyl)-N-nitrosocarbamoyl (CNC)-omega-lysine increased the therapeutic efficacy of both cytostatics up to twofold in the MNU-induced rat mammary carcinoma.202 However, after the end of treatment with diazoxide and alkylating agents, tumors in this group grew faster than in controls. In DMBA-induced rat mammary carcinomas, the combination of diazoxide and medroxyprogesterone acetate moderately increased the remission rate, but clearly shortened the remission duration.203 In contrast, adding 200 mg diazoxide per kg to 5 mg/kg tamoxifen synergistically prolonged the remission duration and decreased tumor weight, although the latter effect was lost at the higher dose of 50 mg/kg tamoxifen (Table 3).
In a clinical pilot study, diazoxide was used at a relatively low dose of 200–300 mg/day.186 For inclusion, the maximal tolerated fasting glucose level was 110 mg/dl, and 180 mg/dl after an oral glucose load with 75 g. Nine breast cancer patients were included, and the best response was seen in a 60-year-old woman, who had glucose levels of only 56–105–115 mg/dl after an oral glucose load. After progression of her cutaneous metastases during tamoxifen, she was supplemented with 200 mg diazoxide per day; fasting glucose levels rose to 90 mg/dl, and surprisingly tamoxifen-induced hyperhydrosis disappeared. Partial remission with this combination ended after 7 months, when liver metastases were detected sonographically. Two months later, both medicaments were withdrawn because of rapidly growing cutaneous metastases and pleural effusion. Another 2 months later, the patient exhibited a rebound response of 4 months duration with disappearance of pleural effusions, partial remission of the cutaneous metastases and stable size of the liver metastases. In two additional patients with prior disease progress, diazoxide treatment resulted in stable disease of 8 (combined with tamoxifen) and 4 months (monotherapy).186
In vitro studies have uncovered other mechanisms of action by which diazoxide suppressed proliferation of human acute leukemic T cells204 and growth of human lung cancer cells.205 However, the growth of human colon cancer cells206 and especially human glioma cells in vitro207, 208 and in nude mice207 was stimulated through diazoxide-mediated opening of K+-channels. The ATP-sensitive K+-channels were thereby found overexpressed in glioma cell lines U87 and U251, and human glioma tissue, and their opening by diazoxide stimulated cell cycle progression and proliferation through activation of the extracellular signal-regulated kinase pathway.207 In principle, diazoxide-associated hyperglycemia may also raise concerns about brain tumor stimulation given the well-established link between hyperglycemia and brain cancer progression.17, 18, 23, 24 However, chronic neonatal diazoxide therapy during postnatal days 2–12 did not induce any lesions or morphological changes of brain anatomy in mice,209 and to our knowledge no glioma or brain metastasis in humans has been reported after treatment with diazoxide.
Conclusions
A large body of preclinical data has indicated that inhibition of the insulin/IGF-1 system has a therapeutic benefit for cancer-bearing animals. However, rodents and man differ in some aspects of their metabolic regulation in response to a certain diet or pharmaceutical intervention targeting the insulin/IGF-1 system. In animals, specifically DR in its various forms (CR/IF/STS, KD, protein restriction) has shown a potential for simultaneously targeting many of the pathways associated with insulin and IGF signaling, usually with no serious, or with even beneficial side effects such as a differential stress response between normal and tumor tissue.
We reviewed human data to obtain the following preliminary conclusions concerning insulin/IGF-1 modulation in humans: (i) DR could be considered as a supportive treatment during cancer therapy due to its probable antitumor effects, and due to its beneficial effects on human metabolism. Considering muscle mass maintenance and the putatively beneficial effects of ketosis, KDs and STS should be compared with chronic CR or protein restriction in clinical studies. (ii) Insulin-lowering drugs such as metformin and diazoxide provide another opportunity for improving cancer outcome in patients. Although their administration is probably easier accomplished than adherence to a DR regime, they do not mimic all the effects of DR. Similar to DR, their anti-neoplastic potential in humans is still insufficiently investigated. At least for metformin it can be expected that current clinical trials will catch upon this. Diazoxide was successfully studied in animals, exhibited first effects in a clinical pilot study and is worth to be further examined. The same may be true for insulin potentiation therapy. These approaches may be useful options in the ambition to exploit the full repertoire of insulin/IGF-1 modulation against cancer.
References
Carrera-Bastos P, Fontes-Villalba M, O´Keefe JH, Lindeberg S, Cordain L . The western diet and lifestyle and diseases of civilization. Res Rep Clin Cardiol 2011; 2: 15–35.
Klement RJ, Gonder U, Orsó E, Paul S, Schilling F, Spitz J . Proceedings of the 2nd annual symposium of the German Society for Paleo Nutrition held in 2014. J Evo Health 2013; 1: 6.
Ruiz-Núñez B, Pruimboom L, Dijck-Brouwer DAJ, Muskiet FAJ . Lifestyle and nutritional imbalances associated with Western diseases: causes and consequences of chronic systemic low-grade inflammation in an evolutionary context. J Nutr Biochem 2013; 24: 1183–1201.
Holly JMP, Zeng L, Perks CM . Epithelial cancers in the post-genomic era: should we reconsider our lifestyle? Cancer Metastasis Rev 2013; 32: 673–705.
Fontana L, Partridge L, Longo VD . Extending healthy life span—from yeast to humans. Science 2010; 328: 321–326.
Cava E, Fontana L . Will calorie restriction work in humans? Aging (Albany NY) 2013; 5: 507–514.
Permert J, Adrian TE, Jacobsson P, Jorfelt L, Fruin AB, Larsson J . Is profound peripheral insulin resistance in patients with pancreatic cancer caused by a tumor-associated factor? Am J Surg 1993; 165: 61–66.
Yoshikawa T, Noguchi Y, Matsumoto A . Effects of tumor removal and body weight loss on insulin resistance in patients with cancer. Surgery 1994; 116: 62–66.
Marat D, Noguchi Y, Yoshikawa T, Tsuburaya A, Ito T, Kondo J . Insulin resistance and tissue glycogen content in the tumor-bearing state. Hepatogastroenterology 1999; 46: 3159–3165.
McCall JL, Tuckey JA, Parry BR . Serum tumour necrosis factor alpha and insulin resistance in gastrointestinal cancer. Br J Surg 1992; 79: 1361–1363.
Doyle SL, Donohoe CL, Lysaght J, Reynolds JV . Visceral obesity, metabolic syndrome, insulin resistance and cancer. Proc Nutr Soc 2012; 71: 181–189.
Shanmugam N, Reddy MA, Guha M, Natarajan R . High glucose–induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes 2003; 52: 1256–1264.
Wen Y, Gu J, Li S-L, Reddy MA, Natarajan R, Nadler JL . Elevated glucose and diabetes promote interleukin-12 cytokine gene expression in mouse macrophages. Endocrinology 2006; 147: 2518–2525.
Dandona P, Chaudhuri A, Ghanim H, Mohanty P . Proinflammatory effects of glucose and anti-inflammatory effects of insulin: relevance to cardiovascular disease. Am J Cardiol 2007; 99: 15B–26B.
Maestu I, Pastor M, Aparicio J, Oltra A, Herranz C, Montalar J et al. Pretreatment prognostic factors for survival in small-cell lung cancer: A new prognostic index and validation of three known prognostic indices on 341 patients. Ann Oncol 1997; 8: 547–553.
Weiser MA, Cabanillas ME, Konopleva M, Thomas Da, Pierce Sa, Escalante CP et al. Relation between the duration of remission and hyperglycemia during induction chemotherapy for acute lymphocytic leukemia with a hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone/methotrexate-cytarabine regimen. Cancer 2004; 100: 1179–1185.
McGirt MJ, Chaichana KL, Gathinji M, Attenello F, Than K, Ruiz AJ et al. Persistent outpatient hyperglycemia is independently associated with decreased survival after primary resection of malignant brain astrocytomas. Neurosurgery 2008; 63: 286–291.
Derr RL, Ye X, Islas MU, Desideri S, Saudek CD, Grossman SA . Association between hyperglycemia and survival in patients with newly diagnosed glioblastoma. J Clin Oncol 2009; 27: 1082–1086.
Lamkin DM, Spitz DR, Shahzad MMK, Zimmerman B, Lenihan DL, DeGeest K et al. Glucose as a prognostic factor in ovarian carcinoma. Cancer 2009; 115: 1021–1027.
Erickson K, Patterson RE, Flatt SW, Natarajan L, Parker BA, Heath DD et al. Clinically defined type 2 diabetes mellitus and prognosis in early-stage breast cancer. J Clin Oncol 2011; 29: 54–60.
Villarreal-Garza C, Shaw-Dulin R, Lara-Medina F, Bacon L, Rivera D, Urzua L et al. Impact of diabetes and hyperglycemia on survival in advanced breast cancer patients. Exp Diabetes Res 2012; 2012: 732027.
Minicozzi P, Berrino F, Sebastiani F, Falcini F, Vattiato R, Cioccoloni F et al. High fasting blood glucose and obesity significantly and independently increase risk of breast cancer death in hormone receptor-positive disease. Eur J Cancer 2013; 49: 3881–3888.
Mayer A, Vaupel P, Struss H-G, Giese A, Stockinger M, Schmidberger H . Strong adverse prognostic impact of hyperglycemic episodes during adjuvant chemoradiotherapy of glioblastoma multiforme. Strahlenther Onkol 2014; 190: 933–938.
Tieu MT, Lovblom LE, McNamara MG, Mason W, Laperriere N, Millar B-A et al. Impact of glycemia on survival of glioblastoma patients treated with radiation and temozolomide. J Neurooncol 2015; 124: 119–126.
Nikiteas NI, Tzanakis N, Gazouli M, Rallis G, Daniilidis K, Theodoropoulos G et al. Serum IL-6, TNFα and CRP levels in Greek colorectal cancer patients: prognostic implications. World J Gastroenterol 2005; 11: 1639–1643.
Coussens LM, Werb Z . Inflammation and cancer. Nature 2002; 420: 860–867.
Lv M, Zhu X, Wang H, Wang F, Guan W . Roles of caloric restriction, ketogenic diet and intermittent fasting during initiation, progression and metastasis of cancer in animal models: a systematic review and meta-analysis. PLoS ONE 2014; 9: e115147.
Cianfarani S . Insulin-like growth factor-II: new roles for an old actor. Front Endocrinol (Lausanne) 2012; 3: 118.
Yu H, Mistry J, Nicar MJ, Khosravi MJ, Diamandis A, Van Doorn J et al. Insulin-like growth factors (IGF-I, free IGF-I, and IGF-II) and insulin- like growth factor binding proteins (IGFBP-2, IGFBP-3, IGFBP-6, and ALS) in blood circulation. J Clin Lab Anal 1999; 13: 166–172.
Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R . Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev 2009; 30: 586–623.
Rajaram S, Baylink DJ, Mohan S . Insulin-like growth factor-binding proteins in serum and other biological fluids: regulation and functions. Endocr Rev 1997; 18: 801–831.
Sonksen P, Sonksen J . Insulin: understanding its action in health and disease. Br J Anaesth 2000; 85: 69–79.
Straus DS . Growth-stimulatory actions of insulin in vitro and in vivo. Endocr Rev 1984; 5: 356–369.
Dhillon AS, Hagan S, Rath O, Kolch W . MAP kinase signalling pathways in cancer. Oncogene 2007; 26: 3279–3290.
Grabacka M, Pierzchalska M, Reiss K . Peroxisome proliferator activated receptor α ligands as anticancer drugs targeting mitochondrial metabolism. Curr Pharm Biotechnol 2013; 14: 342–356.
Meynet O, Ricci J-E . Caloric restriction and cancer: molecular mechanisms and clinical implications. Trends Mol Med 2014; 20: 419–427.
Pollak M . The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer 2012; 12: 159–169.
Nowak K, Eldredge-Hindy H, Champ CE . Metformin: the sweet link between tumor genetics and metabolism? OA Cancer 2014; 2: 7.
Warburg O, Posener K, Negelein E . Über den Stoffwechsel der Carcinomzelle. Biochem Zeitschr 1924; 152: 309–343.
Warburg O . Über den Stoffwechsel der Carcinomzelle. Klin Wochenschr 1925; 4: 12–18.
Warburg O, Wind F, Negelein E . Über den Stoffwechsel der Tumoren im Körper. Klin Wochenschr 1926; 5: 829–838.
Robey RB, Hay N . Is Akt the ‘Warburg kinase’?−Akt-energy metabolism interactions. Semin Cancer Biol 2009; 19: 25–31.
Fine EJ, Feinman RD . Insulin, carbohydrate restriction, metabolic syndrome and cancer. Exp Rev Endocrin Metab 2014; 10: 15–24.
Laron Z . The GH-IGF1 axis and longevity. The paradigm of IGF1 deficiency. Hormones 2008; 7: 24–27.
Guevara-Aguirre J, Rosenbloom AL . Obesity, diabetes and cancer: insight into the relationship from a cohort with growth hormone receptor deficiency. Diabetologia 2015; 58: 37–42.
Osborne CK, Monaco ME, Lippman ME, Kahn CR . Correlation among insulin binding, degradation, and biological activity in human breast cancer cells in long-term tissue culture. Cancer Res 1978; 38: 94–102.
Osborne CK, Monaco ME, Kahn CR, Huff K, Bronzert D, Lippman ME . Direct inhibition of growth and antagonism of insulin action by glucocorticoids in human breast cancer cells in culture. Cancer Res 1979; 39: 2422–2428.
Cezard JP, Forgue-Lafitte ME, Chamblier MC, Rosselin GE . Growth-promoting effect, biological activity, and binding of insulin in human intestinal cancer cells in culture. Cancer Res 1981; 41: 1148–1153.
Sinclair J, McClain D, Taetle R . Effects of insulin and insulin-like growth factor I on growth of human leukemia cells in serum-free and protein-free medium. Blood 1988; 72: 66–72.
Vetter U, Schlickenrieder JH, Zapf J, Hartmann W, Heit W, Hitzler H et al. Human leukemic cells: receptor binding and biological effects of insulin and insulin-like growth factors. Leuk Res 1986; 10: 1201–1207.
Yamanouchi T, Tsushima T, Akanuma Y, Kasuga M, Mizoguchi H, Takaku F . Characteristics of insulin receptors and insulin action in human myelogenous leukemia cell line K-562. Diabetes 1985; 34: 347–352.
Rodeck U, Herlyn M, Menssen HD, Furlanetto RW, Koprowsk H . Metastatic but not primary melanoma cell lines grow in vitro independently of exogenous growth factors. Int J Cancer 1987; 40: 687–690.
Demetrakopoulos GE, Linn B, Amos H . Rapid loss of ATP by tumor cells deprived of glucose: contrast to normal cells. Biochem Biophys Res Commun 1978; 82: 787–794.
Shim H, Chun YS, Lewis BC, Dang CV . A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc Natl Acad Sci USA 1998; 95: 1511–1516.
Li Y, Liu L, Tollefsbol TO . Glucose restriction can extend normal cell lifespan and impair precancerous cell growth through epigenetic control of hTERT and p16 expression. FASEB J 2010; 24: 1442–1453.
Priebe A, Tan L, Wahl H, Kueck A, He G, Kwok R et al. Glucose deprivation activates AMPK and induces cell death through modulation of Akt in ovarian cancer cells. Gynecol Oncol 2011; 122: 389–395.
Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, Spitz DR . Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J 2009; 418: 29–37.
Graham NA, Tahmasian M, Kohli B, Komisopoulou E, Zhu M, Vivanco I . Glucose deprivation activates a metabolic and signaling amplification loop leading to cell death. Mol Syst Biol 2012; 8: 589.
Mathews EH, Stander BA, Joubert AM, Liebenberg L . Tumor cell culture survival following glucose and glutamine deprivation at typical physiological concentrations. Nutrition 2014; 30: 218–227.
Garcı-Jiménez C, Garcia-Martínez JM, Chocarro-Calvo A, De la Vieja A . A new link between diabetes and cancer: enhanced WNT/b-catenin signaling by high glucose. J Mol Endocrinol 2014; 52: R51–R61.
Masur K, Vetter C, Hinz A, Tomas N, Henrich H, Niggemann B et al. Diabetogenic glucose and insulin concentrations modulate transcriptom and protein levels involved in tumour cell migration, adhesion and proliferation. Br J Cancer 2011; 104: 345–352.
Tomas NM, Masur K, Piecha JC, Niggemann B, Zänker KS . Akt and phospholipase Cγ are involved in the regulation of growth and migration of MDA-MB-468 breast cancer and SW480 colon cancer cells when cultured with diabetogenic levels of glucose and insulin. BMC Res Notes 2012; 5: 214.
Kalaany NY, Sabatini DM . Tumours with PI3K activation are resistant to dietary restriction. Nature 2009; 458: 725–731.
Heuson JC, Legros N . Effect of insulin and of alloxan diabetes on growth of the rat mammary carcinoma in vivo. Eur J Cancer 1970; 6: 349–351.
Heuson JC, Legros N . Effect of insulin on DNA synthesis and DNA polymerase activity in organ culture of rat mammary carcinoma, and the inflence of insulin pretreatment and of alloxan diabetes. Cancer Res 1971; 31: 59–65.
Jiang Y-S, Wang F-R . Caloric restriction reduces edema and prolongs survival in a mouse glioma model. J Neurooncology 2013; 114: 25–32.
Marsh J, Mukherjee P, Seyfried TN . Akt-dependent proapoptotic effects of dietary restriction on late-stage management of a phosphatase and tensin homologue/tuberous sclerosis complex 2-deficient mouse astrocytoma. Clin Cancer Res 2008; 9: 7751–7762.
Curry NL, Mino-Kenudson M, Oliver TG, Yilmaz ÖH, Yilmaz VO, Moon JY et al. Pten-null tumors cohabiting the same lung display differential AKT activation and sensitivity to dietary restriction. Cancer Discov 2013; 3: 908–921.
Chaparro RJ, Konigshofer Y, Beilhack GF, Shizuru JA, McDevitt HO, Chien Y-H . Nonobese diabetic mice express aspects of both type 1 and type 2 diabetes. Proc Natl Acad Sci USA 2006; 103: 12475–12480.
Lee C, Safdie FM, Raffaghello L, Wei M, Madia F, Parrella E et al. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res 2010; 70: 1564–1572.
Yamaza H, Komatsu T, Wakita S, Kijogi C, Park S, Hayashi H et al. FoxO1 is involved in the antineoplastic effect of calorie restriction. Aging Cell 2010; 9: 372–382.
Klement RJ, Champ CE . Calories, carbohydrates, and cancer therapy with radiation: exploiting the five R’s through dietary manipulation. Cancer Metastasis Rev 2014; 33: 217–229.
Fine EJ, Miller A, Quadros EV, Sequeira JM, Feinman RD . Acetoacetate reduces growth and ATP concentration in cancer cell lines which over-express uncoupling protein 2. Cancer Cell Int 2009; 9: 14.
Shukla SK, Gebregiworgis T, Purohit V, Chaika NV, Gunda V, Radhakrishnan P et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab 2014; 2: 18.
Nebeling L, Miraldi F, Shurin S, Lerner E . Effects of a ketogenic diet on tumor metabolism and nutritional status in pediatric oncology patients: two case reports. J Am Coll Nutr 1995; 14: 202–208.
Fine EJ, Segal-isaacson CJ, Feinman RD, Herszkopf S, Romano MC, Tomuta N et al. Targeting insulin inhibition as a metabolic therapy in advanced cancer: A pilot safety and feasibility dietary trial in 10 patients. Nutrition 2012; 28: 1028–1035.
Schroeder U, Himpe B, Pries R, Vonthein R, Nitsch S, Wollenberg B . Decline of lactate in tumor tissue after ketogenic diet: in vivo microdialysis study in patients with head and neck cancer. Nutr Cancer 2013; 65: 843–849.
Tisdale MJ, Brennan RA . Loss of acetoacetate coenzyme A transferase activity in tumours of peripheral tissues. Brit J Cancer 1983; 47: 293–297.
Skinner R, Trujillo A, Ma X, Beierle EA . Ketone bodies inhibit the viability of human neuroblastoma cells. J Pediatr Surg 2009; 44: 212–216.
Maurer GD, Brucker DP, Bähr O, Harter PN, Hattingen E, Walenta S et al. Differential utilization of ketone bodies by neurons and glioma cell lines: a rationale for ketogenic diet as experimental glioma therapy. BMC Cancer 2011; 11: 315.
Chang HT, Olson LK, Schwartz KA . Ketolytic and glycolytic enzymatic expression profiles in malignant gliomas: implication for ketogenic diet therapy. Nutr Metab 2013; 10: 47.
Otto C, Klingelhöffer C, Biggermann L, Melkus G, Mörchel P, Jürgens C et al. Analysis of the metabolism of ketone bodies and lactate by gastrointestinal tumor cells in vitro. Aktuel Ernährungsmed 2014; 39: 51–59.
Woolf EC, Scheck AC . The ketogenic diet for the treatment of malignant glioma. J Lipid Res 2015; 56: 5–10.
Cohen ND, Hilf R . Influence of insulin on estrogen-induced responses in the R3230AC mammary carcinoma. Cancer Res 1975; 35: 560–567.
Hilf R, Hissin PJ, Shafie SM . Regulatory interrelationships for insulin and estrogen action in mammary tumors. Cancer Res 1978; 38: 4076–4085.
Hilf R, Livingston JN, Crofton DH . Effects of diabetes and sex steroid hormones on insulin receptor tyrosine kinase activity in R3230AC mammary adenocarcinomas. Cancer Res 1988; 48: 3742–3750.
Salter JM, de Meyer R, Best CH . Effect of insulin and glucagon on tumor growth. Br Med J 1958; 2: 5–7.
Goranson ES, Cinits E, Hercz A . The effect of glucagon on tumor growth. Cancer Res 1959; 19: 512–514.
Pavelić K, Vuk-Pavlović S . Retarded growth of murine tumors in vivo by insulin- and glucagon-stimulated immunity and phagocytosis. J Natl Cancer Inst 1981; 66: 889–892.
Sauer LA, Dauchy RT . Stimulation of tumor growth in adult rats in vivo during acute streptozotocin-induced diabetes. Cancer Res 1987; 47: 1756–1761.
Sauer LA, Nagel WO, Dauchy RT, Miceli LA, Austin JE . Stimulation of tumor growth in adult rats in vivo during an acute fast. Cancer Res 1986; 46: 3469–3475.
Sauer LA, Dauchy RT . Identification of linoleic and arachidonic acids as the factors in hyperlipemic blood that increase [3H] thymidine incorporation in hepatoma 7288CTC perfused in Situ1. Cancer Res 1988; 48: 3106–3111.
Fisher WE, Boros LG, Schirmer WJ . Reversal of enhanced pancreatic cancer growth in diabetes by insulin. Surgery 1995; 118: 453–457.
Hursting SD, Switzer BR, French JE, Kari FW . The growth hormone: Insulin-like growth factor 1 axis is a mediator of diet restriction-induced inhibition of mononuclear cell leukemia in Fischer rats. Cancer Res 1993; 53: 2750–2757.
Dunn SE, Kari FW, French J, Leininger JR, Travios G, Wilson R et al. Dietary restriction reduces insulin-like growth factor I levels, which modulates apoptosis, cell proliferation, and tumor progression in p53-deficient mice. Cancer Res 1997; 57: 4667–4672.
Nogueira LM, Lavigne JA, Chandramouli GVR, Lui H, Barrett JC, Hurstling SD . Dose-dependent effects of calorie restriction on gene expression, metabolism, and tumor progression are partially mediated by insulin-like growth factor-1. Cancer Med 2012; 1: 275–288.
Mitchell SE, Delville C, Konstantopedos P, Hurst J, Derous D, Green C et al. The effects of graded levels of calorie restriction: II. Impact of short term calorie and protein restriction on circulating hormone levels, glucose homeostasis and oxidative stress in male C57BL/6 mice. Oncotarget 2015; 6: 23213–23237.
Harvie MN, Pegington M, Mattson MP, Frystyk J, Dillon B, Evans G et al. The effects of intermittent or continuous energy restriction on weight loss and metabolic disease risk markers: a randomized trial in young overweight women. Int J Obes (Lond) 2011; 35: 714–727.
Harvie M, Wright C, Pegington M, Mcmullan D, Mitchell E, Martin B et al. The effect of intermittent energy and carbohydrate restriction v. daily energy restriction on weight loss and metabolic disease risk markers in overweight women. Brit J Nutr 2013; 110: 1534–1547.
Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R . Reversal of type 2 diabetes: Normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 2011; 54: 2506–2514.
Kitada M, Kume S, Takeda-Watanabe A, Tsuda S, Kanasaki K, Koya D . Calorie restriction in overweight males ameliorates obesity-related metabolic alterations and cellular adaptations through anti-aging effects, possibly including AMPK and SIRT1 activation. Biochim Biophys Acta 2013; 1830: 4820–4827.
Mercken EM, Crosby SD, Dudley W, Jebailey L, Krzysik-Walker S, Villareal DT et al. Calorie restriction in humans inhibits the PI3K/AKT pathway and induces a younger transcription profile. Aging Cell 2013; 12: 645–651.
Fontana L, Weiss EP, Villareal DT, Klein S, Holloszy JO, Holloszy O . Long-term effects of calorie or protein restriction on serum IGF-1 and IGFBP-3 concentrations in humans. Aging Cell 2008; 7: 681–687.
Mahoney LB, Denny CA, Seyfried TN . Calorie restriction in C57BL/6 J mice mimics therapeutic fasting in humans. Lipids Heal Dis 2006; 5: 13.
Irwin ML . Weight loss interventions and breast cancer survival: the time is now. J Clin Oncol 2014; 32: 2197–2199.
Simone BA, Champ CE, Rosenberg AL, Berger AC, Anne RP, Monti DA et al. Selectively starving cancer cells through dietary manipulation: methods and clinical implications. Futur Oncol 2013; 9: 959–976.
Robinson AM, Williamson DH . Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev 1980; 60: 143–187.
Borghjid S, Feinman RD . Response of C57Bl/6 mice to a carbohydrate-free diet. Nutr Metab 2012; 9: 69.
Meidenbauer JJ, Ta N, Seyfried TN . Influence of a ketogenic diet, fish-oil, and calorie restriction on plasma metabolites and lipids in C57BL/6 J mice. Nutr Metab 2014; 11: 23.
Volek JS, Sharman MJ, Love DM, Avery NG, Gómez AL, Scheett TP et al. Body composition and hormonal responses to a carbohydrate-restricted diet. Metabolism 2002; 51: 864–870.
Klement RJ, Frobel T, Albers T, Fikenzer S, Prinzhausen J . A pilot case study on the impact of a self-prescribed ketogenic diet on biochemical parameters and running performance in healthy and physically active individuals. Nutr Med 2013; 1: 10.
Forsythe CE, Phinney SD, Fernandez ML, Quann EE, Wood RJ, Bibus DM et al. Comparison of low fat and low carbohydrate diets on circulating fatty acid composition and markers of inflammation. Lipids 2008; 43: 65–77.
Feinman RD, Pogozelski WK, Astrup A, Bernstein RK, Fine EJ, Westman EC et al. Dietary Carbohydrate restriction as the first approach in diabetes management. Critical review and evidence base. Nutrition 2015; 31: 1–13.
Fabbrini E, Higgins PB, Magkos F, Bastarrachea Ra, Voruganti VS, Comuzzie AG et al. Metabolic response to high-carbohydrate and low-carbohydrate meals in a nonhuman primate model. Am J Physiol Endocrinol Metab 2013; 304: E444–E451.
Klein S, Wolfe RR . Carbohydrate restriction regulates the adaptive response to fasting. Am J Physiol 1992; 262: E631–E636.
Klement RJ . Calorie or carbohydrate restriction? The ketogenic diet as another option for supportive cancer treatment. Oncologist 2013; 18: 1056.
Klement RJ . Mimicking caloric restriction: what about macronutrient manipulation? A response to Meynet and Ricci. Trends Mol Med 2014; 20: 471–472.
Solon-Biet SM, McMahon AC, Ballard JWO, Ruohonen K, Wu LE, Cogger VC et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab 2014; 19: 418–430.
Stemmer K, Zani F, Habegger KM, Neff C, Kotzbeck P, Bauer M et al. FGF21 is not required for glucose homeostasis, ketosis or tumour suppression associated with ketogenic diets in mice. Diabetologia 2015; 58: 2414–2423.
Solon-Biet SM, Mitchell SJ, Coogan SCP, Cogger VC, Gokarn R, McMahon AC et al. Dietary protein to carbohydrate ratio and caloric restriction: comparing metabolic outcomes in mice. Cell Rep 2015; 11: 1529–1534.
Lees EK, Król E, Grant L, Shearer K, Wyse C, Moncur E et al. Methionine restriction restores a younger metabolic phenotype in adult mice with alterations in fibroblast growth factor 21. Aging Cell. 2014; 13: 817–827.
Isley WL, Underwood LE, Clemmons DR . Dietary components that regulate serum somatomedin-C concentrations in humans. J Clin Invest 1983; 71: 175–182.
Isley WL, Underwood LE, Clemmons DR . Changes in plasma somatomedin-C in response to ingestion of diets with variable protein and energy content. JPEN J Parenter Enteral Nutr 1984; 8: 407–411.
Donahue SP, Phillips LS . Response of IGF-1 to nutritional support in malnourished hospital patients: a possible indicator changes in nutritional status. Am J Clin Nutr 1989; 50: 962–969.
Smith WJ, Underwood LE, Clemmons DR . Effects of caloric or protein restriction on insulin-like growth factor-1 (IGF-1) and IGF-binding proteins in children and adults. J Clin Endocrinol Metab 1995; 80: 443–449.
Fraser DA, Thoen J, Bondhus S, Haugen M, Reseland JE, Djoseland O et al. Reduction in serum leptin and IGF-1 but preserved T-lymphocyte numbers and activation after a ketogenic diet in rheumatoid arthritis patients. Clin Exp Rheumatol 2000; 18: 209–214.
Fontana L, Weiss EP, Villareal DT, Klein S, Holloszy O . Long-term effects of calorie or protein restriction on serum IGF-1 and IGFBP-3 concentrations in humans. Aging Cell 2009; 7: 681–687.
Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E . Hepatic fibroblast growth factor 21 is regulated by pparα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab 2007; 5: 426–437.
Li Y, Wong K, Giles A, Jiang J, Lee JW, Adams AC et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology 2010; 146: 539–549.
Asrih M, Altirriba J, Rohner-Jeanrenaud F, Jornayvaz FR . Ketogenic diet impairs FGF21 signaling and promotes differential inflammatory responses in the liver and white adipose tissue. PLoS ONE 2015; 10: e0126364.
Laeger T, Henagan TM, Albarado DC, Redman LM, Bray GA, Noland RC et al. FGF21 is an endocrine signal of protein restriction. J Clin Invest 2014; 124: 3913–3922.
Kim KH, Lee M-S . FGF21 as a mediator of adaptive responses to stress and metabolic benefits of anti-diabetic drugs. J Endocrinol 2015; 226: R1–R16.
Abraham RT . Cell biology. Making sense of amino acid sensing. Science 2015; 347: 128–129.
Jewell JL, Kim YC, Russell RC, Yu F, Park HW, Plouffe SW et al. Differential regulation of mTORC1 by leucine and glutamine. Science 2015; 347: 194–198.
Thomas JD, Zhang Y-J, Wei Y-H, Cho J-H, Morris LE, Wang H-Y et al. Rab1A is an mTORC1 activator and a colorectal oncogene. Cancer Cell 2014; 26: 754–769.
Taylor C, Bartlett JD, van de Graaf CS, Louhelainen J, Coyne V, Iqbal Z et al. Protein ingestion does not impair exercise-induced AMPK signalling when in a glycogen-depleted state: implications for train-low compete-high. Eur J Appl Physiol 2013; 113: 1457–1468.
Draznin B, Wang C, Adochio R, Leitner JW, Cornier M-A . Effect of dietary macronutrient composition on AMPK and SIRT1 expression and activity in human skeletal muscle. Horm Metab Res 2012; 44: 650–655.
Dirx MJM, Zeegers MPA, Dagnelie PC, Van Den Bogaard T, Van Den Brandt PA . Energy restriction and the risk of spontaneous mammary tumors in mice: a meta-analysis. Int J Cancer 2003; 106: 766–770.
Frimberger E, Dagnelie PC, Ries P . Karzinomtherapie durch kontrolliertes Fasten. Dtsch Zschr Onkol 1992; 24: 1–9.
Magee BA, Potezny N, Rofe AM, Conyers RA . The inhibition of malignant cell growth by ketone bodies. Aust J Exp Biol Med Sci 1979; 57: 529–539.
Zhou W, Mukherjee P, Kiebish MA, Markis WT, Mantis JG, Seyfried TN . The calorically restricted ketogenic diet, an effective alternative therapy for malignant brain cancer. Nutr Metab 2007; 4: 5.
Seyfried TN, Sanderson TM, El-Abbadi MM, McGowan R, Mukherjee P . Role of glucose and ketone bodies in the metabolic control of experimental brain cancer. Br J Cancer 2003; 89: 1375–1382.
Otto C, Kaemmerer U, Illert B, Muehling B, Pfetzer N, Wittig R et al. Growth of human gastric cancer cells in nude mice is delayed by a ketogenic diet supplemented with omega-3 fatty acids and medium-chain triglycerides. BMC Cancer 2008; 8: 122.
Stafford P, Abdelwahab MG, Kim DY, Preul MC, Rho JM, Scheck AC . The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr Metab 2010; 7: 74.
Abdelwahab MG, Fenton KE, Preul MC, Rho JM, Lynch A, Stafford P et al. The ketogenic diet is an effective adjuvant to radiation therapy for the treatment of malignant glioma. PLoS ONE 2012; 7: e36197.
Allen BG, Bhatia SK, Buatti JM, Brandt KE, Lindholm KE, Button AM et al. Ketogenic diets enhance oxidative stress and radio-chemo-therapy responses in lung cancer xenografts. Clin Cancer Res 2013; 19: 3905–3913.
Hao G, Chen Y-S, He D-M, Wang H-Y, Wu G-H, Zhang B . Growth of human colon cancer cells in nude mice is delayed by ketogenic diet with or without omega-3 fatty acids and medium-chain triglycerides. Asian Pac J Cancer Prev 2015; 16: 2061–2068.
Morscher RJ, Aminzadeh-Gohari S, Feichtinger RG, Mayr JA, Lang R, Neureiter D et al. Inhibition of neuroblastoma tumor growth by ketogenic diet and/or calorie restriction in a CD1-Nu mouse model. PLoS ONE 2015; 10: e0129802.
Venkateswaran V, Haddad AQ, Fleshner NE, Fan R, Sugar LM, Nam R et al. Association of diet-induced hyperinsulinemia with accelerated growth of prostate cancer (LNCaP) xenografts. J Natl Cancer Inst 2007; 99: 1793–1800.
Ho VW, Leung K, Hsu A, Luk B, Lai J, Shen SY et al. A low carbohydrate, high protein diet slows tumor growth and prevents cancer initiation. Cancer Res 2011; 71: 4484–4493.
Dan TD, Wright CM, Simone NL, Dan E . What benefits could caloric restriction bring to cancer patients? Futur Oncol 2014; 10: 2543–2546.
Safdie FM, Dorff T, Quinn D, Fontana L, Wei M, Lee C et al. Fasting and cancer treatment in humans: a case series report. Aging (Albany NY) 2009; 1: 988–1007.
Zuccoli G, Marcello N, Pisanello A, Servadei F, Vaccaro S, Mukherjee P et al. Metabolic management of glioblastoma multiforme using standard therapy together with a restricted ketogenic diet: case report. Nutr Metab 2010; 7: 33.
Chu-Shore CJ, Thiele EA . Tumor growth in patients with tuberous sclerosis complex on the ketogenic diet. Brain Dev 2010; 32: 318–322.
Schmidt M, Pfetzer N, Schwab M, Strauss I, Kämmerer U . Effects of a ketogenic diet on the quality of life in 16 patients with advanced cancer: a pilot trial. Nutr Metab 2011; 8: 54.
Champ CE, Palmer JD, Volek JS, Werner-Wasik M, Andrews DW, Evans JJ et al. Targeting metabolism with a ketogenic diet during the treatment of glioblastoma multiforme. J Neurooncol 2014; 117: 125–131.
Rieger J, Bähr O, Maurer GD, Hattingen E, Franz K, Brucker D et al. ERGO: a pilot study of ketogenic diet in recurrent glioblastoma. Int J Oncol 2014; 44: 1843–1852.
Oshakbayev KP, Alibek K, Ponomarev IO, Uderbayev NN, Dukenbayeva BA . Weight change therapy as a potential treatment for end-stage ovarian carcinoma. AM J Case Rep 2014; 15: 203–211.
Schwartz K, Chang HT, Nikolai M, Pernicone J, Rhee S, Olson K et al. Treatment of glioma patients with ketogenic diets: report of two cases treated with an IRB-approved energy-restricted ketogenic diet protocol and review of the literature. Cancer Metab 2015; 3: 3.
Kalamian M, Zupec-Kania B, Favara BE, Liepa GU . Ketogenic diet as adjunctive therapy for brain tumors. In: First International Symposium on Dietary Treatments for Epilepsy and other Neurological Disorders. Poster presentation 2008.
Moore K, Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer. Wiley, Hoboken, NJ, USA, 2012, pp 397–400.
Seyfried TN, Flores RE, Poff AM, D’Agostino DP . Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis 2014; 35: 515–527.
Rossi-Fanelli F, Franchi F, Mulieri M, Cangiano C, Cascino A, Ceci F et al. Effect of energy substrate manipulation on tumour cell proliferation in parenterally fed cancer patients. Clin Nutr 1991; 10: 228–232.
Westin T, Stein H, Niedobitek G, Lundholm K, Edström S . Tumor cytokinetic response to total parenteral nutrition in patients with head and neck cancers. Am J Clin Nutr 1991; 53: 764–768.
Rabinovitch R, Grant B, Berkey BA, Raben D, Ang KK, Fu KK et al. Impact of nutrition support on treatment outcome in patients with locally advanced head and neck squamous cell cancer treated with definitive radiotherapy: a secondary analysis of RTOG trial 90-03. Head Neck 2006; 28: 287–296.
Nixon DW, Lawson DH, Kutner MH, Moffitt SD, Ansley J, Heymsfield SB et al. Effect of total parenteral nutrition on survival in advanced colon cancer. Cancer Detect Prev 1981; 4: 421–427.
Cordain L, Miller JB, Eaton SB, Mann N, Holt SHA, Speth JD . Plant-animal subsistence ratios and macronutrient energy estimations in worldwide hunter-gatherer diets. Am J Clin Nutr 2000; 71: 682–692.
Chandrasekera PC, Pippin JJ . Of rodents and men: species-specific glucose regulation and type 2 diabetes research. ALTEX 2013; 31: 157–176.
Ecsedy JA, Holthaus KA, Yohe HC, Seyfried TN . Expression of mouse sialic acid on gangliosides of a human glioma grown as a xenograft in SCID mice. J Neurochem 1999; 73: 254–259.
Lai M, Chandrasekera PC, Barnard ND . You are what you eat, or are you? The challenges of translating high-fat-fed rodents to human obesity and diabetes. Nutr Diabetes 2014; 4: e135.
Sohal RS, Forster MJ . Caloric restriction and the aging process: a critique. Free Radic Biol Med 2014; 73: 366–382.
Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, Navarrete G et al. A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab 2015; 22: 86–99.
Demetrius L . Of mice and men. EMBO Rep 2005; 6 Suppl 1: S39–S44.
Bozzetti F, Zupec-Kania B . Toward a cancer-specific diet. Clin Nutr 2015 (e-pub ahead of print 23 January 2015; doi:10.1016/j.clnu.2015.01.013).
Schlüter G . Toxicology of Acarbose, with special reference to long-term carcinogenecity studies. In: Creutzfeldt W (ed). Acarbose for the Treatment of Diabetes Mellitus. Springer: Berlin, 1988.
McCarty MF, DiNicolantonio JJ . Acarbose, lente carbohydrate, and prebiotics promote metabolic health and longevity by stimulating intestinal production of GLP-1. Open Heart 2015; 2: e000205–e000205.
Warburg O, Wind F, Negelein E . The metabolism of tumors in the body. J Gen Physiol 1927; 8: 519–530.
Brünings W . Beiträge zum Krebsproblem. 1. Mitteilung: Ueber eine diätetisch-hormonale Beeinflussung des Krebses. Münch med Wschr 1941; 88: 117–123.
Brünings W . Beiträge zum Krebsproblem. 2. Mitteilung: Klinische Anwendungen der diätetisch-hormonalen Krebsbeeinflussung (‘Entzuckerungsmethode’). Münch med Wschr 1942; 89: 71–76.
Schulte G, Schütz H . Insulin in der Krebsbehandlung. Münch med Wschr 1942; 89: 648–650.
Weiss J . Über Erfahrungen mit Insulin und kohlenhydratreduzierender Diät bei inkurablen Krebskranken. Med Klin 1957; 52: 1190–1191.
Koroljow S . Two cases of malignant tumors with metastases apparently treated successfully with hypoglycemic coma. Psychiatr Q 1962; 36: 261–270.
Ayre SG, Garcla y Bellon DP, Garcia DP Jr . Insulin, chemotherapy, and the mechanisms of malignancy: the design and the demise of cancer. Med Hypotheses 2000; 55: 330–334.
Alabaster O, Vonderhaar BK, Shafie SM . Metabolic modification by insulin enhances methotrexate cytotoxicity in MCF-7 human breast cancer cells. Eur J Cancer Clin Oncol 1981; 17: 1223–1228.
Fink M, Berger M . Ist eine Optimierung der Tumortherapie durch eine gezielte Beeinflussung des Insulinspiegels möglich? In: Kadach U, Kaufmann M, Kubli F (eds). Aktuelle Onkologie, Band 27: Hormone, Antihormone, Zytostatika zur adjuvanten Therapie des Mammakarzinoms. Zuckschwerdt: München, 1986. 162–176.
Fink M, Abenhardt W, Ostermayr B, Berger M . Insulin and diazoxide for treatment of cancer. Results of clinical pilot studies. Onkologie 1991; 14: 158–164.
Damyanov C, Gerasimova D, Maslev I, Gavrilov V . Low-dose chemotherapy with insulin (insulin potentiation therapy) in combination with hormone therapy for treatment of castration-resistant prostate cancer. ISRN Urol 2012; 2012: 140182.
Sturgess NC, Kozlowski RZ, Carrington Ca, Hales CN, Ashford ML . Effects of sulphonylureas and diazoxide on insulin secretion and nucleotide-sensitive channels in an insulin-secreting cell line. Br J Pharmacol 1988; 95: 83–94.
Trube G, Rorsman P, Ohno-Shosaku T . Opposite effects of tolbutamide and diazoxide on the ATP-dependent K+ channel in mouse pancreatic β-cells. Pflügers Arch 1986; 407: 493–499.
Skoglund G, Ahrén B, Lundquist I . Biochemical determination of islet lysosomal enzyme activities following crinophagy-stimulating treatment with diazoxide in mice. Diabetes Res 1987; 6: 81–84.
Nestler JE, Barlascini CO, Matt DW, Steingold Ka, Plymate SR, Clore JN et al. Suppression of serum insulin by diazoxide reduces serum testosterone levels in obese women with polycystic ovary syndrome. J Clin Endocrinol Metab 1989; 68: 1027–1032.
Nestler JE, Singh R, Matt DW, Clore JN, Blackard WG . Suppression of serum insulin level by diazoxide does not alter serum testosterone or sex hormone-binding globulin levels in healthy, nonobese women. Am J Obstet Gynecol 1990; 163: 1243–1246.
Krassas GE, Kaltsas TT, Pontikides N, Jacobs H, Blum W, Messinis I . Leptin levels in women with polycystic ovary syndrome before and after treatment with diazoxide. Eur J Endocrinol 1998; 139: 184–189.
Tamburrano G, Lala A, Mauceri M, Leonetti F, Libianchi S, Andreani D . Diazoxide infusion test in patients with single benign insulinoma. Horm Res 1983; 17: 141–146.
Hillaire-Buys D, Ribes G, Blayac JP, Loubatières-Mariani MM . A new benzothiadiazine derivative, LN 5330: effects on pancreatic hormones. Diabetologia 1984; 27: 583–586.
Eaton RP, Schade DS . The effect of diazoxide-induced hormonal secretion on plasma triglyceride concentration in the rat. Diabetologia 1980; 18: 301–306.
Goode PN, Farndon JR, Anderson J, Johnston IDa, Morte JA . Diazoxide in the management of patients with insulinoma. World J Surg 1986; 10: 586–591.
Kühnau jr J, Martin W . Mehrjährige Behandlung des Hyperinsulinismus mit Diazoxid. Dtsch med Wschr 1972; 97: 1870–1873.
Meier A, Weidmann P, Glück Z, Keusch G, Grimm M, Minder I et al. Vergleich von oralem Diazoxid und Minoxidil bei therapieresistenter Hypertonie. Klin Wochenschr 1980; 58: 681–687.
Berger MR, Fink M, Feichter GE, Janetschek P . Effects of diazoxide-induced reversible diabetes on chemically induced autochthonous mammary carcinomas in Sprague-Dawley rats. Int J Cancer 1985; 35: 395–401.
Fink M, Berger M . Treatment of the DMBA-induced rat mammary carcinoma by diabetes-induction with diazoxide. J Steroid Biochem 1983; 19: 48S abstract 142.
Klenner T, Berger MR, Zelezny O, Fink M, Schmähl D . Antineoplastic efficacy of melphalan and N-(2-chloroethyl)-N-nitrosocarbamoyl-ω-lysine, in combination with diazoxide or insulin in autochthonous mammary carcinoma of the Sprague-Dawley rat. J Cancer Res Clin Oncol 1990; 116: 45–50.
Berger M, Tilhein B, Fink M, Schmähl B . Therapy of 7,12-DMBA induced rat mammary carcinomas with combinations of diaxozide and tamoxifen or medroxyprogesterone acetate. J Steroid Biochem 1983; 19: 47S abstract 141.
Holmuhamedov E, Lewis L, Bienengraeber M, Holmuhamedova M, Jahangir A, Terzic A . Suppression of human tumor cell proliferation through mitochondrial targeting. FASEB J 2002; 16: 1010–1016.
Ding J, Ge D, Guo W, Lu C . Diazoxide-mediated growth inhibition in human lung cancer cells via downregulation of β-catenin-mediated cyclin D1 transcription. Lung 2009; 187: 61–67.
Abdul M, Hoosein N . Voltage-gated potassium ion channels in colon cancer. Oncol Rep 2002; 9: 961–964.
Huang L, Li B, Li W, Guo H, Zou F . ATP-sensitive potassium channels control glioma cells proliferation by regulating ERK activity. Carcinogenesis 2009; 30: 737–744.
Ru Q, Tian X, Wu Y, Wu R, Pi M, Li C . Voltage-gated and ATP-sensitive K+ channels are associated with cell proliferation and tumorigenesis of human glioma. Oncol Rep 2014; 31: 842–848.
Cox MM, Wendler CC, Erdelyi I, Beck A, Zeiss C, Rivkees SA . Chronic neonatal diazoxide therapy is not associated with adverse effects. Online J Biol Sci 2014; 14: 49–56.
O’Dell SD, Day INM . Molecules in focus: Insulin-like growth factor II (IGF-II). Int J Biochem Cell Biol 1998; 30: 767–771.
Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol 1999; 19: 3278–3288.
Singh P, Alex JM, Bast F . Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1 R) signaling systems: novel treatment strategies for cancer. Med Oncol 2014; 31: 805.
Gallagher EJ, Alikhani N, Tobin-Hess A, Blank J, Buffin NJ, Zelenko Z et al. Insulin receptor phosphorylation by endogenous insulin or the insulin analog AspB10 promotes mammary tumor growth independent of the IGF-I receptor. Diabetes 2013; 62: 3553–3560.
Inoki K, Li Y, Xu T, Guan KL . Rheb GTpase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003; 17: 1829–1834.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
Oncogenesis is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Klement, R., Fink, M. Dietary and pharmacological modification of the insulin/IGF-1 system: exploiting the full repertoire against cancer. Oncogenesis 5, e193 (2016). https://doi.org/10.1038/oncsis.2016.2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/oncsis.2016.2
This article is cited by
-
Nutrient restriction-activated Fra-2 promotes tumor progression via IGF1R in miR-15a downmodulated pancreatic ductal adenocarcinoma
Signal Transduction and Targeted Therapy (2024)
-
Protein intake and cancer: an umbrella review of systematic reviews for the evidence-based guideline of the German Nutrition Society
European Journal of Nutrition (2024)
-
Feasibility and metabolic effects of a 5:2 fasting intervention in women with breast cancer during radiotherapy
Holistic Integrative Oncology (2023)
-
Metabolic factors associated with the prognosis of oligometastatic patients treated with stereotactic body radiotherapy
Cancer and Metastasis Reviews (2023)
-
Randomised controlled trial of intermittent vs continuous energy restriction during chemotherapy for early breast cancer
British Journal of Cancer (2022)
