
Abstract
Abnormal expression of activating/inhibitory receptors leads to natural killer (NK) cells dysfunction in tumor. Here we show that programmed cell death protein 1 (PD-1), a well-known immune checkpoint of T cells, is highly expressed on peripheral and tumor-infiltrating NK cells from patients with digestive cancers including esophageal, liver, colorectal, gastric and biliary cancer. The increased PD-1 expression on NK cells indicates poorer survival in esophageal and liver cancers. Blocking PD-1/PD-L1 signaling markedly enhances cytokines production and degranulation and suppresses apoptosis of NK cells in vitro. PD-1/PD-L1 exerts inhibitory effect through repressing the activation of PI3K/AKT signaling in NK cells. More importantly, a PD-1 blocking antibody was found to significantly suppress the growth of xenografts in nude mice, and this inhibition of tumor growth was completely abrogated by NK depletion. These findings strongly suggested that PD-1 is an inhibitory regulator of NK cells in digestive cancers. PD-1 blockade might be an efficient strategy in NK cell-based tumor immunotherapy.
Similar content being viewed by others


Introduction
Digestive cancers include a heterogeneous group of cancers originating from digestive tract and associated organs. Five of the ten leading causes of cancer death are digestive cancers, which account for over 3 million deaths per year worldwide.1 Chronic inflammation plays a critical role in most digestive cancers, such as hepatitis-related hepatocellular carcinoma (HCC), Helicobacter pylori infection-related gastric cancer and colitis-related colon cancer.2, 3, 4 During the process of uncontrolled inflammation, various immune cells are recruited to the microenvironment and contribute to carcinogenesis.5 Among them, CD8+ T cells and natural killer (NK) cells are critical components. The dysfunction of CD8+ T cells and NK cells has been well described in digestive cancers6, 7, 8, 9 and is regarded as one of the most important mechanisms leading to disease progression.10 In fact, immunotherapy aimed at restoring anti-tumor immunity has joined the ranks of surgery, radiation, chemotherapy and targeted therapy as another pillar of cancer therapy.11
Programmed cell death protein 1 (PD-1) is one of the most important immune checkpoints corresponding to T-cell dysfunction in many solid tumors.12 PD-1 is expressed on activated T cells. Engagement of PD-1 with programmed death ligand 1 (PD-L1) expressed on cancer cells prevents the expansion and function of effector T cells and eventually results in T-cell exhaustion, thereby leading to tumor immune evasion.13 Remarkably, monoclonal antibodies against PD-1 and PD-L1 have been approved by the U.S. Food and Drug Administration and have led to impressive and highly encouraging clinical results in the treatment of melanoma and lung cancer.11 Accumulating data demonstrate increased expression of both PD-1 in T cells and PD-L1 in tumors cells from patients with different types of digestive cancers, including HCC, esophageal squamous cell carcinoma (ESCC), gastric cancer and colorectal cancer.14, 15, 16, 17, 18 Thus, although related treatments have not yet approved, blocking PD-1/PD-L1 interactions may have potential in the treatment of digestive cancers.19
NK cells are important cytotoxic innate immune cells that are involved in the elimination of cancer cells.20 Two main NK cell subsets have been defined on the basis of CD56 and CD16 expression. A CD56brightCD16− NK subset produces abundant cytokines including interferon-γ (IFN-γ) and tumor necrosis factor-α, whereas a CD56dimCD16+ NK subpopulation has high cytolytic activity and releases granules containing perforin and granzymes.21 The activity of both NK cell subsets is precisely controlled by a series of inhibitory and activating receptors, thus endowing NK cells with the ability to eliminate tumor cells.21 However, tumor-induced unbalanced expression of activating/inhibitory receptors hampers NK cell-mediated immune surveillance and promotes tumor progression. As a result, people with advanced cancer often process dysfunctional NK cells,22 and rescuing the function of NK cells in cancer immunotherapy has been attracting substantial attention.23, 24
Several studies have detected PD-1 expression on NK cells in various clinical settings; however, the results have not been consistent.25, 26, 27, 28, 29, 30, 31, 32 PD-1 is highly expressed on peripheral CD56dim NK cells of healthy donors serologically positive for human cytomegalovirus,33 whereas PD-1 expression has been reported on CD56bright NK cells in patients with chronic hepatitis C virus infection.30 Increased PD-1 expression has been demonstrated in chronic viral infection and acute infection with Citrobacter rodentium but not in acute tuberculosis infection.17, 18, 19 Several new studies have also reported enhanced PD-1 expression from tumors including multiple myeloma31 and Kaposi sarcoma.34 Similarly to observations from studies of viral infection, PD-1+ NK cells from Kaposi sarcoma patients display functional hyporesponsiveness, thus suggesting the involvement of PD-1 in NK functional defects in tumor.34 However, whether and how PD-1 contributes to perturbing the anti-tumor functions of NK cells remain unclear. Here we provide the first reported evidence that PD-1 expression on NK cells significantly correlates with poor prognosis in digestive cancers. Moreover, in vitro and in vivo studies clearly demonstrated that PD-1/PD-L1 blockade greatly suppresses the growth of HCC xenografts through promoting NK cells functions.
Results
Increased PD-1 expression on peripheral and tumor infiltrating NK cells from patients with digestive cancers leads to poorer prognosis
We first detected PD-1 expression on peripheral NK cells in several digestive cancers including ESCC, HCC, colorectal cancer, gastric cancer and biliary cancer by flow cytometry. Compared with that in healthy controls, a significant increase in PD-1 expression on CD3−CD56+ NK cells was observed in all cancer patients (Figures 1a and b). Further analysis showed that both CD56dimCD16+ and CD56brightCD16- NK cells from patients with all studied digestive cancer expressed much higher level of PD-1 than those from healthy controls (Figures 1c and d).

PD-1 expression is upregulated on peripheral NK cells isolated from digestive cancer patients and indicates poor prognosis. (a) Lymphocytes were gated according to forward scatter and side scatter. CD3 and CD56 staining was used to identify NK cells. (b) Graph comparing PD-1 expression on NK cells from patients with digestive cancers and healthy individuals. (c) CD3− cells from a representative patient was further gated into two NK-cell subsets on the basis of CD56 and CD16 expression (left). Plots showing PD-1 expression on CD16+CD56dim(right) or CD16-CD56bright(middle) NK cells. (d) Graph comparing PD-1 expression on CD3−CD16-CD56bright(left) and CD3−CD16+CD56dim(right) NK cells in patients with digestive cancers and healthy individuals. (e) Graphs comparing PD-1 expression on CD3−CD56+ NK cells in ESCC patients with different degrees of pathological differentiation (left) or different T (middle) or N stages (right). PD-1 expression level on NK cells is represented as the percentage of PD-1+ NK cells. Cumulative data are shown as mean±s.e.m., analyzed by Student’s t-test. The data were normally distributed and had constant variances. *P⩽0.05, **P⩽0.01, ***P⩽0.001.
We subsequently examined PD-1 expression on tumor-infiltrating NK (TINK) cells. Both immunofluorescence staining and flow cytometry confirmed PD-1 expression on TINK cells from HCC patients (Figures 2a and b). Similarly to our results for peripheral NK cells, PD-1 was detected on both CD56dimCD16+ and CD56brightCD16- subsets of TINK cells (Figure 2c). Furthermore, PD-1 expression on TINK cells was significantly higher than that on infiltrating NK cells from the paired adjacent non-cancerous tissues (Figure 2b). Interestingly, we observed an extremely high expression of PD-1 on tumor-infiltrated CD49a+ NK cells, which have recently been identified as liver resident NK cells in humans35, 36 (Figure 2d).

PD-1 is expressed on tumor-infiltrating NK cells. (a) Immunofluorescence staining was performed in an HCC tissue microarray (n=33). Representative immunofluorescence images showing co-localization of PD-1 on CD56+ NK cells. The clinical characteristics of these patients are listed in Supplementary Table S2. (b) Histograms showing PD-1 expression on CD3−CD56+ NK cells isolated from paratumor and tumor tissues from a representative patient (upper and middle) and pooled data (low). (c) Dot plots showing PD-1 expression on CD16-CD56bright and CD16+CD56dim NK cells infiltrated in tumor tissues. (d) Dot plots showing the percentage of CD49a+ NK cells in tumor tissues (left). Histograms showing high PD-1 expression on CD49a+ tumor-infiltrating NK cells (right). Cumulative data are shown as mean±s.e.m., analyzed by Student’s t-test. The data were normally distributed and had constant variances. *P⩽0.05, **P⩽0.01, ***P⩽0.001.
To further evaluate the involvement of PD-1+ NK cells in tumor development, PD-1 expression on peripheral NK cells from ESCC patients and TINK cells from HCC patients were analyzed according to related clinical characteristics. As shown in Figure 1e, PD-1 expression on peripheral NK cells correlated with the degree of tumor differentiation, tumor lymph nodes metastasis stage and metastasis in ESCC. The level of PD-1 expression on peripheral NK cells from patients with G3 stage cancer was higher than that from patients with G2 or G1 stage cancer (left, Figure 1e). NK cells from ESCC patients with T3~T4 stage cancer exhibited higher PD-1 expression than did patients with T1~T2 stage cancer (middle, Figure 1e). Accordingly, NK cells from patients with lymph node metastasis had higher PD-1 expression than did patients without lymph node metastasis (right, Figure 1e). These data supported the notion that PD-1 expression on CD3−CD56+ NK cells was higher in ESCC patients with poorer prognostic factors. Furthermore, immunofluorescence staining of specimens from 33 HCC patients confirmed the significant correlation of PD-1 expression on CD56+ TINK cells with shorter survival (P=0.008) (Supplementary Figure S1). Thus, PD-1 expression on either peripheral or infiltrating NK cells from patients with digestive cancer was significantly upregulated and the increased expression of PD-1 on NK cells predicted a poorer prognosis.
PD-1/PD-L1 blockade increased IFN-γ production and CD107a expression in NK cells
PD-1 expression can be induced by either cytokine stimulation or infection.37 We next examined whether PD-1 on NK cells from digestive cancer could also be regulated. As shown in Figures 3a and b, both phorbol-12-myristate-13-acetate (PMA) and ionomycin or interleukin (IL)-12 and IL-15 stimulation markedly enhanced PD-1 expression on NK cells from ESCC patients in a time-dependent manner.

PD-1/PD-L1 blockade increased IFN-γ production and CD107a expression in NK cells. (a and b) Plots from a representative ESCC patient (a) and pooled data showing the induced surface expression of PD-1 on peripheral NK cells after PMA and ionomycin (b left panel) or IL-12 and IL-15 (b right panel) stimulation. Pooled data from ESCC patients showing dynamically increasing PD-1 expression after PMA and ionomycin stimulation (b middle panel). (c) NK cells from ESCC patients were stimulated with PMA and ionomycin for 5 h, after which CD107a degranulation(upper panel) was compared between PD-1+ and PD-1− NK cells. Expression of informative surface markers, NKG2A (middle panel) and NKG2D (low panel), was also compared between PD-1+ and PD-1− NK cells. (d–f) After pre-incubation with 2 μg/ml (d) or 8 μg/ml (e) PD-L1 blocking antibodies (aPD-L1) or 2 μg/ml PD-1 blocking antibodies (aPD-1) (f) and matched IgG, NK cells isolated from ESCC patients were stimulated with PMA and ionomycin for 5 h. IFN-γ secretion and CD107a degranulation of NK cells were compared between two groups. Representative flow cytometry analyses from one patient are located in the left and middle panels. Pooled data are located in the right panel. Cumulative data are shown as mean±s.e.m., analyzed by Student’s t-test. The data were normally distributed and had constant variances. *P⩽0.05, **P⩽0.01, ***P⩽0.001.
By analyzing the relationship between PD-1 expression and NK-cell activation, degranulation and cytokine production, we found that PD-1+ NK cells had higher expression of CD107a and NKG2D but not NKG2A than PD-1− NK cells (Figure 3c), a result consistent with findings from a report by Emanuela Marcenaro.33
To directly assess the involvement of PD-1 in NK cell functions, neutralizing antibodies against PD-L1 were used. Treatment with 2 μg/ml or 8 μg/ml PD-L1 blocking antibodies significantly increased the percentages of both IFN-γ+ and CD107a+ NK cells from ESCC patients (Figures 3d and e), thus indicating that PD-1 is an inhibitory regulator of NK cells. This finding was further confirmed with PD-1 blocking antibodies (Figure 3f).
PD-1 ligation preferentially promotes apoptosis of PD-1+ NK cells
Because PD-1 and PD-L1 ligation specifically induced apoptosis in PD-1+CD8+ T cells and leads to the damage of T-cell function,38 we next sought to determine whether PD-1 signaling regulated the apoptosis of NK cells. Compared with that in PD-1− NK cells, the percentage of annexin V+ cells in PD-1+ NK cells was significantly higher (Figure 4a), thus indicating a correlation of apoptosis sensitivity with PD-1 expression in NK cells. Moreover, PD-1 ligation with plate-bound anti-PD-1 greatly enhanced the apoptosis of PD-1+ NK cells up to 50% or more, whereas the apoptosis of PD-1− NK cells did not increase (Figure 4b). Furthermore, blockade with PD-L1 neutralizing antibodies markedly decreased the apoptosis of PD-1+ NK cells but not PD-1− NK cells (Figure 4c). Together, all these results suggested that PD-1 signaling promotes the apoptosis in NK cells.

PD-1/PD-L1 signaling induces apoptosis of PD-1+ NK. (a) Representative dot plots (left panel) depicting the spontaneous apoptosis (annexin-V staining) of PD-1+ and PD-1− NK cells from one ESCC patient and pooled data (right panel). (b) NK cells from ESCC patients were cultured without or with plate-bound anti-PD-1 (aPD-1) (8 μg/ml), and apoptosis (annexin-V staining) of PD-1− and PD-1+ NK cells was compared between the two groups. PMA and ionomycin were applied for 3 h to stimulate activation-induced apoptosis. (c) After pre-incubation with 8 μg/ml IgG or PD-L1 blocking antibodies (aPD-L1), NK cells isolated from ESCC patients were stimulated with PMA and ionomycin and apoptosis of PD-1− and PD-1+ NK cells were compared. Cumulative data are shown as mean±s.e.m., analyzed by Student’s t-test. The data were normally distributed and had constant variances. *P⩽0.05, **P⩽0.01, ***P⩽0.001.
PD-1/PD-L1 blockade augments the activation of the PI3K/AKT signaling pathway in NK cells
One of the well-known downstream signals of PD-1/PD-L1 in primary T cells is the PI3K/AKT signaling pathway.39 PD-1/PD-L1 interaction blocks PI3K activation, suppresses phosphorylation of AKT and subsequently inhibits T-cell function.39, 40 PI3K/AKT signaling in NK cells is important for their responsiveness to cytokine stimulation and their cytotoxic functions.41 Thus we suspected that PD-1 might achieve its effect on NK cells by targeting AKT phosphorylation. Because sorting a sufficient number of NK cells from peripheral blood mononuclear cells requires large amounts of blood, we used NK92 and NKL cells, two human NK-cell lines, to test our hypothesis. After an hour of PMA and ionomycin stimulation, the whole-cell lysates were used to evaluate phosphorylated AKT. As shown in Figure 5a, treatment with either PD-1 blocking antibodies or PD-L1 blocking antibodies significantly enhanced PMA-induced activation of AKT. To further simulate conditions in which NK cells interact with target cells, EC9706 cells, a ESCC cell line, were co-cultured with NK92 or NKL cells. In these co-culture systems, by interaction with PD-L1 expressed on EC9706 cells (Supplementary Figure S2), NK cells received PD-1 signaling. Besides, we also detected PD-L1 expression on NK92 and NKL cells (Supplementary Figure S2), indicating that NK cells might provide PD-L1 as well. In agreement with PMA induced signaling, PD-1 or PD-L1 blockade markedly increased the phosphorylation of AKT in NK92 and NKL cells (Figure 5b). These data suggested that PD-1 may exert its inhibitory effect on NK cells through interfering with AKT activation.

PD-1/PD-L1 blockade enhances the pAKT signaling pathway in NK92 and NKL cells. (a and b) After pre-incubation with 2 μg/ml IgG or matched PD-1/PD-L1 blocking antibodies (aPD-1 or aPD-L1), NK92 or NKL cells were stimulated with PMA and ionomycin (a) or EC9706 target cells for 1 h, and western blot analysis was used to determine AKT phosphorylation. The results in a and b are representative of three independent experiments.
PD-1 blockade inhibited tumor growth through NK cells in nude mice
Because PD-1 blockade enhances NK-cell function, we hypothesized that anti-PD-1 therapy might enhance the anti-tumor function of NK cells in vivo. To avoid the interference of T cells, xenografts were prepared in nude mice with EC9706 cells that did not express PD-1 (Supplementary Figure S3). Anti-PD-1 antibody, which has previously been described as blocking function in vivo,42 was included for in vivo blocking. Compared with IgG control treatment, anti-PD-1 treatment significantly repressed tumor growth in nude mice (Figures 6a and b). Ex vivo studies with NK cells isolated from spleen and tumor tissues showed that anti-PD-1 treatment augmented CD69 expression both on spleen and TINK cells (Figures 6c and d), thus suggesting that PD-1 blockade promoted NK-cell activation, and possibly accounting for the tumor suppression. To further verify the role of NK cells in tumor suppression induced by PD-1 blocking in nude mice, anti-asGM1 was used to deplete NK cells as described in the methods. The NK cell depletion in the spleen and tumor was evaluated by flow cytometry (Figure 6e). As expected, the inhibition of anti-PD-1 treatment on tumor growth was completely abrogated by NK-cell depletion, thus suggesting that anti-PD-1 treatment exerted its effect through NK cells (Figure 6f). These data demonstrated that PD-1 blockade enhanced the function of NK cells and suppressed tumor growth in vivo.

Administration of PD-1 blocking antibodies inhibits tumor growth through enhancing NK-cell activity in nude mice. (a–d) The EC9706 tumor-bearing nude mouse models were established and treated with anti-PD-1 blocking antibodies (aPD-1) or matched IgG. (a) Images of tumors at the time of killing from each group are shown. (b) Tumor size was monitored every 3~4 days. (c) Flow cytometric plots showing CD69 expression on CD3−DX5+ NK cells from spleen and tumor tissues. (d) Summarized data showing CD69 expression on NK cells from spleen. (e–f) NK cells were depleted as previously described in the methods and the tumor-bearing mouse models were then established. Anti-PD-1 antibodies (aPD-1) or IgG was administered as before. (e) Flow cytometric plots showing the successful depletion of NK cells. (f) Images of tumors at the time of killing (left panel) and tumor weight (right panel) from each group are shown. The size of the tumors was also monitored (middle panel). Cumulative data are shown as mean±s.e.m., analyzed by Student’s t-test. The data were normally distributed and had constant variances. *P⩽0.05, **P⩽0.01, ***P⩽0.001.
Discussion
PD-1 is well-known for its critical role in suppressing T-cell activation and proliferation.43 Blocking antibodies against PD-1/PD-L1 have been successfully used as new strategies for promoting T-cell-mediated anti-tumor immunity in clinics.11 Recent data have confirmed PD-1 expression on NK cells in different disease models.30, 33, 34 However, whether PD-1 blockade enhances NK-cell-mediated anti-tumor immunity is still unknown. Here our work systematically and comprehensively demonstrated the important role of PD-1 in the NK-cell-mediated anti-tumor immune response. The data here provide the first direct evidence of the therapeutic effect of PD-1 blockade in NK-cell-based tumor immune therapy.
NK cells are well known as the first line of anti-tumor immunity. Dysfunction of NK cells, including decreased cell number and defects in cytotoxicity, has been reported in many solid tumors.22, 23 Indeed, various strategies aimed at rescuing NK-cell dysfunction or enhance NK-cell cytotoxicity have been reported and have displayed great potential in tumor therapy.21 Our data here strongly suggested PD-1 as a potential target to rescue NK-cell dysfunction in tumor therapy. First, PD-1 expression was significantly upregulated on peripheral and TINK cells in patients with different digestive cancers. More importantly, the increased PD-1 expression on NK cells correlated well with poor prognosis, such as tumor differentiation grades, tumor metastasis and patients survival (Figure 1), thus strongly suggesting the involvement of NK cell-expressed PD-1 in cancer progression. Second, PD-L1 blockade with different doses of blocking antibodies significantly enhanced the degranulation and IFN-γ secretion of cultured NK cells from ESCC patients. Similar effects were also found with PD-1 blockade (Figure 3). Furthermore, cross linkage of PD-1 with plate-bound antibodies promoted apoptosis in PD-1+ NK cells but not PD-1− NK cells, whereas neutralizing antibodies against PD-L1 inhibited the apoptosis of PD-1+ NK cells (Figure 4). Third and more directly, treatment with neutralizing antibodies against PD-1 significantly suppressed the growth of xenografts in nude mice in an NK-cell-dependent manner (Figure 6). Together with previous studies reporting the critical role of PD-1 in CD8+ T-cell exhaustion in many digestive cancers,19, 44 our data offer evidence of the potential of PD-1 blockade in the treatment of digestive cancers by promoting both CD8+ T cells and NK-cells-mediated immunity.
PD-1 expression has been detected on human NK cells from different populations, including healthy donors and patients with tumor or chronic infection.33 PD-1 might be preferentially expressed on activated NK cells because almost all reported PD-1+ NK cells have come from individuals who had been exposed to antigens, such as tumor antigen, viral antigen and transplantation antigen.28, 30, 33 Even in the healthy donors, all the subjects with PD-1+ NK cells were seropositive for human cytomegalovirus, thus suggesting exposure to human cytomegalovirus.33 Accordingly, PD-1+ NK cells displayed higher level of CD69, CD107a, perforin and granzyme than PD-1− NK cells.33 Our results showed that activation by either cytokines or PMA quickly and significantly enhanced PD-1 expression on NK cells from ESCC patients (Figure 3). The phenotype of PD-1+ NK cells appears to be diverse. Marcenaro E has defined PD-1+ NK cells in human cytomegalovirus-positive healthy individuals as fully matured CD57+ NK cells displaying higher level of NKp46 and NKp30 than those in PD-1− NK cells. PD-1+ NK cells from patients with Kaposi sarcoma have been described as activated and mature NK cells which express levels of NKp46 and NKp30 comparable to those in PD-1− NK cells.34 Alternatively, in patients with chronic hepatitis C virus infection, PD-1 expression has been detected on immature CD56bright NK cells.30 Here we demonstrated increased PD-1 expression on both CD56bright and CD56dim NK cells from patients with digestive cancers (Figures 1c and d). More interestingly, in HCC specimens, we detected abundant PD-1 expression on infiltrated CD3−CD49a+CD56+ NK cells, the newly identified hepatic resident NK cells (Figure 2d). To our knowledge, this is the first report demonstrating the presence of PD-1 on resident NK cells, and our results further suggest the potential role of PD-1 in an NK-cell-mediated dysfunctional tumor microenvironment. Accordingly, PD-1 expression on CD56+ TINK cells suggested shorter survival in HCC patients (Supplementary Figure S1). Therefore, it is necessary to explore the mechanisms responsible for upregulation of PD-1 on NK cells.
Our data also demonstrated that PD-1/PD-L1 blockade augmented phosphorylation of AKT in NK92 and NKL cells, thus indicating that PD-1 may exert its inhibitory function on NK cells through interfering with PI3K/AKT signaling (Figure 5). In T cells, co-ligation of PD-1 and its ligands results in rapid activation of Src homology region 2 containing tyrosine phosphatase-2 (SHP-2), thus causing dephosphorylation of key signal transducers of activation signaling, for example activation of AKT is consequently stopped.45 Our data suggested that the biochemical mechanisms by which PD-1 regulates NK and T cells may share much in common. Activation of PI3K/AKT is indispensable in maintaining the appropriate anti-tumor function of immune cells including NK and T cells. However, activation of the PI3K/AKT signaling in tumor cells is instrumental in promoting tumor development and resistance to anticancer therapies.46 Numerous efforts have been made to develop PI3K/AKT/mTOR inhibitors for cancer treatment and several drugs (everolimus and temsirolimus) have been approved by the US. Food and Drug Administration.46 Our data here together with those from previous studies showing that PD-1 promotes deactivation of AKT in CD8+ T cells40 strongly suggest the potential side effects of drugs targeting PI3K/AKT signaling, thus limiting the anti-tumor function of PD-1 blockage in immune therapy. Therefore, precautions should be taken when PI3K/AKT targeted therapy is used, and the function of CD8+ T cells and NK cells should be monitored.
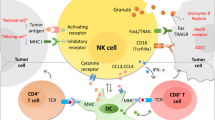
In conclusion, in this report, we show that the PD-1/PD-L1 signaling mediates NK-cell activation and cytotoxicity. PD-1 expression on NK cells is increased in digestive cancer patients, and is further increased when stimulated. Cancer cells tend to overexpress PD-L1. PD-1/PD-L1 ligation inhibits NK-cell anti-tumor effects, thereby protecting tumor cells from being killed (Figure 7). Together, these findings provide novel mechanistic data and expand on previous experience with PD-1/PD-L1 blocking antibodies in the treatment of digestive cancers.

Graphic model as discussed in the text. PD-1 expression on NK cells is enhanced in digestive cancer patients, and is further induced when stimulated. Ligation of PD-1 on NK cells with PD-L1 on tumor cells inhibits activation PI3K/AKT signaling of NK cells and thus suppresses NK–cell-mediated anti-tumor activity. PD-1/PD-L1 blockade treatment rescues NK-cell function and may benefit digestive cancer patients.
Materials and methods
Human subjects and cell lines
A total of 79 subjects, including 42 patients with ESCC, 15 patients with HCC, 6 patients with colorectal cancer, 3 patients with gastric cancer, 1 patient with biliary cancer and 12 healthy individuals, were enrolled in this study. The diagnosis of all patients was confirmed on the basis of surgery and pathology. Blood and tissue samples were collected from the Qilu Hospital of Shandong University between September 2015 and November 2016. The healthy control group was recruited from the Healthy Physical Examination Center of Qilu Hospital. Although the investigators were not blinded during collecting the clinical samples, the involvement of at least two investigators in each step has ensured the validity of the data. The study was approved by the Ethics Committee of Shandong University, and informed consent was acquired from each individual.
EC9706 human ESCC cells were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum. Human NK cell line NK92 cells were cultured in NK92 medium (α-medium containing 10% fetal bovine serum, 10% horse serum, 0.2 mM inositol, 0.1 mM β-mercaptoethanol, 0.02 mM folic acid) containing 100 U/ml recombinant human IL-2. Human NK cell line NKL cells were cultured in 1640 medium with 10% fetal bovine serum containing 100 U/ml human IL-2. EC9706 cells and NK92 cells were purchased from ATCC. NKL cell line was kindly gifted by Professor BQ Jin (Department of Immunology, Fourth Military Medical University, Xi’an, China). The cell lines were recently authenticated.
Peripheral blood mononuclear cells and preparation of tissue infiltrated lymphocytes
Blood samples were processed within 6 h. Peripheral blood mononuclear cells were prepared by centrifugation with Ficoll-Paque Plus (Amersham Biosciences, Piscataway, NJ, USA). Infiltrating lymphocytes were isolated from HCC tissues and matched paratumor tissues as previously described.47
Functional assay
Freshly isolated peripheral blood mononuclear cells were used in an NK-cell cytokine production assay, degranulation assay and blocking assay. For the cytokine production assay, cells were incubated for 5 h with PMA (50 ng/ml) and ionomycin (1 μg/ml) or IL-12 (12 ng/ml) and IL-15 (100 ng/ml), and GolgiStop (1:1000). For the degranulation assay, a fluorescence-conjugated anti-CD107a mAb was added as soon as the cells were stimulated, and then the activated cells were collected and stained for surface molecules. For the blocking assay, cells were pretreated with PD-1 blocking antibodies (R&D systems, Minneapolis, MN, USA) or PD-L1 blocking antibodies (Biolegend, San Diego, CA, USA) or matched IgG for 30 min before receiving stimulation. After being processed, the stained cells were analyzed by flow cytometry (BD, FACS Aria II, San Jose, CA, USA).
Flow cytometric analysis
For surface molecules staining, cells were stained with appropriate mixed fluorescence conjugated mAb or isotype controls for 30 min in the dark. For intracellular staining, activated cells were first stained for surface molecules, then fixed with fixation buffer, permeabilized with permealization buffer, and stained for 30 min in the dark with fluorescence-labeled IFN-γ. All of the fluorescence-labeled antibodies were purchased from Biolegend: PE/Cy7-conjugated anti-human CD56 (318318), Percy/cy5.5-conjugated anti-human CD3 (300328), FITC-conjugated anti-human PD-1 (329904), APC-conjugated anti-human CD107a (328620), BV421-conjugated anti-human IFN-γ (502531), Percp/cy5.5-conjugated anti-human CD16 (302028), APC-conjugated anti-mouse CD3(100235), FITC-conjugated anti-mouse DX5 (108905) and PE/Cy7-conjugated anti-mouse CD69 (104511).
Apoptosis studies
For the PD-1 cross linking assay, 106 peripheral blood mononuclear cells were cultured in 24-well plates (Corning, Corning, NY, USA) in the absence or presence of plate-bound anti-human PD-1(R&D systems) (10 μg/ml). For the PD-L1 blocking assay, cells were pre-incubated with human anti-PD-L1 blocking mAb, at 2 μg/ml for 30 min at 37 °C before being cultured in 24-well plates. PMA and ionomycin stimulation was used for activation-induced apoptosis. Three hours later, the cells were collected and surface stained with APC-labeled CD3-PE, CD56-PE/cy7, and PD-1-PE/dazzle and subsequently with annexin V-FITC. Stained cells were then analyzed using flow cytometry.
Cell lysis and western blotting
Cells were lysed in RIPA lysis buffer containing 1% phenylmethanesulfonyl fluoride and 1% phosphatase inhibitor cocktail. Proteins were resolved by SDS–PAGE and transferred to polyvinylidene fluoride membranes. Rabbit anti-phospho-Akt antibody (CST 4691p), rabbit anti-Akt antibody (CST 4060p), mouse anti-GAPDH antibody (Goodbio,GB11002) and secondary horseradish peroxidase-conjugated antibodies were used for the assay. The proteins were detected using an ECL detection system (Thermo Fisher Scientific, San Jose, CA, USA).
Tissue microarrays and immunofluorescence microscopy
Tissue microarrays containing 33 HCC tissue cores (2 mm in diameter, Supplementary Table S1) were ordered from the Shanghai Outdo biotech company (HLiv-HCC180Sur-05). For immunofluorescence double staining, sections were first deparaffinized and then incubated with retrieval solution (ZSGB-Bio, Beijing, China) for 15 min in a microwave oven. After treatment with 10% goat serum at 37 °C for half an hour, the tissues were incubated with anti-CD56 (Abcam, Cambridge, MA, USA) and anti-PD-1(Abcam) antibodies overnight. The sections were then incubated with FITC or PE-labeled secondary antibodies (ZSJQ-Bio). 4',6-diamidino-2-phenylindole was used for counterstaining. The staining was detected on a Zeiss DM2500 microscope with × 20 objectives.
PD-1 blockade and NK-cell depletion in nude mice with xenografts of EC9706 cells
For the xenograft assay, 3 × 106 EC9706 cells were injected subcutaneously into the right flanks of 5-week-old male nude mice. The minimum sample size, five mice per group, is calculated using Sigma Stat3.5 on the basis of preliminary experimental data (probability level of 0.05 and power of 0.80). Ten mice were numbered from 1 to 10 and randomly divided into two groups with the help of random number table. Anti-PD-1 antibodies (BioXcell, West Lebanon, NH, USA) or IgG (BioXcell) were administered i.p. to mice 1 day after injection, and once every 3–4 days for 2 weeks. Tumor size was measured every 3–4 days with a caliper. The tumor volume was calculated according to the formula: volume=(length × width2)/2. NK-cell depletion was achieved by i.p. injection of 30 μl undiluted anti-asialo GM1 antibodies (Wako Pure Chemical Industries, Osaka, Japan) every 4–5 days, starting 2 days before EC9706 cells engraftment, thus resulting in >80% depletion of CD3− CD49b(DX5)+ NK cells in the nude mice as determined by flow cytometry. Animals were killed 17 days later. NK cells infiltrating in the spleen and tumor were extracted as previously described.48 Although no blinding was carried out during the process, the involvement of at least two investigators in each step has ensured the validity of the data. All of the animal studies were approved by the Animal Care and Use Committee of Shandong University and conducted in compliance with UKCCCR guidelines.
Statistical analysis
The clinical sample size was calculated using Sigma Stat3.5 to ensure the validity of the results. Data are reported as mean values±s.e.m. All data were analyzed using GraphPad Prism 5.0. All data were normally distributed and had constant variances. The statistical significance of differences between two groups was determined by Student’s t-test. P-values <0.05 were considered significant.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A . Global cancer statistics, 2012. CA Cancer J Clin 2015; 65: 87–108.
Giannelli G, Rani B, Dituri F, Cao Y, Palasciano G . Moving towards personalised therapy in patients with hepatocellular carcinoma: the role of the microenvironment. Gut 2014; 63: 1668–1676.
Ferrone C, Dranoff G . Dual roles for immunity in gastrointestinal cancers. J Clin Oncol 2010; 28: 4045–4051.
Lin EW, Karakasheva TA, Hicks PD, Bass AJ, Rustgi AK . The tumor microenvironment in esophageal cancer. Oncogene 2016; 35: 5337–5349.
Zhang L, Song X, Mohri Y, Qiao L . Role of inflammation and tumor microenvironment in the development of gastrointestinal cancers: what induced pluripotent stem cells can do? Curr Stem Cell Res Ther 2015; 10: 245–250.
Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012; 56: 1342–1351.
Wu Y, Kuang DM, Pan WD, Wan YL, Lao XM, Wang D et al. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology 2013; 57: 1107–1116.
Watanabe M, Kono K, Kawaguchi Y, Mizukami Y, Mimura K, Maruyama T et al. NK cell dysfunction with down-regulated CD16 and up-regulated CD56 molecules in patients with esophageal squamous cell carcinoma. Dis Esophagus 2010; 23: 675–681.
Zhang G, Liu WL, Zhang L, Wang JY, Kuang MH, Liu P et al. Involvement of indoleamine 2,3-dioxygenase in impairing tumor-infiltrating CD8 T-cell functions in esophageal squamous cell carcinoma. Clin Dev Immunol 2011; 2011: 384726.
Wargo JA, Reddy SM, Reuben A, Sharma P . Monitoring immune responses in the tumor microenvironment. Curr Opin Immunol 2016; 41: 23–31.
Sharma P, Allison JP . The future of immune checkpoint therapy. Science 2015; 348: 56–61.
Kamphorst AO, Ahmed R . Manipulating the PD-1 pathway to improve immunity. Curr Opin Immunol 2013; 25: 381–388.
Li Y, Liang L, Dai W, Cai G, Xu Y, Li X et al. Prognostic impact of programed cell death-1 (PD-1) and PD-ligand 1 (PD-L1) expression in cancer cells and tumor infiltrating lymphocytes in colorectal cancer. Mol Cancer 2016; 15: 55.
Chen K, Cheng G, Zhang F, Zhang N, Li D, Jin J et al. Prognostic significance of programmed death-1 and programmed death-ligand 1 expression in patients with esophageal squamous cell carcinoma. Oncotarget 2016; 7: 30772–30780.
Saito H, Kuroda H, Matsunaga T, Osaki T, Ikeguchi M . Increased PD-1 expression on CD4+ and CD8+ T cells is involved in immune evasion in gastric cancer. J Surg Oncol 2013; 107: 517–522.
Ma C, Patel K, Singhi AD, Ren B, Zhu B, Shaikh F et al. Programmed death-ligand 1 expression is common in gastric cancer associated with Epstein–Barr virus or microsatellite instability. Am J Surg Pathol 2016; 40: 1496–1506.
Toh JW, de Souza P, Lim SH, Singh P, Chua W, Ng W et al. The potential value of immunotherapy in colorectal cancers: review of the evidence for programmed death-1 inhibitor therapy. Clin Colorectal Cancer 2016; 15: 285–291.
Moreno-Cubero E, Larrubia JR . Specific CD8(+) T cell response immunotherapy for hepatocellular carcinoma and viral hepatitis. World J Gastroenterol 2016; 22: 6469–6483.
Pardoll DM . The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12: 252–264.
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S . Functions of natural killer cells. Nat Immunol 2008; 9: 503–510.
Guillerey C, Huntington ND, Smyth MJ . Targeting natural killer cells in cancer immunotherapy. Nat Immunol 2016; 17: 1025–1036.
Bucklein V, Adunka T, Mendler AN, Issels R, Subklewe M, Schmollinger JC et al. Progressive natural killer cell dysfunction associated with alterations in subset proportions and receptor expression in soft-tissue sarcoma patients. Oncoimmunology 2016; 5: e1178421.
Tarazona R, Sanchez-Correa B, Casas-Aviles I, Campos C, Pera A, Morgado S et al. Immunosenescence: limitations of natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother 2016; 66: 233–245.
Chen Y, Chen B, Yang T, Xiao W, Qian L, Ding Y et al. Human fused NKG2D-IL-15 protein controls xenografted human gastric cancer through the recruitment and activation of NK cells. Cell Mol Immunol 2017; 14: 293–307.
Solaymani-Mohammadi S, Lakhdari O, Minev I, Shenouda S, Frey BF, Billeskov R et al. Lack of the programmed death-1 receptor renders host susceptible to enteric microbial infection through impairing the production of the mucosal natural killer cell effector molecules. J Leukoc Biol 2016; 99: 475–482.
Hassan SS, Akram M, King EC, Dockrell HM, Cliff JM . PD-1, PD-L1 and PD-L2 gene expression on T-cells and natural killer cells declines in conjunction with a reduction in PD-1 protein during the intensive phase of tuberculosis treatment. PLoS One 2015; 10: e0137646.
Norris S, Coleman A, Kuri-Cervantes L, Bower M, Nelson M, Goodier MR . PD-1 expression on natural killer cells and CD8(+) T cells during chronic HIV-1 infection. Viral Immunol 2012; 25: 329–332.
Wiesmayr S, Webber SA, Macedo C, Popescu I, Smith L, Luce J et al. Decreased NKp46 and NKG2D and elevated PD-1 are associated with altered NK-cell function in pediatric transplant patients with PTLD. Eur J Immunol 2012; 42: 541–550.
Chen Y, Wu S, Guo G, Fei L, Guo S, Yang C et al. Programmed death (PD)-1-deficient mice are extremely sensitive to murine hepatitis virus strain-3 (MHV-3) infection. PLoS Pathog 2011; 7: e1001347.
Golden-Mason L, Klarquist J, Wahed AS, Rosen HR . Cutting edge: programmed death-1 expression is increased on immunocytes in chronic hepatitis C virus and predicts failure of response to antiviral therapy: race-dependent differences. J Immunol 2008; 180: 3637–3641.
Benson DM Jr., Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010; 116: 2286–2294.
Huang BY, Zhan YP, Zong WJ, Yu CJ, Li JF, Qu YM et al. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS One 2015; 10: e0134715.
Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: a phenotypic and functional characterization. J Allergy Clin Immunol 2017; 139: 335–346.e3.
Beldi-Ferchiou A, Lambert M, Dogniaux S, Vely F, Vivier E, Olive D et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget 2016; 7: 72961–72977.
Peng H, Jiang X, Chen Y, Sojka DK, Wei H, Gao X et al. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J Clin Invest 2013; 123: 1444–1456.
Hydes T, Abuhilal M, Armstrong T, Primrose J, Takhar A, Khakoo S . Natural killer cell maturation markers in the human liver and expansion of an NKG2C+KIR+ population. Lancet 2015; 385 (Suppl 1): S45.
Kinter AL, Godbout EJ, McNally JP, Sereti I, Roby GA, O'Shea MA et al. The common γ-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol 2008; 181: 6738–6746.
Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med 2006; 203: 2281–2292.
Riley JL . PD-1 signaling in primary T cells. Immunol Rev 2009; 229: 114–125.
Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005; 25: 9543–9553.
Ali AK, Nandagopal N, Lee SH . IL-15-PI3K-AKT-mTOR: a critical pathway in the life journey of natural killer cells. Front Immunol 2015; 6: 355.
Shi L, Chen L, Wu C, Zhu Y, Xu B, Zheng X et al. PD-1 blockade boosts radiofrequency ablation-elicited adaptive immune responses against tumor. Clin Cancer Res 2016; 22: 1173–1184.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515: 568–571.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012; 366: 2455–2465.
Blank C, Gajewski TF, Mackensen A . Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother 2005; 54: 307–314.
Polivka J Jr., Janku F . Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther 2014; 142: 164–175.
Yan W, Liu X, Ma H, Zhang H, Song X, Gao L et al. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut 2015; 64: 1593–1604.
Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol 2016; 17: 816–824.
Acknowledgements
We would like to thank Professor Weiming Zhao from the Department of Pathogenic Biology for the gift of anti-asialoGM1. We would like to thank Mingsheng Zhao from the Department of Immunology for help with the in vivo experiments. This work was supported by grants from the National Science Foundation for Distinguished Young Scholars of China (81425012), NSFC (91529305 and 81371831) and the Shandong Provincial Natural Science Foundation (ZR2015HZ005).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Liu, Y., Cheng, Y., Xu, Y. et al. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 36, 6143–6153 (2017). https://doi.org/10.1038/onc.2017.209
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2017.209
This article is cited by
-
CT-based radiomics nomogram to predict proliferative hepatocellular carcinoma and explore the tumor microenvironment
Journal of Translational Medicine (2024)
-
First-line penpulimab (an anti-PD1 antibody) and anlotinib (an angiogenesis inhibitor) with nab-paclitaxel/gemcitabine (PAAG) in metastatic pancreatic cancer: a prospective, multicentre, biomolecular exploratory, phase II trial
Signal Transduction and Targeted Therapy (2024)
-
Intravenous administration of IL-12 encoding self-replicating RNA-lipid nanoparticle complex leads to safe and effective antitumor responses
Scientific Reports (2024)
-
Advances in PD-1 signaling inhibition-based nano-delivery systems for tumor therapy
Journal of Nanobiotechnology (2023)
-
BRCA1 and BRCA2 deficient tumour models generate distinct ovarian tumour microenvironments and differential responses to therapy
Journal of Ovarian Research (2023)
