
Abstract
Epithelial–mesenchymal transition (EMT) has been recognized as a key element of cell migration and invasion in lung cancer; however, the underlying mechanisms are not fully elucidated. Recently, emerging evidence suggest that miRNAs have crucial roles in control of EMT and EMT-associated traits such as migration, invasion and chemoresistance. Here, we found that miR-218 expression levels were significantly downregulated in lung cancer tissues compared with adjacent non-cancerous tissues, and the levels of miR-218 were significantly associated with histological grades and lymph node metastasis. Overexpression of miR-218 inhibited cell migration and invasion as well as the EMT process. Of particular importance, miR-218 was involved in the metastatic process of lung cancer cells in vivo by suppressing local invasion and distant colonization. We identified Slug and ZEB2 as direct functional targets of miR-218. Inverse correlations were observed between miR-218 levels and Slug/ZEB2 levels in cancer tissue samples. In addition, overexpression of miR-218 in H1299 increased chemosensitivity of cells to cisplatin treatment through suppression of Slug and ZEB2. These findings highlight an important role of miR-218 in the regulation of EMT-related traits and metastasis of lung cancer in part by modulation of Slug/ZEB2 signaling, and provide a potential therapeutic strategy by targeting miR-218 in NSCLC.
Similar content being viewed by others


Introduction
Lung cancer is the most common cancer worldwide. Approximately 80% of lung cancers are non-small cell lung cancer (NSCLC), with high mortality and low 5-year survival rate of <15% after initial diagnosis.1, 2 Owing to recurrence, extensive invasion and metastasis, the clinical outcomes for patients with NSCLC are still dismal. Cisplatin-based chemotherapy is widely used as the first-line chemotherapeutic agent for advanced NSCLC.3 However, the chemotherapy response varies among patients, and the development of resistance is one of the biggest obstacles against successful chemotherapy in clinical application. Previous studies have demonstrated that epithelial–mesenchymal transition (EMT), especially that induced by transforming growth factor β1 (TGF-β1), is a necessary and a key process in metastasis of cancer including NSCLC.4, 5, 6 Cancer cells undergoing EMT may acquire altered traits, including migration, invasion and chemoresistance to cisplatin and epidermal growth factor tyrosine kinase inhibitors in diverse cancers.7, 8 Therefore, it is vital to investigate the molecular mechanisms underlying the EMT and EMT-related traits of NSCLC, which could help to develop novel prognostic biomarkers and an efficient strategy for the treatment of NSCLC.
MicroRNAs (miRNAs) are a class of short noncoding RNAs, approximately 18–24 nucleotides in length, which post-transcriptionally modulate gene expression by either inhibiting translation or inducing mRNA degradation.9 MiRNAs can function as either tumor suppressors or oncogenes, which are involved in various biological and pathological processes such as cell proliferation, apoptosis, differentiation and tumorigenesis.10, 11, 12 Emerging evidence has indicated that miRNAs regulate EMT and EMT-mediated drug resistance. For example, the miR-200 family members have been shown to reduce cell migration and invasion by targeting ZEB1 and ZEB2 in variety cancers including lung cancer.13, 14, 15, 16 Overexpression of miR-216a/217 induced EMT and promoted drug resistance by targeting PTEN and SMAD7.17 Similarly, ectopic expression of miR-200b and miR-15b reversed the phenotype of EMT in human tongue cancer cells, and sensitized them to cisplatin by targeting BMI1.18 These findings suggest that alterations of certain miRNAs expression in cancer cells may contribute to cancer EMT progression.
Deregulation of miR-218 has been reported in different types of tumors.19, 20 In lung cancer, overexpression of miR-218 inhibited cell proliferation, migration and invasion by targeting genes such as epidermal growth factor receptor, HMGB1,21, 22 and was associated with cisplatin chemosensitivity,23 suggested a critical role of miR-218 in lung cancer development. However, the function of miR-218 in lung cancer progression, in particular EMT and subsequently invasion, metastasis and chemoresistance, remains to be investigated. Here, we aimed to evaluate the underlying roles and mechanisms of miR-218 in lung tumorigenesis. We found that the expression level of miR-218 was significantly lower in NSCLC tissues than in the corresponding normal lung tissues, and inversely associated with advanced stage and lymph node metastasis of NSCLC. Overexpression of miR-218 inhibited EMT and EMT-related traits by targeting the EMT regulator Slug and ZEB2 in vitro and in vivo. These observations would provide new insights into the molecular mechanism of lung cancer development as well as new therapeutic strategy in the future.
Results
MiR-218 was downregulated and associated with advanced clinical stage and metastasis in NSCLC
Microarray-based miRNA expression profiling of A549 and A549/TGF-β1 cells revealed a set of differentially expressed miRNA candidates. We found that miR-218 was the most prominent downregulated miRNA in response to TGF-β1 treatment, and selected miR-218 for further analysis (Supplementary Table1). We first assayed the expression levels of miR-218 in 60 pairs of human NSCLC and their corresponding non-cancerous lung tissues. By stem-loop quantitative reverse transcription–PCR, we verified that the expression levels of miR-218 were significantly decreased in human NSCLC compared with those of adjacent non-cancerous lung tissues (Figure 1a). Correlation analysis showed that the miR-218 expression level was reversely correlated to histological grade (Figure 1b). Moreover, when 60 NSCLC samples were classified based on the occurrence of lymph node metastasis, we found that the expression levels of miR-218 were lower in NSCLC with lymph node metastasis compared with those without metastasis (Figure 1c, Supplementary Table2). These results indicated that the reduced expression of miR-218 is associated with highly metastatic capacity of NSCLC cells.

MiR-218 was downregulated and associated with advanced clinical stage and NSCLC metastasis. (a) The relative mRNA levels of miR-218 were detected by qRT–PCR and normalized against an endogenous control (U6 RNA) in 60 pairs of lung cancer tissues. (b) Relative expression levels of miR-218 in different stages of cancer tissues. (c) qRT–PCR analysis of miR-218 expression levels in 60 pairs of primary NSCLC tissues and their corresponding lymph node metastases. Data represent mean±s.d. of three replicates. *Indicated significant difference at P<0.05; **Indicated significant difference at P<0.01.
MiR-218 negatively regulated the migration, invasion and EMT of NSCLC cells
To determine the effect of miR-218 on cancer cell metastasis, we first assessed the expression levels of miR-218 in four human lung cancer cell lines (A549, H1299, PC9 and SPCA-1). The miR-218 expression levels were the lowest in H1299 and the highest in A549 among four lung cancer cell lines (Figure 2a). Meanwhile, the migration and invasion ability of the four cell lines were tested by Transwell assay with or without coated Matrigel. As is shown in Figure 2b, H1299 cells (which were originally derived from a lymph node metastasis of the lung from a patient) showed a strong migration and invasiveness, whereas A549 cells exhibited the lowest ability for migration and invasion, which were inversely correlated with the expression levels of miR-218 in these cells. Then, we chose H1299 and A549 cells for subsequent analysis accordingly. H1299 cells with lower endogenous miR-218 expression levels were applied in gain-of-function studies using miR-218 mimics, whereas A549 cells with higher miR-218 levels were applied in loss-of-function studies using anti-miR-218 inhibitors. Overexpression of miR-218 in H1299 cells reduced cell migration and invasion ability by 50% and 40%, respectively, compared with control cells (Figure 2c). In contrast, inhibition of miR-218 in A549 cells increased cell migration and invasion ability by >3.5- and 4-fold (Figure 2d). In addition, when transfected with miR-218 mimics, H1299 cells underwent morphological change from a spindle shaped to a rounded or cobblestone-like shaped. In contrast, more rounded A549 cells became spindle shaped when transfected with anti-miR-218 inhibitors (Figure 2e). These observations suggested that miR-218 may be involved in the EMT of NSCLC cells. Next, we assessed EMT markers E-cadherin and Vimentin using western blot analysis. The protein levels of E-cadherin and Vimentin were significantly increased and decreased, respectively, in H1299 cells overexpressing miR-218 (Figure 2f). Furthermore, inhibition of miR-218 caused the decrease of E-cadherin and increase of Vimentin expression in A549 cells (Figure 2f). Similar results were obtained by immunofluorescence staining assay (Figure 2g).


MiR-218 negatively regulated the migration, invasion and EMT of NSCLC cells. (a) The expression levels of miR-218 were detected by qRT–PCR in human lung cancer cells (A549, H1299, PC9 and SPCA-1). (b) The levels of cell migration and invasion in indicated human lung cancer cells (A549, H1299, PC9 and SPCA-1) were analyzed using the Transwell chambers assay with or without coated Matrigel. (c) MiR-218 overexpression decreased cell migration and invasion activities in H1299 cells. Indicated cells were subjected to migration and invasion assay. Scale bar, 20 μm. (d) Anti-miR-218 inhibitor increased cell migration and invasion activities in A549 cells. Indicated cells were subjected to migration and invasion assay. Scale bar, 20 μm. (e) Phase contrast images of H1299 and A549 cells in indicated treatment. H1299 transfected with miR-218 underwent morphological change from a spindle-shaped to a rounded or cobblestone-like shaped. In contrast, more rounded A549 cells became spindle-shaped after the treatment with anti-miR-218 inhibitor. (f) Western blot analysis of EMT markers E-cadherin and Vimentin were shown in H1299 and A549 cells, respectively. (g) Immunofluorescence images of EMT markers, E-cadherin and Vimentin were shown in H1299 and A549 cells, respectively. Data represent mean±s.d. of three replicates. *Indicated significant difference at P<0.05; **Indicated significant difference at P<0.01.
It was reported that A549 cells undergo EMT morphological changes upon exposure to TGF-β1.24 To explore whether miR-218 is involved in TGF-β1-induced EMT, we first analyzed expression levels of miR-218 in A549 cells treated by TGF-β1. Upon treatment with TGF-β1, the expression levels of miR-218 were decreased and A549 cells acquired spindle-type cell morphology (Supplementary Figures 1A and B). Overexpression of miR-218 partly reversed TGF-β1-induced morphological change in A549 cell (Supplementary Figure 1B). Furthermore, western blot analysis and qRT–PCR assay showed that A549 cells treated with miR-218 and TGF-β1 stimulation showed diminished E-cadherin expression and increased Vimentin expression (Supplementary Figures 1C and D). These results suggest that miR-218 contributes to TGF-β1-induced EMT in NSCLC cells.
MiR-218 directly targets Slug and ZEB2
To elucidate the molecular mechanism by which miR-218 suppresses the metastasis of NSCLC cells, we used bioinformatics tools to identify its potential target genes. MiR-218 is predicted to target Slug and ZEB2 (Figure 3a), which are known to be relevant to EMT and tumor metastasis.25 In order to prove whether miR-218 directly targets both Slug and ZEB2, 3′-UTR sequences containing wide-type or mutant miR-218-binding sites of Slug and ZEB2 were cloned into the pMIR-REPORT vector, respectively. H1299 cells were co-transfected with reporter plasmids and miR-218 or miR-NC. MiR-218-transfected cells showed a remarkable inhibition of luciferase activities of both Slug and ZEB2 reporters with wild-type binding sites, but not with mutant binding sites (Figure 3b). Western blot analysis showed that overexpression of miR-218 repressed protein levels of both Slug and ZEB2, and vice versa (Figure 3c). These data indicate that miR-218 regulates Slug and ZEB2 expression through directly targeting their 3′-UTR regions.

MiR-218 targets Slug and ZEB2. (a) The miR-218 binding site predicted in the 3′-UTR regions of Slug/ZEB2 mRNAs. Mutant constructs were generated at the seed regions of Slug/ZEB2 3′-UTRs as indicated in red. 3′-UTR fragments of Slug/ZEB2 mRNAs containing wild-type or mutant of the miR-218 binding sequences were cloned into the downstream of the luciferase gene in pMIR-vector. (b) H1299 cells were transfected with pMIR reporter vectors containing either wild-type or mutant Slug/ZEB2 3′-UTRs (indicated as pMIR-Slug/ZEB2-WT and pMIR-Slug/ZEB2-MT) with either miR-NC or miR-218. Luciferase activities were determined 48 h after transfection. (c) The protein levels of Slug and ZEB2 in H1299 and A549 cells were examined by western blot analysis. (d) The expression levels of Slug or ZEB2 in normal tissues and human NSCLC specimens were determined by qRT–PCR analysis, and fold changes were obtained by the ratios of Slug or ZEB2 to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) levels. (e) Spearman′s correlation analysis was used to determine the correlations between the expression levels of Slug or ZEB2 and miR-218 in human NSCLC specimens. Data represent mean±s.d. of three replicates. *Indicated significant difference at P<0.05; **Indicated significant difference at P<0.01.
To explore the possible clinical relevance of these findings, we investigated the expression levels of Slug and ZEB2 in human NSCLC tissues. The mRNA levels of Slug and ZEB2 were found higher in NSCLC tissues than those in non-tumor tissues (Figure 3d). We further analyzed the correlation between Slug/ZEB2 levels and miR-218 expression levels in the same NSCLC specimen. As shown in Figure 3e, Spearman′s correlation analysis demonstrated that both Slug and ZEB2 levels in NSCLC samples were inversely correlated with miR-218 expression levels (Spearman′s correlation r=–0.8352 and Spearman′s correlation r= –0.8398, respectively).
Modulations of miR-218 on EMT and EMT-related invasion and drug resistance through Slug/ZEB2 pathway
We next investigated whether Slug and ZEB2 are functionally important in miR-218-induced alterations in EMT and EMT-related traits of lung cancer cells. H1299 cells were co-transfected with miR-218 or miR-NC together with pCMV-Slug or pCMV-ZEB2 for 48 h. The results showed that overexpression of Slug or ZEB2 without the 3′-UTR partially reversed the morphological change caused by ectopic expression of miR-218 in H1299 cells (Figure 4a). In addition, the protein levels of E-cadherin and Vimentin were restored by overexpression of Slug or ZEB2 complementary DNA (Figure 4b). The cell invasion assay showed that overexpression of Slug or ZEB2 abrogated the reduction of invasion ability caused by ectopic expression of miR-218 in H1299 cells (Figure 4c). It was reported that cancer cells undergoing EMT may acquire the ability of migration and invasion, as well as the increase of chemoresistance.26 As overexpression of miR-218 can regulate the migration and invasion of lung cancer cells, we next examined whether miR-218 has effect on drug resistance in H1299 cells. As expected, overexpression of miR-218 significantly increased cell chemosensitivity in response to cisplatin treatment (Supplementary Figure 2). Ectopic expression of Slug or ZEB2 partially reversed miR-218-induced H1299 cells chemosensitivity to cisplatin (Figures 4d and e). We also found that the combination of miR-218 and cisplatin treatment significantly induced cell apoptosis, whereas forced expression of Slug or ZEB2 partially abolished the apoptosis induced by miR-218 compared with miR-NC group (Figure 4f). These findings suggest that miR-218 regulates the EMT and EMT-related invasion and drug resistance of NSCLC cells through suppression of Slug and ZEB2.

Effect of miR-218 on EMT and drug resistance through Slug/ZEB2 pathway. (a) Image showing the morphological changes of H1299 cells expressing miR-218 mimics or miR-NC or miR-218 in combination with Slug or ZEB2 overexpression. Overexpression of Slug or ZEB2 partially reversed the morphological change caused by ectopic expression of miR-218 in H1299 cells. (b) H1299 cells stably expressing miR-NC or miR-218, or miR-218 in combination with Slug or ZEB2 overexpression were conducted by western blot analysis. (c) Indicated cells were subjected to Matrigel invasion assay. Scale bar, 20 μm. (d and e) H1299 cells stably expressing miR-NC or miR-218, miR-218 in combination with Slug or ZEB2 overexpression were pretreated with 5 μm cisplatin for indicated time points, then subjected to CCK-8 Assay, and apoptosis analysis by flow cytometry (f). Data represent mean±s.d. of three replicates. *Indicated significant difference at P< 0.05 compared with miR-NC control; **At P<0.01 compared with miR-NC control; #At P<0.05 compared with miR-218 plus Slug or ZEB2 overexpression.
The role of miR-218 in tumor growth and metastasis in vivo
We further explore whether miR-218 overexpression can inhibit tumor growth and metastasis in vivo. H1299 cells with low basal expression levels of miR-218 were infected with lentivirus expressing miR-218 or negative control; whereas A549 cells with high basal expression levels of miR-218 were infected with lentivirus expressing anti-miR-218 inhibitor or negative inhibitor control. After the cell selection by puromycin, stable cell lines termed as H1299/miR-NC, H1299/miR-218, A549/miR-NC-inhibitor and A549/miR-218 inhibitor cells were established. Cells were injected into posterior flanks of immune-deficient mice, and tumor sizes were to be measured 12 days after injection. Compared with miR-218 group, miR-NC group developed significantly larger tumors started from days 12 to 22 (Figure 5a). Inhibition of miR-218 resulted in bigger tumors than those in control mice (Figures 5a and b). Consistent with in vitro data, the protein levels of Slug and ZEB2 in tumor tissues from miR-218 overexpression group were much lower than those of miR-NC group analyzed by western blot analysis, whereas levels of Slug and ZEB2 in anti-miR-218 inhibitor group were higher compared with the control (Figure 5c). Taken together, these results suggest that miR-218 suppresses the expression levels of both Slug and ZEB2, and inhibits tumor growth in vivo.

The role of miR-218 in tumor growth in vivo. (a) H1299/miR-NC, H1299/miR-218, A549/miR-NC-inhibitor and A549/miR-218 inhibitor cells (5 × 106 cells) were dispersed in 100 μl of serum-free RPMI 1640 medium, and subcutaneously injected into both sides of posterior flanks of the nude mice (n=6). Tumors were measured every 3 days after they were apparently detectable at day 12. Tumor volumes were calculated using the following formula: volume=0.5 × (length × width2). (b) Tumor growth assay and average weights of tumors from indicated groups (bar=2 mm). (c) The total proteins were extracted from xenografts and subjected to western blot analysis to test levels of Slug and ZEB2 as indicated. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) levels were used as the internal control. Data represent mean±s.d. of three replicates. *Indicated significant difference at P<0.05; **Indicated significant difference at P<0.01.
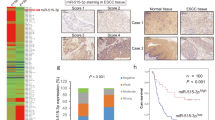
Local invasion and tumor growth at a distant site are the key steps in tumor metastasis.27 To determine if miR-218 could also inhibit metastatic process in vivo, we stably overexpressed miR-218 or miR-NC in H1299, the cell line with the highest metastatic potential, then established an orthotopic lung cancer xenograft model in nude mice. The mice were killed 50 days after cell implantation. The metastatic foci counted based on fluorescent areas of tumors, the mice in miR-NC group showed extensive invasion and contra-lateral lung metastasis, whereas the mice in miR-218 group had far less metastasized foci (Figures 6a and b). Furthermore, hematoxylin and eosin staining in tissues revealed the presence of more metastatic tumor nodules in the lungs of the miR-NC group than those in miR-218 group (Figure 6c). The ISH assay showed that miR-218 expression was barely detected in the majority of lung cancer tissues in miR-NC group, but very high levels of miR-218 in the miR-218 group. Similarly, immunohistochemical staining of lung cancer tissues showed that the expression levels of Slug and ZEB2 in miR-218-overexpressing group were significantly decreased compared with miR-NC group (Figure 6d). These findings suggested that overexpression of miR-218 was able to suppress tumor local invasion and metastatic ability.

The role of miR-218 in tumor growth and metastasis in vivo. (a) We established an orthotopic lung cancer xenograft model in nude mice. The trimmed tumor pieces of 1 mm in diameter were transplanted into lung tissues by surgical orthotopic implantation. The fluorescent areas in lungs of mice indicated were tumor tissues with H1299-red fluorescent protein (RFP) cells 7 weeks after the transplantation. (b) We counted the incidence of lung metastasis in the H1299/miR-NC and H1299/miR-218 xenografts based on the fluorescent areas showing the metastatic foci in the lung tissues of the mice. (c) Hematoxylin and eosin (H&E) staining was conducted, and arrowheads indicated metastatic tumor nodules. Magnification, × 40; scale bar, 200 μm. (d) In situ hybridization analysis showed miR-218 expression was not detected in the majority of miR-NC tissues with blue staining, and very high levels of miR-218 in miR-218-expressing tissues with strong brown signals in the cytoplasm. The expression levels of Slug and ZEB2 were analyzed in tumor tissues using immunohistochemistry. Magnification, × 400; scale bar, 50 μm.
Discussion
Deregulation of miRNAs has been observed in various cancers;28, 29 they either function as oncogenes or tumor-suppressor genes to be involved in tumorigenesis and cancer progression.30, 31 In this study, we showed that the expression level of miR-218 was significantly lower in human lung cancer tissues than that of adjacent normal tissues. Meanwhile, the downregulated miR-218 was significantly associated with histological grade and lymph node metastasis. The functional analysis revealed that ectopic expression of miR-218 markedly repressed the migration, invasion and EMT of lung cancer cells. Interestingly, we found that the expression levels of miR-218 were further decreased in lung cancer cells during TGF-β1-induced EMT process. However, the mechanism underlying TGF-β1-induced miR-218 repression is not understood yet. It was reported that EZH2, a catalytic subunit of PRC2, mediated epigenetic silencing of miR-218 via histone methylation in pancreatic ductal adenocarcinoma cells.32, 33 Besides, the TGF-β signaling pathways have been associated with PRC2 family.34, 35 Therefore, we speculated that TGF-β1 may suppress the expression of miR-218 via recruiting EZH2 to the promoter of miR-218 gene. To the best of our knowledge, this is the first study indicating that TGF-β1 inhibits miR-218 expression in lung cancer. Further investigation is needed to explore epigenetic modifications of miR-218 gene by TGF-β signaling.
EMT is a progressive biological phenomenon that epithelial cells gradually acquire a mesenchymal (fibroblast-like) cell phenotype, resulting in enhanced invasion and metastasis.36 Studies showed that EMT is relevant in the progression of lung cancer toward invasion and metastasis.37 Loss of E-cadherin expression is a hallmark of the EMT process, and transcription factors that can regulate E-cadherin transcription such as Snai1, Slug, ZEB1, ZEB2 and Twist1 are the focus of investigation.38, 39, 40 Recently, miRNAs have been shown to be powerful regulators of EMT through EMT-related signaling pathways and transcription factors.41, 42, 43 Beside miR-200 family, miR-155 facilitated TGF-β1-induced EMT by targeting RhoA in breast cancer cells.44 Meanwhile, it was demonstrated that miR-33b inhibited cells growth, invasion and EMT by suppressing Wnt/β-catenin/ZEB1 signaling in lung adenocarcinoma.45 In this study, we found that lower expression levels of E-cadherin and higher expression levels of Vimentin were detected in the cells that underwent EMT triggered by miR-218 inhibition. Ectopic expression of miR-218 decreased the invasive capacity of lung cancer cells. Meanwhile, we identified that Slug and ZEB2 are functional targets in miR-218-mediated EMT inhibition, which is clinical relevant as evidenced by the inverse correlations between miR-218 and Slug/ZEB2 expression levels in clinical tumor samples. These in vitro results indicated that miR-218 expression is closely related to invasion and metastasis by regulating EMT. We further found that miR-218 significantly decreased local invasion and the contra-lateral lung metastasis by using an orthotopic human lung cancer xenograft model. It is generally believed that activation of EMT promoted carcinoma cells to dissociate from each other and to metastasize to distant organs.46, 47, 48 Reversion of EMT is essential to establish macro-metastasis, which clearly supported the role of an EMT in dissemination and the necessity of a subsequent MET for colonization.49 However, controversial findings existed regarding the role of EMT in the process of metastasis using experimental system in vivo. For example, it was indicated that engineered cancer cells to express a Cre-switchable fluorescent marker so that the expression of red fluorescent protein can be switched to green fluorescent protein upon the activation of mesenchymal-specific markers.50 By using this EMT-tracking system, the authors concluded that EMT is not required for metastasis. The findings were supported by a similar study in genetically engineered mouse models of pancreatic ductal adenocarcinoma, demonstrating that the deletion of Twist1 or Snai1 could not reduce cancer initiation and metastasis.51 These two studies showed that EMT may not be as indispensable as it was previously assumed in cancer metastasis. Given the characteristics of the different tumors and the complexity of the process of metastasis, EMT may not be the essential factor in metastasis, but could still be very important in tumor development. As discussed in the same study, although EMT driven by Snail or Twist was not necessary for pancreatic cancer progression, possibilities remained that other EMT genes are responsible for metastasis. More studies using in vivo system are needed in order to clarify the roles of EMT and MET during cancer progression and metastasis in specific contexts in the future.
It is known that EMT is associated with drug resistance, resulting in recurrence and metastasis after initial standard chemotherapeutic treatments in various cancers.52, 53 The dysregulation of transcription factors associated with EMT progress is correlated with higher tumor aggressive behavior and resistance to chemotherapy drugs. For example, knockdown of Slug in gefitinib-resistant cells sensitized cells to gefitinib and enhanced gefitinib-induced apoptosis through Bim upregulation and caspase-9 activation.54 ZEB2 is also involved in multi-drug resistance in small cell lung cancer and cisplatin resistance in ovarian cancer.55, 56 Consistent with these results, we found that miR-218 sensitized cells to cisplatin treatment through inhibition of Slug and ZEB2. This finding reveals a new role of miR-218/Slug/ZEB2 pathway in EMT-related drug resistance, and provides a clue to use miR-218 mimic in cancer treatment. In summary, our study demonstrated that downregulation of miR-218, a frequent event in lung cancer, is associated with progression and metastasis of NSCLC. MiR-218 regulates EMT and EMT-related traits by suppression of Slug and ZEB2 expression, thus inhibits tumor growth and metastasis. In addition, overexpression of miR-218 sensitizes lung cancer cells in response to cisplatin treatment (Figure 7). With more understanding of its functions, miR-218 may be used as a potential prognostic biomarker and therapeutic target for NSCLC treatment.

Schematic diagram. TGF-β1 signaling promoted EMT, tumor growth and metastasis by altering expression levels of downstream genes including miR-218. In cancer cells, activated TGF-β1 inhibited miR-218 expression, which in turn activated expression of the key EMT promoting transcription factors Slug and ZEB2 that repressed the expression of E-cadherin, and induced the expression of Vimentin, which in turn promotes EMT and metastasis of disseminated tumor cells in lung.
Materials and methods
Human lung cancer specimens
Human lung cancer samples and normal tissues were obtained from the Department of Oncology, the First Affiliated Hospital of Nanjing Medical University, Nanjing, China. Tissue samples were collected at surgery, immediately frozen in liquid nitrogen and stored until total RNAs or proteins were extracted. This study was approved by the institutional review board and the ethics committee of Nanjing Medical University and written informed consent was obtained from all participants.
MiRNA microarray analysis
Total RNAs from A549 and A549 cells with TGF-β1 treatment were isolated with Trizol reagent (Invitrogen, Carlsbad, CA, USA), and miRNA fractions were further purified using a mirVana miRNA isolation kit (Ambion, Austin, TX, USA). The isolated miRNAs from two different cell lines were labeled with Hy3 using the miRCURY Array Labeling kit (Exiqon, Vedbaek, Denmark) and hybridized, respectively, on a miRCURY LNA microRNA Array (v 8.0, Exiqon) as was described.57 Microarray images were acquired using a Genepix 4000B scanner (Axon Instruments, Union City, CA, USA), processed and analyzed using Genepix Pro 6.0 software (Axon Instruments).
Cell culture
Human lung cancer cell lines A549, H1299, PC9 and SPCA-1 were purchased from ATCC (Manassas, VA, USA) and maintained in RPMI 1640, and human embryonic kidney 293T (HEK-293T) cells were cultured in Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal bovine serum and antibiotics (100 units/ml penicillin and 100 mg/ml streptomycin). Cells were incubated at 37 °C in a humidified atmosphere of 5% CO2 in air.
Lentivirus packaging and stable cell lines
The lentiviral packaging kit was purchased from Open Biosystems (Huntsville, AL, USA). Lentivirus carrying hsa-miR-218, hsa-miR-negative control (miR-NC), hsa-miR-218 inhibitor and hsa-miR-NC-inhibitor were packaged following the manufacturer’s manual. Lentivirus was packaged in HEK-293T cells. Thirty-six hours after the co-transfection, supernatants were collected and incubated with cells to be infected for 24 h in the presence of polybrene (2.5 μg/ml). After infection, puromycin (1.5 μg/ml) was used to select stably transduced cells.
RNA extraction, reverse transcription and real-time RT–PCR
RNA was isolated from harvested cells or human tissues with Trizol reagent according to the manufacturer′s instruction (Invitrogen). To measure expression levels of miR-218, stem-loop-specific primer method was used. Expression of U6 was used as an endogenous control. To determine the mRNA levels of Slug, ZEB2, E-cadherin and Vimentin, total RNAs were reversely transcribed by oligodT primer using RT Reagent Kit (Vazyme, Nanjing, China). Housekeeping gene GAPDH was used as internal control. The complementary DNAs were amplified by qRT–PCR using AceQqPCR SYBR Green Master Mix (Vazyme) on a 7900HT system, and fold changes were calculated by relative quantification (2−ΔΔCt). Primers were listed in Supplementary Table 3.
Western blot
Cells were washed with ice-cold phosphate-buffered saline buffer, scraped from the dishes and centrifuged at 12 000 r.p.m., 4 °C for 15 min. Cell lysates were prepared using RIPA buffer supplemented with protease inhibitors (100 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1% deoxycholate acid, 0.1% sodium dodecyl sulfate, 2 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, 2 mM DTT, 2 mM leupeptin, 2 mM pepstatin). The supernatants were collected and protein concentration was determined using BCA assay (Beyotime, Jiangsu, China). Tumor tissues from human and nude mice were grinded into powder in liquid nitrogen with RIPA buffer, and the total tissue proteins were extracted as described above. Aliquots of protein lysates were fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to a polyvinylidene difluoride membrane (Roche, Basel, Switzerland), and subjected to western blot analysis according to the manufacturer’s instruction. ECL Detection System (Thermo Scientific, Rockford, IL, USA) was used for signal detection. Antibodies against E-cadherin and Slug were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibody against glyceraldehyde 3-phosphate dehydrogenase was from Bioworld Technology (Atlanta, GA, USA), and antibodies against ZEB2 and Vimentin were from Abcam (Cambridge, UK), respectively.
Cell migration and invasion assays
Cell migration and invasion assays were analyzed using the Transwell chambers assay (Costar, Corning Inc., Corning, NY, USA), with or without coated Matrigel (BD Biosciences, San Jose, CA, USA). Cells were plated at a density of 5 × 104 per well in the upper chamber without serum. The lower chamber of the Transwell device was filled with 500 μl RPMI 1640 supplemented with 10% fetal bovine serum. After incubation for 12–24 h, noninvading cells were removed from the top well with a cotton swab, whereas the bottom cells were fixed with 3% paraformaldehyde, stained with 0.1% crystal violet, and photographed in three independent fields for each well. They were finally extracted with 33% acetic acid and detected quantitatively using a standard microplate reader (at 570 nm). Three independent experiments were conducted in triplicate.
Luciferase reporter assay
Cells were seeded in triplicate in 24-well plates and allowed to settle for 24 h. Indicated plasmids plus pRL-TK Renilla plasmid were transfected into the cells using Lipofectamine 2000 Reagent (Life Technologies, Carlsbad, CA, USA). Twenty-four hours after transfection, dual-luciferase reporter assay (Promega, Madison, WI, USA) was performed according to the manufacturer’s instructions. Primers were listed in Supplementary Table 3.
Immunofluorescence
Cells were transfected with miRNA oligonucleotides or inhibitors, as described in the section of oligonucleotide transfection, plated onto glass coverslips in Millicell EZ SLIDES (Merck Millipore, Bedford, MA, USA) and stained after 96 h. For E-Cadherin and Vimentin staining, cells were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100 and probed with rabbit-anti-E-Cadherin antibody (1:200; Proteintech Technology, Chicago, IL, USA) or mouse-anti-Vimentin antibody (1:1000; Proteintech Technology) overnight at 4 °C. The primary antibody was detected using anti-rabbit-Alexa594 or anti-mouse-Alexa488-conjugated antibodies (1:200; Invitrogen). To detect nuclei, cells were co-stained with 4,6-diamidino-2-phenylindole. Fluorescence was observed and imaged using a ZEISS ScopeA1 microscope (Carl Zeiss, Jena, Germany).
In vitro chemosensitivity array
Cancer cells were seeded at a density of 5000 cells per well in a 96-well plate overnight. Freshly prepared cisplatin (Sigma, St Louis, MO, USA) was added with the final concentration ranging from 1 to 32 μM. Forty-eight hours later, cell viability was assayed by CCK-8 kit.
Apoptosis assay
Apoptosis was measured by flow cytometry as described before.58 For Annexin V staining, 5 μl phycoerythrin-Annexin V, 5 μl propidium iodide (BD Pharmingen, San Jose, CA, USA) and 400 μl 1 × binding buffer were added to the samples, which were incubated for 15 min at room temperature in the dark. Then, the samples were analyzed by flow cytometry (FACSCanto II, BD Biosciences) within 1 h. The data were analyzed using FlowJo software (LLC, Ashland, OR, USA). Three experiments were performed in triplicate.
In situ hybridization
The slides were treated according to the manufacturer′s instruction, and hybridized with a 10 pmol probe (DIG labeled oligonucleotide, Exiqon, Vedbaek, Denmark) complementary to miR-218. After incubation with anti-DIG-HRP Fab fragments conjugated to horseradish peroxidase, the hybridized probes were detected by incubating with 3′3-diaminobenzidine solution and nuclei were counterstained with hematoxylin.
Animal studies
BALB/c nude mice (4-week-old) were used for animal studies. H1299 cells with stably overexpression miR-218 and A549 cells with miR-218-inhibition were established, injected subcutaneously into each side of the posterior flank of the same nude mouse. Twenty-two days later, all mice were killed. Tumor sizes were evaluated by the formula: volume (mm3)=(width2(mm2) × length (mm))/2, then the tumor were transplanted into lungs of nude mice. Total proteins were extracted for western blot.
An orthotopic human lung cancer xenograft model was established for in vivo tumor invasion and metastasis analysis. Briefly, H1299 cells stably overexpressing miR-218 and miR-NC were used to generate tumors as above. The tumor specimens for orthotopic implantation were derived from tumor tissues that grew subcutaneously in nude mice. Tumor tissues were trimmed of necrotic and any non-tumor portion and cut into small pieces of 1 mm in diameter. The trimmed tumor pieces were transplanted into lung tissues by surgical orthotopic implantation as was described.59, 60, 61, 62 At the 50 day, intraliver dissemination and lung metastasis were determined by fluorescent areas using Image-Pro Plus software 6.0 (Media Cybernetics Inc., Bethesda, MD, USA), and conducted by pathological examinations. Tumors were formalin-fixed, paraffin-embedded, and sectioned at 5 μm for Slug and ZEB2 immunohistochemical staining under the standard procedure as described before.63 All procedures were approved by the Animal Care and Use Committee of Nanjing Medical University, Nanjing, China.
Statistics
All experiments were performed three times and data were analyzed with GraphPad Prism 5 (La Jolla, CA, USA). The correlation between miR-218 expression and Slug as well as ZEB2 levels in human NSCLC tissues were analyzed using Spearman′s rank test. Qualitative variables were compared using Fisher's exact test. Statistical evaluation for data analysis was determined by t-test. The differences were considered to be statistically significant at P< 0.05.
References
Siegel R, Naishadham D, Jemal A . Cancer statistics, 2013. CA Cancer J Clin 2013; 63: 11–30.
Heist RS, Engelman JA . SnapShot: non-small cell lung cancer. Cancer Cell 2012; 21: 448 e442.
Scagliotti GV, Parikh P, von Pawel J, Biesma B, Vansteenkiste J, Manegold C et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol 2008; 26: 3543–3551.
Wu YC, Tang SJ, Sun GH, Sun KH . CXCR7 mediates TGFbeta1-promoted EMT and tumor-initiating features in lung cancer. Oncogene 2015; 35: 2123–2132.
Zhang J, Tian XJ, Zhang H, Teng Y, Li R, Bai F et al. TGF-beta-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci Signal 2014; 7: ra91.
Nasarre P, Gemmill RM, Potiron VA, Roche J, Lu X, Baron AE et al. Neuropilin-2 Is upregulated in lung cancer cells during TGF-beta1-induced epithelial-mesenchymal transition. Cancer Res 2013; 73: 7111–7121.
Miow QH, Tan TZ, Ye J, Lau JA, Yokomizo T, Thiery JP et al. Epithelial-mesenchymal status renders differential responses to cisplatin in ovarian cancer. Oncogene 2015; 34: 1899–1907.
Izumchenko E, Chang X, Michailidi C, Kagohara L, Ravi R, Paz K et al. The TGFbeta-miR200-MIG6 pathway orchestrates the EMT-associated kinase switch that induces resistance to EGFR inhibitors. Cancer Res 2014; 74: 3995–4005.
Bartel DP . MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–297.
Lin S, Gregory RI . MicroRNA biogenesis pathways in cancer. Nat Rev Cancer 2015; 15: 321–333.
Hammond SM . An overview of microRNAs. Adv Drug Deliv Rev 2015; 87: 3–14.
Valeri N, Braconi C, Gasparini P, Murgia C, Lampis A, Paulus-Hock V et al. MicroRNA-135b promotes cancer progression by acting as a downstream effector of oncogenic pathways in colon cancer. Cancer Cell 2014; 25: 469–483.
Park SM, Gaur AB, Lengyel E, Peter ME . The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 2008; 22: 894–907.
Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun 2014; 5: 5241.
Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev 2009; 23: 2140–2151.
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10: 593–601.
Xia H, Ooi LL, Hui KM . MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology 2013; 58: 629–641.
Sun L, Yao Y, Liu B, Lin Z, Lin L, Yang M et al. MiR-200b and miR-15b regulate chemotherapy-induced epithelial-mesenchymal transition in human tongue cancer cells by targeting BMI1. Oncogene 2012; 31: 432–445.
Tu Y, Gao X, Li G, Fu H, Cui D, Liu H et al. MicroRNA-218 inhibits glioma invasion, migration, proliferation, and cancer stem-like cell self-renewal by targeting the polycomb group gene Bmi1. Cancer Res 2013; 73: 6046–6055.
Guan H, Wei G, Wu J, Fang D, Liao Z, Xiao H et al. Down-regulation of miR-218-2 and its host gene SLIT3 cooperate to promote invasion and progression of thyroid cancer. J Clin Endocrinol Metab 2013; 98: E1334–E1344.
Zhang C, Ge S, Hu C, Yang N, Zhang J . MiRNA-218, a new regulator of HMGB1, suppresses cell migration and invasion in non-small cell lung cancer. Acta Biochim Biophys Sinica 2013; 45: 1055–1061.
Zhu K, Ding H, Wang W, Liao Z, Fu Z, Hong Y et al. Tumor-suppressive miR-218-5p inhibits cancer cell proliferation and migration via EGFR in non-small cell lung cancer. Oncotarget 2016; 7: 28075–28085.
Xie J, Yu F, Li D, Zhu X, Zhang X, Lv Z . MicroRNA-218 regulates cisplatin (DPP) chemosensitivity in non-small cell lung cancer by targeting RUNX2. Tumour Biol 2015; 37: 1197–1204.
Ko H, So Y, Jeon H, Jeong MH, Choi HK, Ryu SH et al. TGF-beta1-induced epithelial-mesenchymal transition and acetylation of Smad2 and Smad3 are negatively regulated by EGCG in human A549 lung cancer cells. Cancer Lett 2013; 335: 205–213.
Tsai JH, Yang J . Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev 2013; 27: 2192–2206.
Voulgari A, Pintzas A . Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta 2009; 1796: 75–90.
Chaffer CL, Weinberg RA . A perspective on cancer cell metastasis. Science 2011; 331: 1559–1564.
Garzon R, Calin GA, Croce CM . MicroRNAs in cancer. Annu Rev Med 2009; 60: 167–179.
Feng B, Zhang K, Wang R, Chen L . Correction: Non-small-cell lung cancer and miRNAs: novel biomarkers and promising tools for treatment. Clin Sci 2015; 129: 673.
Sachdeva M, Mito JK, Lee CL, Zhang M, Li Z, Dodd RD et al. MicroRNA-182 drives metastasis of primary sarcomas by targeting multiple genes. J Clin Invest 2014; 124: 4305–4319.
Okada N, Lin CP, Ribeiro MC, Biton A, Lai G, He X et al. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev 2014; 28: 438–450.
Li CH, To KF, Tong JH, Xiao Z, Xia T, Lai PB et al. Enhancer of zeste homolog 2 silences microRNA-218 in human pancreatic ductal adenocarcinoma cells by inducing formation of heterochromatin. Gastroenterology 2013; 144: 1086–1097 e1089.
Wang B, Liu Y, Luo F, Xu Y, Qin Y, Lu X et al. Epigenetic silencing of microRNA-218 via EZH2-mediated H3K27 trimethylation is involved in malignant transformation of HBE cells induced by cigarette smoke extract. Arch Toxicol 2016; 90: 449–461.
Oktyabri D, Tange S, Terashima M, Ishimura A, Suzuki T . EED regulates epithelial-mesenchymal transition of cancer cells induced by TGF-beta. Biochem Biophys Res Commun 2014; 453: 124–130.
Zhou X, Zang X, Ponnusamy M, Masucci MV, Tolbert E, Gong R et al. Enhancer of Zeste homolog 2 inhibition attenuates renal fibrosis by maintaining Smad7 and phosphatase and tensin homolog expression. J Am Soc Nephrol 2015; 27: 2092–2108.
Kraljevic Pavelic S, Sedic M, Bosnjak H, Spaventi S, Pavelic K . Metastasis: new perspectives on an old problem. Mol Cancer 2011; 10: 22.
Jin Y, Li F, Zheng C, Wang Y, Fang Z, Guo C et al. NEDD9 promotes lung cancer metastasis through epithelial-mesenchymal transition. Int J Cancer 2014; 134: 2294–2304.
Mallini P, Lennard T, Kirby J, Meeson A . Epithelial-to-mesenchymal transition: what is the impact on breast cancer stem cells and drug resistance. Cancer Treat Rev 2014; 40: 341–348.
Nagaraju GP, Long TE, Park W, Landry JC, Taliaferro-Smith L, Farris AB et al. Heat shock protein 90 promotes epithelial to mesenchymal transition, invasion, and migration in colorectal cancer. Mol Carcinog 2015; 54: 1147–1158.
Puisieux A, Brabletz T, Caramel J . Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol 2014; 16: 488–494.
Cai J, Guan H, Fang L, Yang Y, Zhu X, Yuan J et al. MicroRNA-374a activates Wnt/beta-catenin signaling to promote breast cancer metastasis. J Clin Invest 2013; 123: 566–579.
Hahn S, Jackstadt R, Siemens H, Hunten S, Hermeking H . SNAIL and miR-34a feed-forward regulation of ZNF281/ZBP99 promotes epithelial-mesenchymal transition. EMBO J 2013; 32: 3079–3095.
Yang L, Yang J, Li J, Shen X, Le Y, Zhou C et al. MircoRNA-33a inhibits epithelial-to-mesenchymal transition and metastasis and could be a prognostic marker in non-small cell lung cancer. Sci Rep 2015; 5: 13677.
Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS et al. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol 2008; 28: 6773–6784.
Qu J, Li M, An J, Zhao B, Zhong W, Gu Q et al. MicroRNA-33b inhibits lung adenocarcinoma cell growth, invasion, and epithelial-mesenchymal transition by suppressing Wnt/beta-catenin/ZEB1 signaling. Int J Oncol 2015; 47: 2141–2152.
Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L et al. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011; 19: 372–386.
Kalluri R, Weinberg RA . The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119: 1420–1428.
Thiery JP, Acloque H, Huang RY, Nieto MA . Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139: 871–890.
Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J . Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012; 22: 725–736.
Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015; 527: 472–476.
Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015; 527: 525–530.
Zhu X, Shen H, Yin X, Long L, Xie C, Liu Y et al. miR-186 regulation of Twist1 and ovarian cancer sensitivity to cisplatin. Oncogene 2015; 35: 323–332.
Wilson C, Nicholes K, Bustos D, Lin E, Song Q, Stephan JP et al. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget 2014; 5: 7328–7341.
Chang TH, Tsai MF, Su KY, Wu SG, Huang CP, Yu SL et al. Slug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitor. Am J Respir Crit Care Med 2011; 183: 1071–1079.
Fang S, Zeng X, Zhu W, Tang R, Chao Y, Guo L . Zinc finger E-box-binding homeobox 2 (ZEB2) regulated by miR-200b contributes to multi-drug resistance of small cell lung cancer. Exp Mol Pathol 2014; 96: 438–444.
Haslehurst AM, Koti M, Dharsee M, Nuin P, Evans K, Geraci J et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012; 12: 91.
Castoldi M, Schmidt S, Benes V, Noerholm M, Kulozik AE, Hentze MW et al. A sensitive array for microRNA expression profiling (miChip) based on locked nucleic acids (LNA). RNA 2006; 12: 913–920.
Valeri N, Gasparini P, Braconi C, Paone A, Lovat F, Fabbri M et al. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2). Proc Natl Acad Sci USA 2010; 107: 21098–21103.
Fu XY, Besterman JM, Monosov A, Hoffman RM . Models of human metastatic colon cancer in nude mice orthotopically constructed by using histologically intact patient specimens. Proc Natl Acad Sci USA 1991; 88: 9345–9349.
Yang M, Jiang P, Sun FX, Hasegawa S, Baranov E, Chishima T et al. A fluorescent orthotopic bone metastasis model of human prostate cancer. Cancer Res 1999; 59: 781–786.
Katz MH, Takimoto S, Spivack D, Moossa AR, Hoffman RM, Bouvet M . An imageable highly metastatic orthotopic red fluorescent protein model of pancreatic cancer. Clin Exp Metastasis 2004; 21: 7–12.
An Z, Wang X, Astoul P, Danays T, Moossa AR, Hoffman RM . Interferon gamma is highly effective against orthotopically-implanted human pleural adenocarcinoma in nude mice. Anticancer Res 1996; 16: 2545–2551.
Liu LZ, Zheng JZ, Wang XR, Jiang BH . Endothelial p70 S6 kinase 1 in regulating tumor angiogenesis. Cancer Res 2008; 68: 8183–8188.
Acknowledgements
This work was supported in part by National Natural Science Foundation of China (81472944, 81320108019, 81302182, 81270736 and 81502170); The Research Special Fund for Public Welfare Industry of Health (201402008); by National Institutes of Health grants (R01ES020868 and R01CA193511); and by American Cancer Society (RSG NEC -129306).
Author contributions
Z-MS, LW, HS, C-FJ, XG, D-ML, Y-YW, H-RS, M-HP and WL performed experiments. Z-MS, LW, HS, Y-QS, L-ZL, SCP, JH and B-HJ designed the studies. Z-MS, LW, JH and B-HJ wrote the manuscript. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Shi, ZM., Wang, L., Shen, H. et al. Downregulation of miR-218 contributes to epithelial–mesenchymal transition and tumor metastasis in lung cancer by targeting Slug/ZEB2 signaling. Oncogene 36, 2577–2588 (2017). https://doi.org/10.1038/onc.2016.414
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.414
This article is cited by
-
Interaction between Wnt/β-catenin signaling pathway and EMT pathway mediates the mechanism of sunitinib resistance in renal cell carcinoma
BMC Cancer (2024)
-
Mesenchymal stem cell-derived exosomes enriched with miR-218 reduce the epithelial–mesenchymal transition and angiogenesis in triple-negative breast cancer cells
European Journal of Medical Research (2023)
-
Two microRNAs of plasma-derived small extracellular vesicles as biomarkers for metastatic non-small cell lung cancer
BMC Pulmonary Medicine (2023)
-
Exosomal miR-218 derived from mesenchymal stem cells inhibits endothelial-to-mesenchymal transition by epigenetically modulating of BMP2 in pulmonary fibrosis
Cell Biology and Toxicology (2023)
-
Prospects of microRNAs as therapeutic biomarkers in non-small cell lung cancer
Medical Oncology (2023)
