
Abstract
BRAF inhibitor (BRAFi) therapy for melanoma patients harboring the V600E mutation is initially highly effective, but almost all patients relapse within a few months. Understanding the molecular mechanisms underpinning BRAFi-based therapy is therefore an important issue. Here we identified a previously unsuspected mechanism of BRAFi resistance driven by elevated Hedgehog (Hh) pathway activation that is observed in a cohort of melanoma patients after vemurafenib treatment. Specifically, we demonstrate that melanoma cell lines, with acquired in vitro-induced vemurafenib resistance, show increased levels of glioma-associated oncogene homolog 1 and 2 (GLI1/GLI2) compared with naïve cells. We also observed these findings in clinical melanoma specimens. Moreover, the increased expression of the transcription factors GLI1/GLI2 was independent of canonical Hh signaling and was instead correlated with the noncanonical Hh pathway, involving TGFβ/SMAD (transforming growth factor-β/Sma- and Mad-related family) signaling. Knockdown of GLI1 and GLI2 restored sensitivity to vemurafenib-resistant cells, an effect associated with both growth arrest and senescence. Treatment of vemurafenib-resistant cells with the GLI1/GLI2 inhibitor Gant61 led to decreased invasion of the melanoma cells in a three-dimensional skin reconstruct model and was associated with a decrease in metalloproteinase (MMP2/MMP9) expression and microphthalmia transcription factor upregulation. Gant61 monotherapy did not alter the drug sensitivity of naïve cells, but could reverse the resistance of melanoma cells chronically treated with vemurafenib. We further noted that alternating dosing schedules of Gant61 and vemurafenib prevented the onset of BRAFi resistance, suggesting that this could be a potential therapeutic strategy for the prevention of therapeutic escape. Our results suggest that targeting the Hh pathway in BRAFi-resistant melanoma may represent a viable therapeutic strategy to restore vemurafenib sensitivity, reducing or even inhibiting the acquired chemoresistance in melanoma patients.
Similar content being viewed by others

Introduction
Melanoma represents the deadliest of all skin cancers (currently accounting for more than 75% of skin cancer-related deaths).1 About 50% of melanomas harbor activating V600E mutations in the serine/threonine kinase BRAF, which drives melanoma initiation and progression through the mitogen-activated protein kinase (MAPK) pathway.2 The MAPK pathway is a bona fide therapeutic target in melanoma with small-molecule BRAF and MEK inhibitors (BRAFi and MEKi) demonstrating significant survival advantage in patients whose melanomas harbor the BRAF driver mutation.3, 4, 5 Although the results with BRAFi have been very promising, practically all of the patients treated thus far have developed resistance.6 Preclinical studies have shown resistance to be mediated through a diverse array of mediators that lead to reactivation of MAPK, such as NRAS and MEK mutations, receptor tyrosine kinase upregulation or elevated COT expression.7 A role has also been reported for an increase of phosphoinositide 3-kinase (PI3K/AKT) signaling,8, 9 which can arise through phosphatase and tensin homolog loss10 and platelet-derived growth factor receptor-α upregulation.11 The identification of MAPK reactivation as a major mediator of resistance led to the development of BRAFi-MEKi combinations, which are associated with a longer overall survival than single-agent BRAFi therapy. Despite the successes of the combination therapy vs BRAF monotherapy, resistance still occurs.12, 13
The development-associated Hedgehog (Hh) signaling pathway has been implicated in a variety of malignancies, including melanoma.14 In canonical Hh signaling, sonic Hh (SHH) inhibits the suppressor of fused, and activates a complex formed by patched-1 and smoothened (SMO), thus releasing SMO to enable glioma-associated oncogene homolog (GLI) protein regulation of target genes.15 GLI1 and GLI2 are transcription factor members of the Gli-Kruppel family and their overexpression is linked to the development of multiple tumor types.16, 17 However, some tumors express GLI1 and GLI2 in the absence of any activating mutations, suggesting that Hh signaling can also be activated through alternative pathways.16 GLI1 and GLI2 activation by noncanonical Hh pathways already have been described, such as by the PI3K/AKT pathway,18 and by transforming growth factor-β/Sma- and Mad-related family (TGFβ/SMAD) pathway.19 TGFβ/SMAD noncanonical Hh signaling is a potential driver in melanoma16 and GLI2/TGF-β cooperate to repress microphthalmia transcription factor (MITF) expression.20 In the current study, we demonstrate a role for TGFβ/SMAD-driven noncanonical Hh signaling in vemurafenib resistance in melanoma patient samples and in vitro models of acquired BRAFi resistance.
Results
GLI1 and GLI2 expression is increased in vemurafenib-resistant melanoma cell lines in vitro
To examine the potential role of GLI1/GLI2 in BRAFi-resistant melanoma, we initially characterized their expression in normal skin cells and melanoma cell lines. In normal keratinocytes and fibroblasts, basal GLI1/GLI2 mRNA expression was higher compared with that in all melanoma cell lines examined, whereas normal melanocytes had GLI1/GLI2 mRNA expression levels equal to or lower than melanoma cell lines (Supplementary Figures S1A and B). GLI1 mRNA and protein expression were higher in almost all naïve primary melanoma cells (WM35, WM278, WM793 and WM1552c) compared with metastatic lines (WM9, WM1617, 1205Lu, UACC62, SK-MEL-19, SK-MEL-28 and SK-MEL-29). This profile was also found for GLI2 expression, but to a lesser extent (Supplementary Figure S1C).
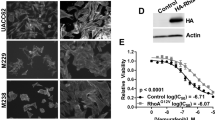
We next generated isogenic pairs of vemurafenib-sensitive and -resistant cell lines (Supplementary Figure S2A). Acquired vemurafenib resistance was validated by the ability of the cells to maintain their MEK and ERK phosphorylation levels in the presence of the drug and by the observed shift in the inhibitory concentration 50% in the resistant cell lines (IC50; Supplementary Figure S2B), and as observed in our previous study.21 It was noted that GLI1 mRNA expression was elevated in 8/9 (89%) and GLI2 mRNA expression was elevated in 4/9 (44%) of vemurafenib-resistant melanoma cell lines in comparison with the naïve counterpart (Figures 1a and b, respectively). These data were further confirmed at the protein level (Figure 1c). Taken together, these data demonstrated that vemurafenib-resistant melanoma cells showed increased expression of GLI1 and GLI2 compared with drug-naïve cell lines (Figures 1d and e, respectively).

GLI1 and GLI2 expression after vemurafenib resistance acquirement in melanoma cell lines. (a) Comparison between GLI1 mRNA expression in naïve and vemurafenib-resistant melanoma cell lines. (b) Comparison between GLI2 mRNA expression in naïve and vemurafenib-resistant melanoma cell lines. (c) Comparison between GLI1 and GLI2 protein expression in naïve and vemurafenib-resistant melanoma cell lines. (d) Comparison between GLI1 mRNA expression in naïve (primary and metastatic) and vemurafenib-resistant melanoma cell lines. (e) Comparison between GLI2 mRNA expression in naïve (primary and metastatic) and vemurafenib-resistant melanoma cell lines. Values are expressed as mean±s.d. Significance is indicated by *P<0.05, **P<0.01 and ***P<0.001.
Increased expression of GLI1 and GLI2 is observed in melanoma patients failing BRAFi therapy
We next determined the clinical relevance of the increased GLI1/GLI2 expression seen upon acquisition of BRAFi resistance. Eleven melanoma tissue samples were analyzed (six samples pre-vemurafenib treatment and five samples post-vemurafenib treatment), from two different patients. The resistance profiles were confirmed by clinical progression of the lesions on vemurafenib treatment. These specimens were confirmed by two independent pathologists (Supplementary Table S1). The sites of melanoma were identified in both patients, by 1–6 (pre-treatment) and by 1-5R (postrelapse) (R1–R5; Figure 2a).

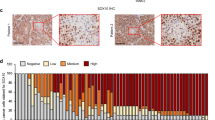
GLI1 and GLI2 expression after vemurafenib resistance in patient melanoma samples. (a) Tumor localization (red circles) in patient number 1 and 2 before (pre-vemurafenib) and after (post-vemurafenib) treatment. Melanoma samples 1, 2, 3, 4 and 5 from patient no. 1 and sample 6 from patient number. 2 were extracted before vemurafenib treatment; melanoma samples R1, R2 and R3 from patient number. 1 and samples R4 and R5 from patient no. 2 were extracted after acquired resistance to vemurafenib. (b) Hematoxylin and eosin staining, and GLI1 and GLI2 immunostaining in samples 1 and R3 from patient number. 1, and samples 6 and R4 from patient number. 2. (c) Incidence of GLI1 and GLI2 protein expression in all patient‘s melanoma samples pre- and post-vemurafenib treatment. Values are expressed as mean±s.d. Significance is indicated by *P<0.05, **P<0.01 and ***P<0.001.
GLI1/GLI2 immunostaining was performed in all melanoma samples (Figure 2b), in regions previously selected by hematoxylin/eosin (H&E) and morphological analysis. Before vemurafenib treatment, 0/6 (0%) and 1/6 (17%) of patients’ samples were positive for GLI1 and GLI2 expression, respectively. Following the acquisition of vemurafenib resistance, 5/5 (100%) and 2/5 (40%) of samples were positive for GLI1 and GLI2 expression, respectively (Figure 2c).
Noncanonical Hh pathway is involved in acquisition of vemurafenib resistance
GLI1 and GLI2 are members of both the canonical and noncanonical Hh pathways. As the expression of SMO, suppressor of fused, patched-1 (Figure 3a) and SHH mRNA (data not shown) did not increase following the acquisition of vemurafenib resistance, we focused upon the noncanonical members of the Hh pathway. Expression of soluble TGFβ1 and its membrane receptor TGFβRII were significantly upregulated in both cell lines with BRAFi resistance, and in the UACC62 vemurafenib-resistant cell line, the membrane receptor TGFβRI mRNA was also increased in comparison with the naïve counterpart (Figure 3b). Moreover, SMAD3 expression was increased in both vemurafenib-resistant cell lines, and SMAD2 mRNA expression increased in the UACC62 vemurafenib-resistant cell line (Figure 3c). SMAD4 mRNA expression was not altered in either of the cell lines. The increased expression of components of the TGFβ/SMAD pathway was confirmed at the protein level (Figures 3d and e). Moreover, addition of TGF-β1 led to increases in GLI1 and GLI2 mRNA expression, with more robust increases occurring in the vemurafenib-resistant SK-MEL-28 cell line (Figure 3f). A link between the TGF-β pathway and noncanonical GLI1/GLI2 signaling was suggested by the observation that SIS3 (an SMAD3 inhibitor) treatment led to a significant inhibition of GLI1 and GLI2 expression in both SK-MEL-28 and UACC62 vemurafenib-resistant melanoma cell lines (Figure 3g).

Noncanonical Hh pathway and acquisition of vemurafenib resistance. (a) Comparison between SMO, suppressor of fused (SUFU) and patched-1 (PTCH1) mRNA expression in SK-MEL-28- and UACC62-naïve and vemurafenib-resistant melanoma cell lines. (b) Comparison of TGFβ1, TGFβRI and TGFβRII mRNA expression in SK-MEL-28- and UACC62-naïve and vemurafenib-resistant melanoma cell lines. (c) Comparison between SMAD2, SMAD3 and SMAD4 mRNA expression in SK-MEL-28- and UACC62-naïve and vemurafenib-resistant melanoma cell lines. (d) Comparison of TGFβ1, TGFβRI, TGFβRII, SMAD2 and SMAD3 protein expression in SK-MEL-28- and UACC62-naïve and vemurafenib-resistant melanoma cell lines. (e) Densitometry of the western blot bands showed in (d and f) GLI1 and GLI2 mRNA expression in SK-MEL-28-naïve and -resistant cells after the addition of TGFβ1 (10 ng/ml) for 24–72 h. (g) GLI1 and GLI2 mRNA expression in SK-MEL-28R and UACC62R cells after SIS3 treatment (10 μM). Values are expressed as mean±s.d. Significance is indicated by *P<0.05, **P<0.01 and ***P<0.001 compared with untreated naïve cells.
Knockdown of GLI1/GLI2 leads to both senescence and increased drug sensitivity
To determine the role of GLI1/GLI2 expression in the resistance phenotype, we performed lentivirus-mediated short hairpin RNA (shRNA) knockdown in the SK-MEL-28 cell line (which expressed high levels of GLI1 and GLI2). First, we used a multiplicity of infection (MOI) of 10 and observed 75% and 91% of mRNA and protein inhibition for shGLI1 and shGLI2, respectively (Figures 4a–c). These knockdowns were extremely efficient, but were also associated with induction of senescence (Figure 4d) as determined by staining for senescence-associated β-galactosidase. We therefore performed gene silencing with an MOI of 5 and found 25% and 55% of mRNA and protein inhibition for shGLI1 and shGLI2, respectively (Figures 4e–g). Although the efficiency of gene silencing was less, an MOI of 5 was used in the subsequent assays to evaluate the role of GLI proteins in vemurafenib resistance. After gene silencing, cells were treated for 72 h with vemurafenib, with GLI1 knockdown having a small but significant increase in vemurafenib sensitivity (IC50 of 26.1μM compared with 29.6 μM in shControl cells). Furthermore, GLI2 knockdown caused an even greater increase in drug sensitivity (IC50 of 15.6μM compared with 29.6 μM in shControl cells), which may be explained by GLI2 knockdown reducing GLI1 and GLI2 expression (Figure 4h). Moreover, GLI1 and GLI2 knockdown led to a decrease in MMP2/9 mRNA expression in SK-MEL-28-resistant cells (29% and 16% for shGLI1–20 and 51% for shGLI2, respectively) (Figure 4i).

Knockdown of GLI1/GLI2 and the induction of growth arrest and senescence in SK-MEL-28R melanoma cells. (a) mRNA expression of GLI1 and GLI2 after gene silencing by shGLI1 and shGLI2 with MOI=10. (b) Protein expression of GLI1 and GLI2 after gene silencing (shGLI1 and shGLI2) with MOI=10. (c) Densitometry of the western blot bands shown in (b). (d) Senescence-associated β-galactosidase activity after gene silencing of GLI1 (shGLI1) and GLI2 (shGLI2) with MOI=5 or MOI=10. (e) mRNA expression of GLI1 and GLI2 after gene silencing by shGLI1 and shGLI2 with MOI=5. (f) Protein expression of GLI1 and GLI2 after gene silencing (shGLI1 and shGLI2) with MOI=5. (g) Densitometry of the western blot bands shown in (f and h). Inhibitory concentration 50% (IC50) of vemurafenib after gene silencing of GLI1 (shGLI1) and GLI2 (shGLI2) with MOI=5. (i) MMP2 and MMP9 mRNA expression after gene silencing by shGLI1 and shGLI2 with MOI=5. Values are expressed as mean±s.d. Significance is indicated by *P<0.05, **P<0.01 and ***P<0.001.
Gant61 induces antiproliferative and cytotoxic effects in vemurafenib-resistant melanoma cells
We then sought to verify that GLI1/GLI2 expression in vemurafenib-resistant cells was regulated through the noncanonical Hh pathway. We treated the SK-MEL-28- and UACC62-naïve and -resistant melanoma cell lines with either cyclopamine (inhibitor of SMO) or Gant61 (inhibitor of GLI1/GLI2). Although cyclopamine did not alter cell viability in naïve or resistant lines (Figure 5a), Gant61 led to a 20% decrease in viability in naïve cells and a 60% decrease of viability in resistant cells (Figure 5b).

Effects of Hh inhibitors in viability and proliferation of vemurafenib-resistant melanoma cells. (a) SK-MEL-28- and UACC62-naïve and vemurafenib-resistant melanoma cell line viability after cyclopamine treatment (10 μM) for 24, 48 and 72 h. (b) SK-MEL-28- and UACC62-naïve and vemurafenib-resistant melanoma cell line viability after Gant61 treatment (10 μM) for 24, 48 and 72 h. (c) Clonogenic assay with UACC62R, SK-MEL-28R and R3 cells treated chronically with TGFβ1 (10 or 5 ng/ml), Gant61 (10 or 1 μM) or cyclopamine (10 or 1 μM) for 14 days; cells were stained with crystal violet. (d) Colony area of UACC62R, SK-MEL-28R and R3 cells from the clonogenic assay. (e) Total number of colonies of UACC62R, SK-MEL-28R and R3 cells from the clonogenic assay. (f) Cell morphology and viability of UACC62R, SK-MEL-28R and R3 cells treated with TGFβ1 (5 ng/ml), Gant61 (10 μM), cyclopamine (10 μM) or SIS3 (10 μM) for 48 h. (g) Apoptosis evaluation of UACC62R, SK-MEL-28 R and R3 cells treated with Gant61 (10 μM) for 48 h by flow cytometry detection of Annexin V staining. (h) GLI1, GLI2 and MITF mRNA expression in UACC62R, SK-MEL-28R and R3 cells after Gant61 treatment (10 μM for 48 h). (i) CDH1 and CDH2 mRNA expression in UACC62R, SK-MEL-28 R and R3 cells after Gant61 treatment (10 μM for 48 h). (j) MMP2, MMP9 and MMP14 mRNA expression in UACC62R, SK-MEL-28R and R3 cells after Gant61 treatment (10 μM by 48 h). Values are expressed as mean±s.d. Significance is indicated by *P<0.05, **P<0.01 and ***P<0.001.
We evaluated the colony-forming ability of vemurafenib-resistant SK-MEL-28, UACC62 and R3 cells (extracted from patient no. 1) after long-term treatment with TGFβ1, Gant61 and cyclopamine at different concentrations (Figure 5c). TGFβ1 was associated with significantly more colonies in vemurafenib-resistant SK-MEL-28 cells (SK-MEL-28R). All resistant cells treated with Gant61 showed significantly decreased colony area and fewer total colonies at both 1 and 10 μM of the drug, showing the dose-dependent effect. Cyclopamine treatment was associated with a decrease in colony area in only vemurafenib-resistant UACC62 cells (UACC62R) and a slight decrease in the total number of colonies in all the cell lines at the highest concentration tested (Figures 5d and e). A role for GLI1/GLI2 in the TGF-β1-mediated process of therapeutic escape was suggested by the ability of Gant61 to inhibit the increase in colony formation seen following TGF-β1 treatment (Supplementary Figures S3A and B).
TGFβ1 treatment was also associated with a slight, nonsignificant, decrease in cell viability after 48 h of treatment (up to 12%), whereas Gant61 induced a substantial decrease (up to 80%) and cyclopamine induced a moderate decrease in cell viability (up to 26%; Figure 5f). Another noncanonical Hh inhibitor, SIS3 (SMAD3 inhibitor), also demonstrated a significant reduction in cell viability (up to 64%; Figure 5f), but was also associated with high cytotoxicity to normal skin keratinocytes (Supplementary Figure S4A). This elevated cytotoxicity was related to its high basal SMAD3 mRNA expression (Supplementary Figure S4B). Further investigations into the mechanisms of cell death revealed that Gant61 treatment induced apoptosis in the resistant melanoma cells (Figure 5g) and decreased the Bcl2/Bax ratio (data not shown). Cell death was strongly associated with direct GLI1/GLI2 downregulation, as well as MITF upregulation in the resistant melanoma cells (Figure 5h). To determine the relationship between the GLI proteins and MITF expression in vivo, we next examined their expression in The Cancer Genome Atlas (TCGA) melanoma cohort (Supplementary Figure S5A). These data showed a clear inverse correlation between GLI2/MITF, with MITF expression being markedly reduced in melanomas expressing high levels of GLI2. The relationship with GLI1 was less obvious, indicating that GLI2, rather than GLI1, has a major role in repressing MITF expression in human melanoma specimens. This analysis revealed a positive correlation between GLI1/GLI2/epidermal growth factor receptor (EGFR) and AXL, and that this was negatively correlated with MITF expression. The expression of EGFR showed an 18-fold (Supplementary Figure S5B) increase in the resistant SK-MEL-28 cell line relative to its naïve counterpart, with MITF expression being decreased more than 3-fold (Supplementary Figure S5C). Pharmacological inhibition of GLI1/2 following Gant61 treatment was associated with decreased EGFR expression and increased MITF expression in these resistant cells.
Consistent with our observation that GLI mediated invasiveness, we further observed altered expression of EMT markers following Gant61 treatment, with studies showing an upregulation of CDH1 (E-cadherin) and a downregulation of CDH2 (N-cadherin) (Figure 5i). These changes were even observed in the cells that had previously received TGF-β1 treatment (Supplementary Figure S3C). Taken together, these data suggested a link between GLI signaling and the adoption of an EMT-like phenotype, downstream of TGF-β. Multiple invasion markers, such as matrix metalloproteinases 2/9 (MMP2/MMP9), were also downregulated after Gant61 treatment in three different resistant cell lines (Figure 5j).
Invasive potential decrease after Gant61 treatment in 3D reconstructed melanoma skin model
Because Gant61 treatment inhibited MMPs expression in vitro, we next investigated this phenomenon in a more physiologically relevant three-dimensional (3D) organotypic skin culture model. These studies showed that untreated naïve melanoma cells were highly invasive and crossed the basement membrane into the dermal compartment in reconstructed human skins (RHS) (Figures 6a and b). Treatment with Gant61 led to an increase in the dispersal of the naïve cells (Figures 6c and d) with no apparent tumor foci seen. Non-treated resistant cells (Figures 6e and f) showed more melanoma cell foci than Gant61-treated resistant cells (Figures 6g and h). It is worth noting that the MMP2/MMP9 expression was elevated in untreated naïve cells (Figures 6i and j) and repressed after Gant61 treatment (Figures 6k and l). This same increase in MMP2/MMP9 expression was also found in resistant melanoma cells (Figures 6m and n). On the other hand, Gant61 treatment also inhibited MMP2/MMP9 protein expression in resistant RHS (Figures 6o and p). Moreover, reduction in melanoma growth was also confirmed by the decrease in thickness seen after Gant61 treatment in both RHS with naïve and resistant melanoma cells (Figure 6q).

Invasive potential of melanoma cells after Gant61 treatment in 3D RHS. Reconstructed skin with SK-MEL-28-naïve melanoma cells stained with H&E: (a) control sample (x20 magnification); (b) control sample (x40 magnification); (c) Gant61 treatment sample (x20 magnification); (d) Gant61 treatment sample (x40 magnification). Reconstructed skin with SK-MEL-28 vemurafenib-resistant melanoma cells stained with H&E: (e) control sample (x20 magnification); (f) control sample (x40 magnification); (g) Gant61 treatment sample (x20 magnification); (h) Gant61 treatment group (x40 magnification). Reconstructed skin with SK-MEL-28-naïve melanoma cells from the control group immunostained for detection of (i) MMP2 and (j) MMP9. Reconstructed skin with SK-MEL-28-naïve melanoma cells from Gant61 treatment group immunostained for detection of (k) MMP2 and (l) MMP9. Reconstructed skin with SK-MEL-28 vemurafenib-resistant melanoma cells from the control group immunostained for detection of (m) MMP2 and (n) MMP9. Reconstructed skin with SK-MEL-28 vemurafenib-resistant melanoma cells from Gant61 treatment group immunostained for detection of (o) MMP2 and (p) MMP9. (q) Melanoma compartment thickness values (pixels). (r) GLI1 mRNA expression in reconstructed skin with naïve or vemurafenib-resistant melanoma cells after Gant61 treatment. (s) GLI2 mRNA expression in reconstructed skin with naïve or vemurafenib-resistant melanoma cells after Gant61 treatment. (t) MITF mRNA expression in reconstructed skin with naïve or vemurafenib-resistant melanoma cells after Gant61 treatment. (u) MMP2 mRNA expression in reconstructed skin with naïve or vemurafenib-resistant melanoma cells after Gant61 treatment. (v) MMP2 mRNA expression in reconstructed skin with naïve or vemurafenib-resistant melanoma cells after Gant61 treatment. The reconstructed skin with SK-MEL-28 vemurafenib-resistant melanoma cells was maintained in the presence of 6 μM of vemurafenib and 10 μM of GANT61 during 12 days of culture. The black arrows indicate the tumor foci. Values are expressed as mean±s.d. Significance is indicated by *P<0.05, **P<0.01 and ***P<0.001.
In RHS seeded with drug-naïve melanoma cells, there was a nonsignificant decrease in MMP2/MMP9 expression after Gant61 treatment (5–10%); however, robust MMP2/MMP9 mRNA downregulation (65% and 41%, respectively) was found when using resistant melanoma cells in comparison with the non-treated controls (Figures 6r and s). Furthermore, the decrease in melanoma invasion inhibition is correlated with lower GLI1/GLI2 downregulation after Gant61 treatment (Figures 6t and u) and increased MITF expression (Figure 6v). These results show that GLI1/GLI2 inhibition by Gant61 treatment induces apoptosis, in addition to decreasing survival and invasion markers.
Vemurafenib resistance can be modulated and delayed with alternating Gant61 treatment
We next determined whether vemurafenib resistance could be abrogated or delayed through the use of vemurafenib and/or Gant61 under a variety of dosing schedules (Figure 7a). In all cases, the reversal of vemurafenib resistance was determined by the shift in the IC50 values to the drug (Figure 7b).

Vemurafenib resistance after alternating vemurafenib and Gant61 treatment. (a) Treatment schedule of vemurafenib (v) and/or Gant61 (g) and/or untreated (−) SK-MEL-28- and UACC62-naïve melanoma cells. (b) Inhibitory concentration 50% (IC50) of vemurafenib after 48 h of treatment in different protocols of vemurafenib resistance modulation. (c) GLI1 and GLI2 mRNA expression in UACC62 melanoma cells after each cycle of vemurafenib and/or Gant61 treatment. (d) GLI1 and GLI2 mRNA expression in SK-MEL-28 melanoma cells after each cycle of vemurafenib and Gant61 treatment. Values are expressed as mean±s.d. Significance is indicated by *P<0.05, **P<0.01 and ***P<0.001 compared with vemurafenib-resistant group.
It was found that coadministration of Gant61 and vemurafenib in naïve cell lines prevented the increase in vemurafenib IC50 values typically seen after chronic drug treatment.
It was noted that Gant61 treatment alone did not alter sensitivity to vemurafenib when compared with untreated naïve cells. The concomitant treatment with vemurafenib/Gant61 or only vemurafenib dosed on alternate days conferred resistance to vemurafenib in a manner equivalent to continuous vemurafenib monotherapy. Alternate dosing of vemurafenib and Gant61 induced less resistance and was associated with a lower IC50 value. The delay in resistance was likely to be a result of GLI1 and GLI2 downregulation in the group receiving alternating vemurafenib and Gant61 treatment (Figures 7c and d). No change in GLI1 or GLI2 expression was seen in the continuous or alternating vemurafenib treatment groups. Overall, these findings demonstrate that vemurafenib resistance can be modulated or delayed by alternating treatment with BRAFi and GLI inhibitor.
Discussion
Hh signaling promotes survival, a selective growth advantage to tumor cells16 and has been implicated in multidrug resistance.22, 23 Although some groups have suggested that Hh signaling may be involved in drug resistance, such as that mediated through platelet-derived growth factor receptor-α upregulation,11 the role of this pathway in vemurafenib resistance remains unexplored. We found that GLI1/GLI2 expression increased upon the acquisition of BRAFi resistance. These results were found in both cultured cell lines and in clinical melanoma samples. These findings may be mediated through MAPK pathway reactivation in the resistant cells, which can directly result in increased GLI transcriptional activity to support motility, invasion and tumorigenesis.15
In addition to GLI1/GLI2 expression, we found upregulation of other members of the noncanonical Hh pathway, including TGFβ1, TGFβRII and SMAD3 in the resistant cells. The activation of the noncanonical Hh pathway by TGFβ increases GLI1/GLI2 expression, in part, because GLI1 is a direct transcriptional target of GLI2.24 GLI2 activation potentiates GLI1 expression and promotes the invasion and metastatic processes.25 Accordingly, vemurafenib-resistant melanoma cell lines with increased GLI2 expression also showed elevated GLI1 expression. Furthermore, other factors that are known to promote Hh pathway activation were also found in the melanoma cells,15, 26 that is, EGFR amplification in SK-MEL-28 melanoma cells, loss of phosphatase and tensin homolog in UACC62 melanoma cells and p53 mutations in both melanoma cell lines.27 Taken together, this provides consistent evidence that the Hh pathway is activated in vemurafenib-resistant cells.
There is already some suggestion that GLI signaling may be involved in melanoma initiation, possibly by mediating the escape from cellular senescence.26 Our findings demonstrated that drug-resistant SK-MEL-28 cells exhibited an increase in senescence-associated β-galactosidase staining after effective GLI1 and GLI2 gene silencing. After partial GLI2 knockdown, resistant cells presented increased sensibility to vemurafenib, with a twofold reduction in vemurafenib IC50. The Hh pathway has previously been implicated in resistance to other chemotherapeutics, such as cisplatin and taxanes in ovarian cancer.28, 29 Moreover, GLI2 knockdown increases paclitaxel chemosensitivity in prostate tumor cells.30 Besides increasing vemurafenib sensitivity, both GLI1 and GLI2 knockdown decreased MMP2 and MMP9 gene expression in vemurafenib-resistant cells; however, shGLI1 was more effective in abrogating MMP2 than shGLI2. Some papers described the role of GLI1 and MMPs contributing to a more invasive behavior,31, 32 showing that modulating GLI1 expression can directly affect MMP activation. Furthermore, modulation of GLI1 expression may be linked to many noncanonical Hh pathways, thus inhibition of GLI1 could decrease some growth factors such as TGF-β, as well as decreasing GLI2 expression.33
Vemurafenib-resistant cells treated with cyclopamine did not show altered cell viability or any changes in the total number of colonies at low concentrations. These findings were expected as cyclopamine is a classical Hh canonical SMO inhibitor,34 whereas in vemurafenib resistance appeared more dependent on the noncanonical Hh pathway. Regardless of these findings, cyclopamine is not an ideal choice for clinical development as it is both teratogenic and unable to reverse the adoption of an EMT-like phenotype.35
Elevated TGFβ expression is known to be a poor prognostic factor in melanoma and is associated with tumor progression.36 Moreover, TGFβ and MMP2/9 release are potent regulators of the ‘vicious cycle’ in tumor microenvironment that results in cell invasion and metastasis.37 It is already known that vemurafenib treatment induces TGFβ release from melanoma cells38 and this release can induce expression of EGFR, which contributes to BRAFi and MEKi resistance.39, 40, 41 TGFβ signaling can also mediate resistance by activation of the MAPK pathway and by inhibition of chemotherapy-induced apoptosis in response to a variety of cancer drugs.42, 43 As expected, in cells with noncanonical Hh pathway activation, TGFβ1 treatment significantly increased colony number, indicating an elevated tumorigenic potential.44
Gant61 decreases both GLI gene/protein expression45 and can induce cell cycle arrest, apoptosis and modulation of cell migration/EMT by decreasing MMP2/9 levels.46 In this study, Gant61 treatment induced a decrease in cell viability, colony area and total number of colonies in resistant melanoma cells (as previously described in other cancer cells).25 The clonogenic assay evaluates the capacity of each cell to drive cell division to determine the effectiveness of cytotoxic agents and the capacity to produce individual colonies.47 The decrease in colony area and colony number after Gant61 treatment was dose-dependent with the cell lines demonstrating some variation in their sensitivity to this agent, as shown with cyclopamine and TGF-β1 treatments. Moreover, Gant61 treatment repressed colony formation and modulated EMT by switch of cadherins expression in vemurafenib-resistant cells treated with TGF-β1. The link between EMT and GLI regulation has been established previously48 and TGF-β is known to induce this phenotypic switch,49 showing the cross-talk and downstream effects of GLI modulation by TGFβ in the context of EMT.
Gant61 also increased MITF expression in vemurafenib-resistant melanoma cells, and reversed the invasive profile50 that was previously reported to be induced by MAPKi therapy in melanoma.51 The inverse correlation found between low GLI and high MITF expression was identified previously in melanoma samples,52, 53 and may be linked to the adoption of less aggressive behavior.52, 54, 55, 56, 57, 58 We further examined the TCGA melanoma cohort to determine the relationship between GLI protein expression and the invasion signatures described by Hoek et al.57 or Verfaillie et al.58 In TCGA melanoma samples, we found a signature profile between GLI2-high/MITF-low. This analysis also showed a strong correlation between GLI1/GLI2/EGFR/AXL. The identification of a resistant EGFR/AXL signature may allow patients with intrinsic resistance to BRAFi56 to be identified prospectively. Additionally, the MITF-low/AXL-high levels are often correlated with drug resistance phenotype in melanoma54 and can be resistant to RAFi and MEKi, singly or in combination.55
In RHS, GLI1/GLI2 were downregulated and MITF was upregulated after Gant61 treatment, which is in accordance with the monolayer assays. The in vitro RHS model constitutes both dermal and epidermal layers, and hence it is appropriate to study the invasive potential of skin cancers, allowing assessment of growth and progression of melanoma cells; additionally, this model permits the evaluation of synthesis and release of soluble factors, such as MMPs.59, 60 Gant61 induced a significant decrease of MMP expression in both two-dimensional and 3D models, indicating the loss of invasive potential and, consequently, an inhibition of tumor dissemination. All these observations provide evidence that the RHS can be effectively used in the evaluation of stromal cell migration/invasion and, ultimately, in the screening of antitumor drugs.60, 61, 62, 63, 64
Our findings also demonstrated that GLI1/GLI2 modulation could be a useful strategy to prevent drug resistance, at least in part. Alternating pre-treatment with vemurafenib and Gant61 significantly reduced IC50 values of subsequent vemurafenib treatment in naïve melanoma cells and could represent a promising approach to prevent the onset of vemurafenib resistance. It should be noted, however, that Gant61 did not completely reverse the resistant phenotype, again illustrating the complexity of drug resistance in melanoma and highlighting the redundancy between multiple signaling pathways. The modulation of vemurafenib chemosensitivity resulting from suppression of GLI1/GLI2 expression did not occur under treatment protocols involving continuous or alternating monotherapy with vemurafenib. It was, however, noted that alternating vemurafenib and Gant61 treatment could suppress GLI expression, delaying or decreasing vemurafenib resistance. The continuous treatment with vemurafenib induced a resistance profile even in combination with others inhibitors, for example, BRAFi+MEKi,65 BRAFi+ERKi66 and BRAFi+PI3K/mTORi67 because of resistance mechanisms mainly caused by tumor heterogeneity. Furthermore, a discontinuous dosing strategy can modulate the drug-resistant profile of these cells, which may contribute to extend the vemurafenib response in melanoma patients with BRAF mutations.68, 69
An important feature of anticancer agents is the ability to induce cell death (usually apoptosis).70 Here we found that GLI downregulation induced apoptosis and this event may have contributed to the increased sensitivity of melanoma cells to vemurafenib. Hh signaling is already implicated in chemotherapeutic resistance in multiple cancers, such as in gastric cancer stem cells and basal cell carcinoma.71, 72 Therefore, some studies also describe in vitro the potential of Gant61 in increasing the sensitivity of chemotherapeutic agents such as vincristine-resistant leukemia cells,73 and rapamycin in myeloid leukemia cells.74 Gant61 has also been shown to increase the chemosensitivity in CD34+-enriched acute myeloid leukemia progenitor cells.75
Gant61 is one of the most efficient inhibitors of GLI-DNA binding45 with the potential to target cell viability, proliferation, apoptosis, DNA damage repair and EMT. Many animal studies have shown a decrease in tumor growth and an increase in apoptosis after Gant61 treatment in different tumors such as prostate, neuroblastoma and lung.45, 76, 77 Furthermore, our data suggest that Gant61 could be a useful tool to overcome or delay resistance after single-agent treatment. Studies have already reported the pharmacokinetics and toxicity of Gant61,46 indicating its future translational potential. In this context, the durability of vemurafenib antitumoral response may be improved through modifications in the dosing schedule.68
It is possible that a relationship of synthetic lethal drug combination exists between GLI and BRAFi, leading to MAPK reactivation in vemurafenib-resistant tumors. Taken together, our data demonstrated an unprecedented mechanism of vemurafenib resistance by GLI1/GLI2 upregulation, shedding light on the development of Hh pathway inhibitors as a promising strategy for melanoma treatment.
Materials and methods
Reagents
The salts used and the MCDB153 culture medium were purchased from Merck and Sigma Chemical Co. (St Louis, MO, USA). The culture medium Dulbecco's modified Eagle's medium, Leibovitz's L-15, fetal bovine serum and trypsin were purchased from Gibco (Grand Sland, NY, USA). Vemurafenib, cyclopamine and Gant61 were purchased from Selleck Chemicals (Houston, TX, USA). Antibodies for western blotting and immunochemistry were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), Cell Signaling Technology (Danvers, MA, USA), Sigma Chemical Co. and Abcam (Cambridge, UK).
Melanoma cell line cultures
Melanoma cell lines SK-MEL-19, SK-MEL-28, SK-MEL-29, UACC62, WM9, WM35, WM278, WM793, WM1552c, WM1617 and 1205Lu were kindly provided by Dr Marisol Soengas (Centro Nacional de Investigaciones Oncological, Madrid, Spain) and Dr Enilza Maria Espreáfico (Ribeirão Preto Medical School, University of São Paulo, São Paulo, Brazil). The identities of the cell lines were confirmed through STR validation analysis.21 The R3 melanoma cell line was obtained from patient no. 1 after acquisition of vemurafenib resistance proved by tumor recurrence.
Melanoma cell lines SK-MEL-19, SK-MEL-28, SK-MEL-29, UACC62 and R3 were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and antibiotics. Melanoma cell lines WM9, WM35, WM278, WM793, WM1552c, WM1617 and 1205Lu were cultured in MCDB153/Leibovitz's L-15 (4:1) supplemented with 2% fetal bovine serum, insulin 5 μg/ml and CaCl2 1.6 mM.78 All melanoma cells were maintained in a humidified incubator at 37 °C containing 5% of CO2.
Cell culture and generation of BRAFi resistance
To derive vemurafenib-resistant cell lines, naïve cells were seeded at low cell density and treated with vemurafenib at 0.5–6 μM every 3 days for 4–6 weeks, and thus colonies were then isolated.8, 21 SK-MEL-19-, WM278-, WM793-, WM1552c-, WM1617- and 1205Lu-resistant cells were derived by successive titration of vemurafenib up to 3 μM and SK-MEL-28-, SK-MEL-29- and UACC62-resistant cells up to 6μM. Resistant cell lines were replenished with 3 or 6μM vemurafenib every 2–3 days.
Primary skin cell cultures
Normal human skin cells were obtained from donated foreskin samples from the University of São Paulo Hospital (HU-USP:943/09; CEP/FCF/USP:534). Cells were isolated and cultivated as described previously by our group.60, 79 Fibroblasts and melanocytes were maintained in a humidified incubator at 37 °C containing 5% of CO2, whereas keratinocytes at 7.5% CO2.
Patient samples
Melanoma samples of patients from the Barretos Cancer Hospital (Barretos, Brazil) pre- and post-vemurafenib treatment were obtained after signing of patients in the informed consent form. Melanoma samples were analyzed and confirmed by two different pathologists. This study was approved by the Ethics in Research Committee of the School of Pharmaceutical Sciences of the University of São Paulo (849.540/2014) and the Barretos Cancer Hospital Ethic Committee (890/2014).
Quantitative real-time PCR
Total RNA were isolated using Qiagen’s RNeasy Mini Kit (Qiagen, Valencia, CA, USA). TaqMan Gene Expression Assays primer/probes were used (Supplementary Table S2). The glyceraldehyde 3-phosphate dehydrogenase/β-actin data were used for normalizing interest genes. Quantitative reverse transcriptase–PCR reactions were carried out as described previously.10
Western blotting
Proteins were extracted and blotted for as described previously.80 Total and phosphoproteins were analyzed (Supplementary Table S3). After analysis, western blots were stripped once and reprobed for glyceraldehyde 3-phosphate dehydrogenase/β-actin/vinculin to show even protein loading.
RNA interference
The SK-MEL-28 vemurafenib-resistant melanoma cell line was plated (2.5 × 104 cells per well) to the transfection with lentiviral particles by shRNA technique for GLI1 or GLI2 genes or a negative control (shGLI1, shGLI2 or shControl, respectively) with two or three different hairpin sequences (Supplementary Table S4) according to the manufacturer's protocol. The cells were incubated with polybrene at 5 μg/ml for 12 h at 37°C, then the lentiviral particles were added (MOI=10 or 5) in the culture medium and selection with puromycin was performed (Santa Cruz Biotechnology). The shControl was performed as a negative control (scrambled shRNA sequence). The shRNAs generated were validated for gene and protein by real-time PCR and western blotting, respectively.
Cell viability assay (MTT assay)
Cells were plated in triplicate wells (1 × 104 cells per well) and treated with increasing concentrations of vemurafenib (0–48 μM) or drug diluent (dimethyl sulfoxide) for 24/48/72 h. Cell viability was determined using MTT assay as described previously.81
Senescence β-galactosidase
Cells were seeded in six-well plates at a density of ~2.5 × 104 cells per well after shRNA. Then, the cells were fixed and stained using a senescence β-galactosidase cytochemical detection.82 Plates were incubated overnight at 37 °C and the cells were detected for blue staining under a bright field.83
Colony formation
Cells (1 × 104/ml) were grown overnight before being treated with TGFβ1, cyclopamine or Gant61 for 4 weeks as described previously.84 Relative colony area density was determined by the ImageJ 1.49n software (NIH, Bethesda, MD, USA) with ColonyArea plugin and colony number was determined by the Molecular imaging software Bruker 7.1.1.20220v (Bruker BioSpin Corp., Billerica, MA, USA).
Flow cytometry
Cells were plated into 6-well tissue culture plates at 60% confluency and left to grow overnight before being treated with Gant61 for 48 h. Annexin V and propidium iodide staining were carried out as described previously.84
TCGA transcriptomic analysis
See Supplementary Materials and methods.
Reconstructed skin in vitro (RHS)
RHS were prepared in two steps. First, the dermal compartment was prepared using type-1 collagen gel and 1.5 × 104 human fibroblasts/dermis. After polymerization, 25 × 104 human keratinocytes, 0.83 × 104 human melanocytes and 8.3 × 104 melanoma cells (SK-MEL-28-naïve or vemurafenib-resistant) were seeded on top of each lattice and the skins were kept submerged in the culture medium for 24 h. Subsequently, the culture was raised and maintained at the air–liquid interface for 12 days to allow complete keratinocytes stratification and differentiation.64, 79 The RHS containing resistant cells were treated with vemurafenib every 72 h and the RHS containing naïve or resistant cells were treated with Gant61 in the last 72 h of culture period.
Hematoxylin and eosin staining and immunohistochemistry
The samples were fixed, followed by dehydration, cleaning for paraffin inclusion and stained with H&E for morphological analysis and with different antibodies to immunochemistry assays (Supplementary Table S5). All images were obtained by optical microscopy and analyzed by the NIS-Elements software (Nikon Instruments, Melville, NY, USA). Measurement of melanoma area thickness was carried out using three images with five independent measurements of the melanoma areas thickness from three different experimental conditions.79
Statistical analysis
Results are expressed as mean±s.d of triplicate of three independent experiments. Data were analyzed using one-way analysis of variance and significant mean differences were determined using multiple comparisons by the Tukey–Kramer test at the P<0.05 level. Significant differences between the control and treated groups are indicated by ***P<0.001, **P<0.01 and *P<0.05.
References
Tsao H, Chin L, Garraway LA, Fisher DE . Melanoma: from mutations to medicine. Genes Dev 2012; 26: 1131–1155.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417: 949–954.
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012; 367: 1694–1703.
Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 2012; 366: 707–714.
Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014; 371: 1877–1888.
Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363: 809–819.
Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010; 468: 968–972.
Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010; 468: 973–977.
Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1 R/PI3K. Cancer Cell 2010; 18: 683–695.
Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res 2011; 71: 2750–2760.
Sabbatino F, Wang Y, Wang X, Flaherty KT, Yu L, Pepin D et al. PDGFRalpha up-regulation mediated by sonic hedgehog pathway activation leads to BRAF inhibitor resistance in melanoma cells with BRAF mutation. Oncotarget 2014; 5: 1926–1941.
Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov 2014; 4: 61–68.
Menzies AM, Wilmott JS, Drummond M, Lo S, Lyle M, Chan MM et al. Clinicopathologic features associated with efficacy and long-term survival in metastatic melanoma patients treated with BRAF or combined BRAF and MEK inhibitors. Cancer 2015; 121: 3826–3835.
Dennler S, Andre J, Alexaki I, Li A, Magnaldo T, ten Dijke P et al. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res 2007; 67: 6981–6986.
Ruiz I, Altaba A, Sanchez P, Dahmane N . Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer 2002; 2: 361–372.
Javelaud D, Alexaki VI, Dennler S, Mohammad KS, Guise TA, Mauviel A . TGF-beta/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res 2011; 71: 5606–5610.
Matise MP, Joyner AL . Gli genes in development and cancer. Oncogene 1999; 18: 7852–7859.
Zhou J, Zhu G, Huang J, Li L, Du Y, Gao Y et al. Non-canonical GLI1/2 activation by PI3K/AKT signaling in renal cell carcinoma: a novel potential therapeutic target. Cancer Lett 2016; 370: 313–323.
Dennler S, Andre J, Verrecchia F, Mauviel A . Cloning of the human GLI2 Promoter: transcriptional activation by transforming growth factor-beta via SMAD3/beta-catenin cooperation. J Biol Chem 2009; 284: 31523–31531.
Pierrat MJ, Marsaud V, Mauviel A, Javelaud D . Expression of microphthalmia-associated transcription factor (MITF), which is critical for melanoma progression, is inhibited by both transcription factor GLI2 and transforming growth factor-beta. J Biol Chem 2012; 287: 17996–18004.
Sandri S, Faiao-Flores F, Tiago M, Pennacchi PC, Massaro RR, Alves-Fernandes DK et al. Vemurafenib resistance increases melanoma invasiveness and modulates the tumor microenvironment by MMP-2 upregulation. Pharmacol Res 2016; 111: 523–533.
Sims-Mourtada J, Izzo JG, Ajani J, Chao KS . Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene 2007; 26: 5674–5679.
Zhou H, Zhang Z, Liu C, Jin C, Zhang J, Miao X et al. B4GALT1 gene knockdown inhibits the hedgehog pathway and reverses multidrug resistance in the human leukemia K562/adriamycin-resistant cell line. IUBMB Life 2012; 64: 889–900.
Ikram MS, Neill GW, Regl G, Eichberger T, Frischauf AM, Aberger F et al. GLI2 is expressed in normal human epidermis and BCC and induces GLI1 expression by binding to its promoter. J Invest Dermatol 2004; 122: 1503–1509.
Javelaud D, Pierrat MJ, Mauviel A . Crosstalk between TGF-beta and hedgehog signaling in cancer. FEBS Lett 2012; 586: 2016–2025.
Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V et al. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci USA 2007; 104: 5895–5900.
Niehr F, von Euw E, Attar N, Guo D, Matsunaga D, Sazegar H et al. Combination therapy with vemurafenib (PLX4032/RG7204) and metformin in melanoma cell lines with distinct driver mutations. J Transl Med 2011; 9: 76.
Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, Alvarez RD et al. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin Cancer Res 2012; 18: 869–881.
Steg AD, Katre AA, Bevis KS, Ziebarth A, Dobbin ZC, Shah MM et al. Smoothened antagonists reverse taxane resistance in ovarian cancer. Mol Cancer Ther 2012; 11: 1587–1597.
Narita S, So A, Ettinger S, Hayashi N, Muramaki M, Fazli L et al. GLI2 knockdown using an antisense oligonucleotide induces apoptosis and chemosensitizes cells to paclitaxel in androgen-independent prostate cancer. Clin Cancer Res 2008; 14: 5769–5777.
Fan YH, Ding J, Nguyen S, Liu XJ, Xu G, Zhou HY et al. Aberrant hedgehog signaling is responsible for the highly invasive behavior of a subpopulation of hepatoma cells. Oncogene 2016; 35: 116–124.
Fan HX, Wang S, Zhao H, Liu N, Chen D, Sun M et al. Sonic hedgehog signaling may promote invasion and metastasis of oral squamous cell carcinoma by activating MMP-9 and E-cadherin expression. Med Oncol 2014; 31: 41.
Palle K, Mani C, Tripathi K, Athar M . Aberrant GLI1 activation in DNA damage response, carcinogenesis and chemoresistance. Cancers 2015; 7: 2330–2351.
Chen JK, Taipale J, Cooper MK, Beachy PA . Inhibition of Hedgehog signaling by direct binding of cyclopamine to smoothened. Genes Dev 2002; 16: 2743–2748.
Lei J, Ma J, Ma Q, Li X, Liu H, Xu Q et al. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol Cancer 2013; 12: 66.
Javelaud D, Alexaki VI, Mauviel A . Transforming growth factor-beta in cutaneous melanoma. Pigment Cell Melanoma Res 2008; 21: 123–132.
Tauro M, Laghezza A, Loiodice F, Agamennone M, Campestre C, Tortorella P . Arylamino methylene bisphosphonate derivatives as bone seeking matrix metalloproteinase inhibitors. Bioorg Med Chem 2013; 21: 6456–6465.
Fedorenko IV, Wargo JA, Flaherty KT, Messina JL, Smalley KS . BRAF Inhibition generates a host–tumor niche that mediates therapeutic escape. J Invest Dermatol 2015; 135: 3115–3124.
Menon DR, Wels C, Bonyadi Rad E, Joshi S, Knausz H, Lade-Keller J et al. TGF-beta1 and TNF-alpha differentially regulate Twist1 mediated resistance towards BRAF/MEK inhibition in melanoma. Pigment Cell Melanoma Res 2013; 26: 912–916.
Girotti MR, Pedersen M, Sanchez-Laorden B, Viros A, Turajlic S, Niculescu-Duvaz D et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov 2013; 3: 158–167.
Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014; 508: 118–122.
Eichhorn PJ, Rodon L, Gonzalez-Junca A, Dirac A, Gili M, Martinez-Saez E et al. USP15 stabilizes TGF-beta receptor I and promotes oncogenesis through the activation of TGF-beta signaling in glioblastoma. Nat Med 2012; 18: 429–435.
Huang S, Holzel M, Knijnenburg T, Schlicker A, Roepman P, McDermott U et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell 2012; 151: 937–950.
Tirino V, Camerlingo R, Bifulco K, Irollo E, Montella R, Paino F et al. TGF-beta1 exposure induces epithelial to mesenchymal transition both in CSCs and non-CSCs of the A549 cell line, leading to an increase of migration ability in the CD133+ A549 cell fraction. Cell Death Dis 2013; 4: e620.
Lauth M, Bergstrom A, Shimokawa T, Toftgard R . Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci USA 2007; 104: 8455–8460.
Gonnissen A, Isebaert S, Haustermans K . Targeting the Hedgehog signaling pathway in cancer: beyond smoothened. Oncotarget 2015; 6: 13899–13913.
Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C . Clonogenic assay of cells in vitro. Nat Protocols 2006; 1: 2315–2319.
Li X, Deng W, Nail CD, Bailey SK, Kraus MH, Ruppert JM et al. Snail induction is an early response to Gli1 that determines the efficiency of epithelial transformation. Oncogene 2006; 25: 609–621.
Zavadil J, Bottinger EP . TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005; 24: 5764–5774.
Lasfar A, Cohen-Solal KA . Resistance to transforming growth factor beta-mediated tumor suppression in melanoma: are multiple mechanisms in place? Carcinogenesis 2010; 31: 1710–1717.
Johannessen CM, Johnson LA, Piccioni F, Townes A, Frederick DT, Donahue MK et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013; 504: 138–142.
Javelaud D, Alexaki VI, Pierrat MJ, Hoek KS, Dennler S, Van Kempen L et al. GLI2 and M-MITF transcription factors control exclusive gene expression programs and inversely regulate invasion in human melanoma cells. Pigment Cell Melanoma Res 2011; 24: 932–943.
Alexaki VI, Javelaud D, Van Kempen LC, Mohammad KS, Dennler S, Luciani F et al. GLI2-mediated melanoma invasion and metastasis. J Natl Cancer Inst 2010; 102: 1148–1159.
Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 2014; 5: 5712.
Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov 2014; 4: 816–827.
Dugo M, Nicolini G, Tragni G, Bersani I, Tomassetti A, Colonna V et al. A melanoma subtype with intrinsic resistance to BRAF inhibition identified by receptor tyrosine kinases gene-driven classification. Oncotarget 2015; 6: 5118–5133.
Hoek KS, Schlegel NC, Brafford P, Sucker A, Ugurel S, Kumar R et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res 2006; 19: 290–302.
Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat Commun 2015; 6: 6683.
Smalley KS, Lioni M, Herlyn M . Life isn't flat: taking cancer biology to the next dimension. In vitro Cell Dev Biol Anim 2006; 42: 242–247.
Brohem CA, Massaro RR, Tiago M, Marinho CE, Jasiulionis MG, de Almeida RL et al. Proteasome inhibition and ROS generation by 4-nerolidylcatechol induces melanoma cell death. Pigment Cell Melanoma Res 2012; 25: 354–369.
Yu H, McDaid R, Lee J, Possik P, Li L, Kumar SM et al. The role of BRAF mutation and p53 inactivation during transformation of a subpopulation of primary human melanocytes. Am J Pathol 2009; 174: 2367–2377.
Fernandez Y, Miller TP, Denoyelle C, Esteban JA, Tang WH, Bengston AL et al. Chemical blockage of the proteasome inhibitory function of bortezomib: impact on tumor cell death. J Biol Chem 2006; 281: 1107–1118.
Chioni AM, Grose R . Organotypic modelling as a means of investigating epithelial-stromal interactions during tumourigenesis. Fibrogen Tissue Rep 2008; 1: 8.
Brohem CA, Cardeal LB, Tiago M, Soengas MS, Barros SB, Maria-Engler SS . Artificial skin in perspective: concepts and applications. Pigment Cell Melanoma Res 2011; 24: 35–50.
Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep 2013; 4: 1090–1099.
Carlino MS, Todd JR, Gowrishankar K, Mijatov B, Pupo GM, Fung C et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 2014; 8: 544–554.
Penna I, Molla A, Grazia G, Cleris L, Nicolini G, Perrone F et al. Primary cross-resistance to BRAFV600E-, MEK1/2- and PI3K/mTOR-specific inhibitors in BRAF-mutant melanoma cells counteracted by dual pathway blockade. Oncotarget 2016; 7: 3947–3965.
Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013; 494: 251–255.
Long GV, Weber JS, Infante JR, Kim KB, Daud A, Gonzalez R et al. Overall survival and durable responses in patients with BRAF V600-mutant metastatic melanoma receiving dabrafenib combined with trametinib. J Clin Oncol 2016; 34: 871–878.
Green DR, Walczak H . Apoptosis therapy: driving cancers down the road to ruin. Nat Med 2013; 19: 131–133.
Xu M, Gong A, Yang H, George SK, Jiao Z, Huang H et al. Sonic hedgehog-glioma associated oncogene homolog 1 signaling enhances drug resistance in CD44(+)/Musashi-1(+) gastric cancer stem cells. Cancer Lett 2015; 369: 124–133.
Meani RE, Lim SW, Chang AL, Kelly JW . Emergence of chemoresistance in a metastatic basal cell carcinoma patient after complete response to hedgehog pathway inhibitor vismodegib (GDC-0449). Austral J Dermatol 2014; 55: 218–221.
Queiroz KC, Ruela-de-Sousa RR, Fuhler GM, Aberson HL, Ferreira CV, Peppelenbosch MP et al. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 2010; 29: 6314–6322.
Pan D, Li Y, Li Z, Wang Y, Wang P, Liang Y . Gli inhibitor GANT61 causes apoptosis in myeloid leukemia cells and acts in synergy with rapamycin. Leuk Res 2012; 36: 742–748.
Long B, Wang LX, Zheng FM, Lai SP, Xu DR, Hu Y et al. Targeting GLI1 suppresses cell growth and enhances chemosensitivity in CD34+ enriched acute myeloid leukemia progenitor cells. Cell Physiol Biochem 2016; 38: 1288–1302.
Wickstrom M, Dyberg C, Shimokawa T, Milosevic J, Baryawno N, Fuskevag OM et al. Targeting the hedgehog signal transduction pathway at the level of GLI inhibits neuroblastoma cell growth in vitro and in vivo. Int J Cancer 2013; 132: 1516–1524.
Huang L, Walter V, Hayes DN, Onaitis M . Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin Cancer Res 2014; 20: 1566–1575.
O'Connell MP, Marchbank K, Webster MR, Valiga AA, Kaur A, Vultur A et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov 2013; 3: 1378–1393.
Pennacchi PC, de Almeida ME, Gomes OL, Faiao-Flores F, de Araujo Crepaldi MC, Dos Santos MF et al. Glycated reconstructed human skin as a platform to study the pathogenesis of skin aging. Tissue Eng Part A 2015; 21: 2417–2425.
Smalley KS, Contractor R, Haass NK, Lee JT, Nathanson KL, Medina CA et al. Ki67 expression levels are a better marker of reduced melanoma growth following MEK inhibitor treatment than phospho-ERK levels. Br J Cancer 2007; 96: 445–449.
Faiao-Flores F, Suarez JA, Soto-Cerrato V, Espona-Fiedler M, Perez-Tomas R, Maria DA . Bcl-2 family proteins and cytoskeleton changes involved in DM-1 cytotoxic effect on melanoma cells. Tumour Biol 2013; 34: 1235–1243.
Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O . Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protocols 2009; 4: 1798–1806.
Gary RK, Kindell SM . Quantitative assay of senescence-associated beta-galactosidase activity in mammalian cell extracts. Anal Biochem 2005; 343: 329–334.
Paraiso KH, Fedorenko IV, Cantini LP, Munko AC, Hall M, Sondak VK et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer 2010; 102: 1724–1730.
Acknowledgements
We thank Dr Walter Turato for help with technical advice and support in colony formation studies and Renata Albuquerque for the FACS analyses. This work was supported by Fapesp-grant number 2012/04194-1, 2013/05172-4, 2014/24400-0 and 2015/10821-7, CNPq-grant number 150447/2013-2 and 471512/2013-3 and PRODOC-grant no 3193-32/2010. Work in the lab of KS Smalley was supported by the National Institutes of Health grants R01 CA161107, R21 CA198550, and Skin SPORE grant P50 CA168536.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Faião-Flores, F., Alves-Fernandes, D., Pennacchi, P. et al. Targeting the hedgehog transcription factors GLI1 and GLI2 restores sensitivity to vemurafenib-resistant human melanoma cells. Oncogene 36, 1849–1861 (2017). https://doi.org/10.1038/onc.2016.348
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.348

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}