
Abstract
DOCK proteins are guanine nucleotide exchange factors for Rac and Cdc42 GTPases. DOCK1 is the founding member of the family and acts downstream of integrins via the canonical Crk-p130Cas complex to activate Rac GTPases in numerous contexts. In contrast, DOCK5, which possesses the greatest similarity to DOCK1, remains sparingly studied. Here we establish that DOCK5 has a non-redundant role in regulating motile and invasive capacities of epithelial cells. DOCK1 is constitutively associated with sites of integrin attachment termed focal adhesions (FAs). In contrast, we demonstrate that DOCK5 recruitment to FAs in Hela cells is restricted by GIT2, an established regulator of FA signaling. We determine that GIT2 is targeted to FAs in response to Rho-ROCK signaling and actomyosin contractility. Accordingly, inhibition of ROCK activity or MLC function promotes enrichment of DOCK5 in membrane protrusions and nascent cell–substratum adhesions. We further demonstrate that GIT2 inhibits the interaction of DOCK5 with Crk. Moreover, we show that depletion of GIT2 promotes DOCK5-dependent activation of the Crk-p130Cas signaling cascade to promote Rac1-mediated lamellipodial protrusion and FA turnover. The antagonism between GIT2 and DOCK5 extends to non-transformed MCF10A mammary epithelial cells, with DOCK5 ‘dialing-up’ and GIT2 ‘dialing-down’ invasiveness. Finally, we determine that DOCK5 inhibition attenuates invasion and metastasis of MDA-MB-231 cells and prolongs life span of mice injected with these cells. Collectively, our work identifies DOCK5 as a key regulator of epithelial invasion and metastasis, and demonstrates that suppression of DOCK5 by GIT2 represents a previously unappreciated mechanism for coordination of Rho and Rac GTPases.
Similar content being viewed by others


Introduction
Cell motility requires coordinated actin polymerization and cell adhesion. Directional cell motility moreover necessitates the establishment of front-to-back cell polarity with a protrusive leading edge and a retractive trailing edge. At the leading edge, nascent cell–matrix adhesions tether membrane protrusions to the substrate, whereas mature adhesions behind the leading edge firmly attach the cell membrane to actin stress fibers. These front and rear domains are regulated by Rac and RhoA GTPases, respectively. Moreover, Rac and RhoA antagonize each other, thus reinforcing polarized activation of these GTPases at distinct regions of the cell.1
Activation of Rho GTPases relies on guanine nucleotide exchange factors (GEFs) to promote exchange of GDP for GTP. More than 80 RhoGEFs exist, thus underscoring the precise spatiotemporal control of Rho activation in cell motility.2 The DOCK family comprises 11 GEFs, which are frequently referred to as ‘atypical’ to distinguish them from the larger Dbl family.2 DOCKs are classified into four subgroups all characterized by the presence of two evolutionarily conserved DOCK homology region (DHR) domains: the lipid-binding DHR-1 and the catalytic DHR-2.3, 4
The DOCK-A branch comprises DOCK1, DOCK2 and DOCK5, which function as Rac-specific GEFs.2 DOCK-A members also possess a proline-rich carboxy-terminal domain that binds to the SH2-SH3 adaptor molecule Crk, which mediates targeting to focal adhesions (FAs).5, 6 Assembly of a Crk-p130Cas-DOCK1 complex promotes Rac activation in response to integrin engagement.7 Numerous studies have linked DOCK1 to cellular processes that require coordinated cell adhesion and actin remodeling, such as axon guidance, myoblast fusion, apoptotic cell engulfment and cell migration.2 Recently, oncogenic signaling pathways were demonstrated to regulate DOCK1 activity through phosphorylation, implicating DOCK1 as a key factor in transformation and metastasis.8, 9 DOCK2 is predominantly expressed in hematopoietic tissues where it regulates chemotactic migration and lymphocyte trafficking.10
DOCK5 is the least studied member of the DOCK family, perhaps as result of a dispensable role in murine gross embryonic development and an apparently redundant role in cell migration.11, 12 DOCK5-deficient mice exhibit increased bone mass because of a requirement for DOCK5 in osteoclast adhesion.13 In addition, DOCK5 promotes myoblast fusion, mast cell degranulation and neutrophil chemotaxis.14, 15 Here we identify a role for DOCK5 in epithelial cell motility and invasion. Mechanistically, we show that the FA-associated protein GIT2 suppresses interaction of Crk with DOCK5, thus preventing activation of a Crk-p130Cas-DOCK5 signaling cascade required for plasma membrane protrusion and FA turnover. Moreover, our data indicate that the inhibitory activity of GIT2 requires Rho- and tension-dependent targeting to cell–matrix adhesions. Furthermore, we demonstrate that GIT2 and DOCK5 function as a rheostat in control of invasive capacities in the non-transformed mammary epithelial cell line MCF10A. Finally, we show that in the invasive breast cancer line MDA-MB-231, depletion of DOCK5 attenuates invasiveness resulting in reduced tumor burden and increased survival time in mice challenged intravenously with these cells.
Results
GIT2 represses DOCK5 function
GIT2 is targeted to FAs via its association with paxillin, where it has an important role in regulating cell motility.16 Along with others, our group have found that GIT2 functions to restrict the function of Rac, as loss of GIT2, or its paralog GIT1, results in deregulated Rac signaling in numerous cell types.17, 18, 19, 20, 21
We previously reported that GIT2 suppresses Crk-dependent lamellipodial protrusion.19 The Crk adaptor protein is a constituent of the canonical paxillin-Crk-p130Cas-DOCK1 signaling module required for Rac activation. We hypothesized that DOCK1 may be a target of GIT2 repression.5, 6, 7 To test this hypothesis, GIT2 was depleted alone or in combination with DOCK1 (Supplementary Figure S1a). Surprisingly, DOCK1 is not required for cell spreading induced by GIT2-kd (Supplementary Figure S1b). As DOCK5 is the most closely related protein to DOCK1, we speculated that GIT2 may modulate the function of DOCK5 instead. We generated a highly specific antiserum against the divergent C-terminal tail of DOCK5 (Supplementary Figure S1c). Using this antiserum, we determined that DOCK5 is readily detected in numerous cell lines, including HeLa cells (Supplementary Figure S1d). Next, DOCK5 was targeted alone or together with GIT2 by short hairpin RNA (shRNA)-mediated gene silencing (Supplementary Figure S1a). Depletion of DOCK5 completely abrogates enhanced membrane protrusiveness (Figure 1a) and cell spreading (Figure 1b and Supplementary Figure S1b) elicited by GIT2-kd. These effects were reproduced with an additional four shRNAs targeting distinct sequences in the DOCK5 message (Supplementary Figures S2a–c). Furthermore, in HeLa cells, knockdown of DOCK5 alone inhibits lamellipodial extension and cell spreading (Figures 1a and b), as well as cell adhesion (Figure 1c).

GIT2 represses DOCK5-regulated and Rac1-dependent cell spreading, as well as FA turnover. HeLa cells were transfected with the indicated shRNA expression vectors and selected in 2.5 μg/ml of puromycin for 72 h. Cells were then cultured for an additional 24 h in the absence of puromycin. (a) Phase contrast images of control, GIT2-kd, DOCK1-kd, DOCK5-kd and GIT2-kd+DOCK5-kd cells cultured on collagen. (b) Quantification of cell area occupied by control, GIT2-kd, DOCK5-kd and GIT2-kd+DOCK5-kd HeLa cells 1 h after plating on collagen. (c) Control and DOCK5-kd cells were plated on collagen and assayed for adhesion after 1 h. All values are means±s.d. (n=3). (d) Quantification of the distribution of areas occupied by control, GIT2-kd, GIT2-kd+Rac1-kd and GIT2-kd+Rac3-kd HeLa cells 1 h after plating on collagen. (e) Specificity of NewEast Biosciences antibody for Rac-GTP. Recombinant Rac1 was loaded with either GDP or GTPγS followed by immunoprecipitation with monoclonal antibody to Rac1-GTP and western blotting. (f) Localization of Rac-GTP in control, GIT2-kd, DOCK5-kd and GIT2-kd+DOCK5-kd cells 1 h after plating on collagen. (g) FA turnover in live HeLa cells expressing paxillin-GFP. Representative confocal images illustrate FA formation over times indicated in control, GIT2-kd, DOCK5-kd and GIT2+DOCK5-kd cells grown on glass. (h) Rate (min−1) of assembly, disassembly and turnover of FAs as measured by change in GFP fluorescence over time for control, GIT2-kd, DOCK5-kd and GIT2+DOCK5-kd Hela cells, ***P<0.001, **P<0.002, analysis of variance with Tukey–Kramer post hoc testing.
HeLa cells express Rac1 and Rac3. To identify the Rac form, which GIT2 represses, we depleted GIT2 together with Rac1 or Rac3. Rac1-kd effectively abrogates cell spreading elicited by GIT2 depletion, whereas Rac3-kd has no effect (Figure 1d). To determine the effects of GIT2 and/or DOCK5 depletion on the subcellular localization of activated Rac, we stained HeLa cells with an antibody specific for the GTP-bound form of Rac (Figure 1e). The staining revealed strong enrichment in peripheral membrane ruffles elicited by knockdown of GIT2 (Figure 1e). Such staining was also observed to a lesser extent in control cells, but never in cells depleted of DOCK5 alone or in combination with GIT2 (Figure 1f).
Next, to establish whether GIT2- and DOCK5-modulated changes in lamellipodial protrusion and cell spreading were associated with altered FA turnover, we generated Hela cells expressing paxillin-green fluorescent protein (GFP) and performed live cell imaging (Figure 1g). Depletion of GIT2 in these cells elicits strong increases in the rates of FA assembly, disassembly and turnover that are abrogated by DOCK5-kd (Figure 1h). Moreover, depletion of DOCK5 alone reduces these rates significantly relative control cells (Figure 1h).
Engagement of integrins activates the tyrosine kinases FAK and Src, which phosphorylate paxillin on Y31 and Y118, thereby facilitating binding of Crk and assembly of the Crk-p130Cas-DOCK1 complex.17, 22, 23, 24 We therefore tested a possible role for GIT2 in regulation of this signaling cascade. Knockdown of GIT2 enhances phosphorylation of paxillin at Y118 and p130Cas at Y410 (Figures 2a and b). GIT2-kd also promotes autophosphorylation of FAK at Y397 (Figures 2a and b). DOCK5 appears to be a key activator of this pathway, as DOCK5-deficient cells show impaired phosphorylation of p130Cas, paxillin and FAK (Figures 2a and b). Importantly, DOCK5 is essential for the induced phosphorylation of these proteins in response to GIT2-kd (Figures 2a and b).

GIT2 modulates a Crk-p130Cas-DOCK5 signaling pathway required cell spreading. (a) Puromycin selected control, GIT2-kd, DOCK5-kd or DOCK5 and GIT2-kd cells were plated on collagen-coated plates for 2.5 h. Next, whole-cell lysates were processed for western blotting to detect GIT2, DOCK1, DOCK5 and ERK as loading control, as well as phosphorylated forms and total levels of p130Cas, FAK and paxillin. (b) Quantification of phospho-p130Cas, -paxillin, and -FAK levels from a. All values are means±s.d. (n=3). *P<0.05; **P<0.02; ***P<0.01 by unpaired two-tailed Student's t-test. (c) DOCK5 binds to Crk in a GIT2-regulated manner. HeLa cells with or without GIT2 depletion were plated on collagen-coated plates for 2.5 h. The cells were then lysed and the samples processed for immunoprecipitation with anti-Crk antibody and western blotting to detect Crk, p130Cas and Crk in immunoprecipitates and whole-cell lysates. (d) Quantification of the amount of DOCK5 associated with Crk in control and GIT2-kd cells. Values are means±s.d. (n=3). ***P<0.01 by unpaired two-tailed Student's t-test. (e) Cells with or without knockdown of GIT2 were transfected overnight with a Myc-tagged form of the CBD of DOCK5 comprises amino acids 1712–1871. Transfected cells were plated on collagen for 1 h and labeled to detect Myc (green) and Arp3 (red). (f) Quantification of cell area from e. All values are means±s.d. (n=3). ***P<0.01 by unpaired two-tailed Student's t-test.
To determine if GIT2 modulates the interaction between Crk and DOCK5 and/or p130Cas, we conducted co-immunoprecipitation assays with anti-Crk antibody. Knockdown of GIT2 elicited a significant increase in the interaction between DOCK5 and Crk without affecting the association of p130Cas and Crk (Figures 2c and d). Next, to test a requirement for the interaction between DOCK5 and Crk in promoting cell spreading, we expressed a Myc-tagged form of the DOCK5 Crk-binding domain (DOCK5-CBD; amino acids 1712–1871), which would be expected to act as a dominant inhibitory molecule. Although low level expression of DOCK5-CBD alone exerts minor effects on control cells, there is a significant reduction of cell spreading in GIT2-depleted cells (Figures 2e and f). Collectively, the data support a role for GIT2 as a suppressor of a Crk-p130Cas-DOCK5 signaling pathway in control of FA dynamics and Rac1-regulated cell motility. Moreover, these results establish that DOCK5 has an essential and non-redundant role in membrane protrusiveness at the cell periphery.
Localization of DOCK5 is controlled by GIT2
To elucidate how GIT2 may regulate DOCK5 function we examined the localization of exogenous DOCK1 and DOCK5 in HeLa cells. Although DOCK1 constitutively localizes to FAs (Figure 3a), DOCK5 is found in discrete structures that closely surround but do not overlap with FAs (Figure 3a). In spreading control cells, exogenous DOCK5 was localized peripheral to paxillin-enriched FAs. In contrast, we observed robust colocalization of DOCK5 with paxillin in FAs of spreading GIT2-kd cells (Figure 3b). These results show that GIT2 restricts access of DOCK5 to FAs.

DOCK5 localization is controlled by GIT2. (a) Localization of DOCK1 (green) or DOCK5 (green) together with paxillin (red) in HeLa cells transfected with DOCK1 or DOCK5 expression constructs. (b) Localization of DOCK5 (green) and paxillin (red) in control and GIT2-depleted cells transfected with DOCK5 expression construct and plated on collagen for 1 h. (c) HeLa cells were transfected with control, GIT2, and DOCK5 shRNA expression vectors and selected in puromycin as described in Figure 1. Pooled knockdown cells were plated on collagen for 1 h and labeled to detect DOCK5 (green) and paxillin (red). (d) Pooled knockdown cells plated on collagen for 1 h were labeled to detect DOCK5 (green) and Arp3 (red). All scale bars represent 10 μm each.
We next examined the localization of endogenous DOCK5 in spreading HeLa cells. In control cells, DOCK5 is located at the tip of cellular protrusions (Figure 3c). Strikingly, GIT2-kd enhances membrane recruitment of DOCK5 and colocalization with paxillin at the cell periphery (Figure 3c). Interestingly, DOCK5-depleted cells exhibit fewer and much larger FAs, that is, the direct opposite of what is observed upon GIT2 knockdown (Figure 3c).
Our findings indicated that activation of a Crk-p130Cas-DOCK5 signaling pathway in response to GIT2 depletion promotes Rac1 activation at the cell periphery. Our DOCK5 and Rac-GTP antibodies require denaturing and non-denaturing fixation, respectively. Therefore, to determine whether endogenous DOCK5 was specifically associated with Rac-regulated lamellipodia, we localized Arp3, which operates downstream of Rac to promote actin polymerization. In control cells, DOCK5 and Arp3 colocalize in isolated membrane protrusions at the cell periphery (Figure 3d). Depletion of GIT2 results in a marked enhancement of Arp3 at the cell periphery where Arp3 associates with DOCK5-labeled lamellipodia (Figure 3d). Importantly, DOCK5 is required for generation of Arp3-rich membrane protrusions in response to GIT2 knockdown (Figure 3d). These results are consistent with a role for GIT2 in limiting DOCK5-dependent activation of Rac1 at the cell periphery.
DOCK5 is regulated by a Rho-modulated signaling cascade
In motile cells, Rac and Rho antagonize each other’s activities to mediate polarized assembly of the actin cytoskeleton.1 We previously demonstrated that endogenous GIT2 localizes to Rho-stabilized FAs.19 Taken together with our present data, it raises the possibility that DOCK5 is regulated in a Rho-dependent manner. To test this hypothesis, we treated cells with the myosin II inhibitor blebbistatin, which inhibits Rho-dependent cell retraction and causes rapid disassembly of FAs. Moreover, inhibition of actomyosin contractility activates Rac-dependent membrane extension but the mechanism by which tension inhibits Rac is not well defined.1
HeLa cells show enhanced membrane protrusion in the presence of blebbistatin (Figure 4a). In contrast, treatment of DOCK5-kd cells with blebbistatin results in cell collapse (Figure 4a). Blebbistatin causes the disassembly of mature FAs and the redistribution of paxillin to small adhesions located throughout the cell (Figure 4b). At later times, paxillin can be observed in nascent complexes concentrated at the protrusive edge of the cell (Figure 4b). There is marked enrichment of DOCK5 in lamellipodia of blebbistatin-treated cells, where it colocalizes with paxillin and Arp3 (Figures 4b and c). In stably adherent cells, DOCK5 depletion elicits a substantial increase in FA size, again implicating DOCK5 in FA turnover (Figure 4b). Similar to control cells, blebbistatin treatment of DOCK5-kd cells results in FA disassembly and paxillin redistribution. However, in contrast to control cells, DOCK5-kd cells fail to assemble membrane protrusions with nascent adhesions (Figure 4b). Moreover, DOCK5-depleted cells exhibit an inability to maintain p130Cas phosphorylation in the presence of blebbistatin, which supports a requirement for DOCK5 in activation of the Crk-p130Cas signaling pathway (Figure 4d). Blebbistatin impairs activation of FAK and phosphorylation of its Y118 target site in paxillin (Figure 4d), thus confirming previous reports that FAK activation is dispensable for cell spreading in this context.25, 26 These results indicate that Rho, via actin-myosin contractility, inhibits a Crk-p130Cas-DOCK5-Rac1 signaling pathway to suppress lamellipodial protrusion.

Activity of DOCK5 is repressed by myosin II contractility. (a) HeLa cells were transfected with control or DOCK5 shRNA expression vectors and cultured in puromycin to select for expression of the resistance marker present in the vector. Cells were treated with the myosin II inhibitor blebbistatin (50 μM) or vehicle for 12 h and examined by phase contrast microscopy. (b) Localization of DOCK5 (green) and paxillin (red) in control and DOCK5-kd cells treated with blebbistatin for the indicated times. Scale bar indicates 10 μm. (c) Control cells treated with blebbistatin for 0 or 4 h were labeled to detect DOCK5 (green) and Arp3 (red). Scale bar represents 10 μm. (d) Western blot analysis of control and DOCK5-knockdown cells treated with blebbistatin for 0, 1, 2 and 4 h to detect phosphorylated forms and total levels of p130Cas, paxillin and FAK.
Unbiased biochemical analysis has established that GIT2 is a highly tension-dependent FA constituent.27 Accordingly, endogenous GIT2 localizes to mature FAs, consistent with a requirement for RhoA in its targeting.19 Nevertheless, it has been reported that GIT2 is recruited in a Rac- and not Rho-dependent manner to nascent adhesions.28 To address this conundrum, we treated HeLa cells with blebbistatin for 20 min to initiate FA disassembly. Blebbistatin causes a rapid loss of GIT2 from FAs, preceding complete redistribution of paxillin (Figure 5a). Pharmacological inhibition of ROCKs with the small molecule Y27632 also elicits a rapid loss of GIT2 from FAs (Figure 5a). We additionally tested the effect of Rac interference on GIT2 localization. HeLa cells express Rac1 and Rac3, which we depleted separately or together (Figure 5b). Knockdown of Rac1 and/or Rac3 did not prevent GIT2 targeting to FAs (Figures 5c and c’). Taken together, our results support a model whereby recruitment of GIT2 to FAs is promoted by Rho-dependent cystokeletal tension and leads to suppression of DOCK5, thus suggesting a novel mechanism for Rho to antagonize the function of Rac.

Activity of myosin II is required for localization of GIT2 to FAs. (a) HeLa cells were treated for 20 min with vehicle, blebbistatin (50 μM) or Y27632 (10 μM). Cells were labeled to detect GIT2 (green) or paxillin (red). Scale bar indicates 10 μm. (b) Hela cells were transfected with control, Rac1, Rac3, or Rac1 and Rac3 siRNA. Cells were lysed 48 h post-transfection and whole-cell lysates were immunoblotted with antibodies specific for Rac1, Rac3 and ERK. (c) HeLa cells depleted of Rac1, Rac3, or Rac1 and Rac3 were labeled to detect paxillin (green) and GIT2 (red). Insets in panel (c’), highlight areas of colocalization between GIT2 and paxillin. Scale bar represents 10 μm.
GIT2 and DOCK5 control mammary epithelial cell motility and invasiveness
We previously showed that the loss of GIT2 greatly enhances the invasiveness of non-transformed mammary MCF10A cells.19 To assess the role of DOCK5 in cell migration, we localized DOCK5 in wounded MCF10A cell monolayers. Endogenous DOCK5 is rapidly recruited to the wound edge, where it colocalizes with paxillin, Arp3, and cortactin (Figure 6a). Stable DOCK5 depletion markedly reduces wound healing, indicating that DOCK5 is required for regulated motility (Supplementary Figure S3a). Next, we used a previously described method for inducible knockdown to target GIT2 and DOCK5 in MCF10A cells (Figure 6b).29 Knockdown of GIT2 increases migration through Matrigel-coated invasion chambers when epidermal growth factor (EGF) is provided as a chemoattractant (Figure 6c). Enhanced invasiveness observed in GIT2-kd cells is entirely dependent on DOCK5 (Figure 6c), indicating that GIT2 serves as an essential repressor of DOCK5 in this context.

DOCK5 is required for mammary epithelial cell invasion and wound repair. (a) MCF10A cells were cultured on fibronectin. Cell monolayers were labeled to detect DOCK5 (green) and Arp3, cortactin or paxillin (red) 6 h after wounding. Scale bar indicates 20 μm. (b) MCF10A cells selected to conditionally express control, GIT2, or GIT2 and DOCK5 shRNA were treated with doxcycline for 72 h. Lysates were immunoblotted to detect GIT2, DOCK5, DOCK1 and ERK. (c) To quantify changes in MCF10A invasion, control (black), GIT2-kd (red), or GIT2+DOCK5-kd (blue) cells were harvested and added to the top chamber of Matrigel-coated transwell filters and incubated for 12 h in the presence of 10 ng/ml of EGF as a chemoattractant. Cells that migrated to the bottom chamber were quantified. Values are means±s.d. (n=3). (d) MDA-MB-231 cells transiently transfected with siRNA targeting DOCK5 and DOCK1 were lysed and immunoblotted. (e) siRNA control (black), DOCK5-kd (red) and DOCK1-kd (blue) MDA-MB-231 cells were assayed for invasion through Matrigel-coated filters in the presence of 10% serum as a chemoattractant. Values are means ±s.d. (n=3). (f) Immunolocalization of DOCK5 (green) and Arp3 (red) in MDA-MB-231 control and DOCK5-kd cells. Scale bar represents 10 μM. (g) Whole-cell lysates of MDA-MB-231 cells were processed for western blotting to detect GIT2, DOCK1, DOCK5 and ERK as loading control, as well at phosphorylated forms and total levels of p190Cas, FAK and paxillin. (h) Quantification of phospho-p130Cas, -paxillin and -FAK levels from g. (i) MDA-MB-231 cells stably transduced with empty lentiviral vector (control), lentiviral expression constructs encoding wild-type DOCK5 or catalytically inactive DOCK5(ISP) were assayed for invasion through Matrigel-coated filters in the presence of 10% serum as a chemoattractant. All values in this figure are means±s.d. from a minimum of n=3 or more experiments. *P<0.05; **P<0.02; ***P<0.01 by unpaired two-tailed Student's t-test.
DOCK1 functions downstream of EGF receptors ErbB-2 (a.k.a HER2) to mediate invasion and metastasis of mammary carcinoma cells.30 We used small interfering RNA (siRNA)-mediated gene knockdown to target DOCK1 and DOCK5 in the highly invasive mammary carcinoma cell line MDA-MB-231 (Figure 6d). Consistent with observations in the mammary carcinoma cell line T47D,30 knockdown of DOCK1 impairs invasiveness of MDA-MB-231 cells in response to EGF (Supplementary Figure S3b). DOCK5 depletion also reduces invasiveness significantly, revealing a previously unanticipated role for DOCK5 in this process (Supplementary Figure S3b). Interestingly, although MDA-MB-231 cells appeared slightly more dependent on DOCK1 than DOCK5 for EGF-dependent invasion, the reverse is true when serum is used as a chemoattractant (Figure 6e and Supplementary Figure S3c). These results were validated with an additional three siRNAs targeting DOCK5 (Supplementary Figures S3d and e). Similar to MCF10A and HeLa cells, DOCK5 localizes to the protrusive edge of migrating MDA-MB-231 cells, whereas its loss impairs lamellipodial protrusion (Figure 6f). Moreover, depletion of DOCK5 from MDA-MB-231 cells markedly reduces phosphorylation of p130Cas, paxillin, and FAK (Figures 6g and h). Finally, we performed invasion assays with MDA-MB-231 cells overexpressing wild-type DOCK5 or catalytically inactive DOCK5(ISP). In contrast to wild-type DOCK5, overexpression of DOCK5(ISP) yielded a ~50% inhibition of invasion (Figure 6i). This result shows a role for enzymatic activity of DOCK5 to promote mammary carcinoma cell invasion.
DOCK5 promotes metastasis of mammary carcinoma cells
To test a requirement for DOCK5 in invasiveness in vivo, we performed tail vein injection experiments with MDA-MB-231 cells in nude mice. Cohorts of mice were injected with either control cells, a pool of DOCK5-kd cells, or clonal cell populations with DOCK5-kd (Figure 7a). Four weeks later (day 28), the mice injected with control cells were killed because of respiratory distress (Supplementary Table S1). In contrast, all mice injected with DOCK5-depleted cells were viable and without external signs of disease with exception of one mouse that was killed because of an unrelated liver abnormality. At this time (day 28), three mice injected with a pool of DOCK5-depleted cells, as well as one mouse from each of the DOCK5-kd clones, were killed for histological analysis.

DOCK5 is required for metastasis of MDA-MB-231 cells. (a) Western blot analysis was used to determine DOCK5 levels in MDA-MB-231 cell lines. MDA-MB-231 cells were retrovirally transduced with either control or DOCK5 shRNA expression vectors. A pooled population of DOCK5-kd cells and two independent clonal DOCK5-kd cell lines were generated. (b) Representative pictures of lungs from mice killed 28 days after intravenous injection of control or DOCK5-knockdown (clone A) cell lines. (c) Average lung weights of control (12 mice), pool (3 mice), clone A (1 mouse) and clone B (1 mouse) injected mice killed on day 28. Values are means±s.d. ***P<0.02 by unpaired two-tailed Student's t-test. (d) Hematoxylin and eosin staining of lungs harvested on day 28 from mice intravenously injected with the indicated cell lines. Arrows mark the perimeter of scattered extravascular tumors present in mice injected with pooled, clone A and clone B DOCK5-kd cells. Scale bar indicates 100 μM. (e) Kaplan–Meier survival plots representing results of intravenous tail vein injection of MDA-MB-231 vector control cells, as well as DOCK5 shRNA-expressing pool and clonal cell populations. Median overall survival: control (28 days; n=12 mice), pool (44 days; 7 mice), clone A (52 days; 7 mice) and clone B (58.7 days; 7 mice). (f) P-values calculated with a log-rank test and resulting from the pairwise comparison of survival times for the designated mouse cohorts.
Lungs harvested from mice injected with control MDA-MB-231 cells exhibited major increases in size and weight compared with lungs from mice injected with DOCK5-kd cells (Figures 7b and c). The lungs from mice injected with control cells were compacted with tumor cells (Figure 7d and Supplementary Figure S4a). In contrast, lungs from mice injected with a pool of DOCK5-kd cells contained numerous discrete tumors dispersed within normal lung tissue, whereas much smaller tumors were detected in lungs from mice injected with clonal populations of DOCK5-kd cells (Figure 7d and Supplementary Figure S4a). The remaining mice in the study were subsequently monitored and terminated upon of signs of disease (Supplementary Table S1). The results are shown in the Kaplan–Meier plot in Figure 7e. Mice injected with a pool of DOCK5-kd cells lived significantly longer than controls, but shorter than mice injected with clonal populations of DOCK5-kd cells (Figure 7f). Survival correlated with the efficiency of DOCK5-kd (Figure 7a). Collectively, these results establish that DOCK5 promotes metastasis of MDA-MB-231 cells.
To quantify the effect of DOCK5 depletion on tumor burden, we performed a shorter experiment where all mice were terminated on day 21, before any external signs of disease. Lungs for these mice were processed for stereological analysis (Supplementary Figure S4b). Select sections were moreover stained with antibody specific for human cytokeratin to confirm that tumor elements were indeed comprised of MDA-MB-231 cells (Figure 8a). Quantification revealed that DOCK5-kd elicits a significant decrease in tumor burden from 40% in mice injected with control MDA-MB-231 cells to 7% in mice injected with a pooled population of DOCK5-depleted cells and 2–4% in mice injected with clones of DOCK5-deficient cells (Figures 8a and b). The differences in tumor burden between mice injected with control and DOCK5-kd cells does not appear to result from effects on cell proliferation or cell adhesion (Figures 8c and d). In contrast, formation of membrane ruffles was attenuated in the pool of DOCK5-kd cells and virtually abolished in the clonal DOCK5-kd cell lines (Figure 8e). The latter finding is consistent with the conclusion that reduced tumor burden in mice injected with DOCK5-kd cells results from diminished motile and invasive capacities.

DOCK5-regulated metastasis of MDA-MB-231 is mediated through effects on motile/invasive capacities. (a) Representative images of tumor burden in lungs of nude mice 21 days after injection with control, DOCK5-kd pool, DOCK5-kd clone A and DOCK5-kd clone B cells. Scale bar represents 500 μm. The insets show adjacent sections of a small tumor stained with hematoxylin and eosin and human cytokeratin, respectively. Scale bar represents 50 μm. (b) Quantification of tumor burden in these mouse cohorts as described in Materials and methods (n=4 mice in each cohort). **P<0.02 by unpaired two-tailed Student's t-test. (c) Cell proliferation rates in control, DOCK5-kd pool, DOCK5-kd clone A, and DOCK5-kd clone B MDA-MB-231 cells (n=3). (d) Quantification of cell adhesion, as performed in Figure 1c using 40 μg/ml of collagen, in control, DOCK5-kd pool, DOCK5-kd clone A, and DOCK5-kd clone B MDA-MB-231 cells (n=3). (e) Morphology of control, DOCK5-kd pool, DOCK5-kd clone A and DOCK5-kd clone B MDA-MB-231 cells after overnight plating on collagen. The fraction of cells with membrane ruffles ⩾30 μm in length was quantified from 125 cells from each condition.
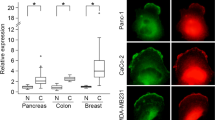
Finally, expression analysis of HER2 mouse breast tumors identified DOCK1, DOCK5, DOCK6 and DOCK7 among the most abundantly expressed Rho GEFs, indicating that multiple DOCK members may have a role in capacities associated with oncogenic cell transformation.30 We probed human breast cancer cell protein arrays and found that DOCK5 was widely expressed (Supplementary Figure S5). However, we found no consistent differences in expression between tumor sample and matched adjacent normal tissue. Thus, any changes in DOCK5 function in human cancer are more likely the result of modulated activity, as opposed to increased expression.
Discussion
DOCK family members are emerging as pivotal regulators of cellular processes involving adhesion and the actin cytoskeleton. Here we identify DOCK5 as an essential regulator of epithelial motility and invasiveness. Moreover, in an animal model of metastasis, inhibition of DOCK5 reduces tumor burden and significantly increases survival time. DOCK5 and DOCK1 are co-expressed in numerous cell types. Our results indicate that DOCK1 and DOCK5 function in a non-redundant manner to modulate serum- and EGF-dependent invasiveness.
DOCK1 and DOCK5 contribute to the generation of distinct actin structures. Here we demonstrate that DOCK5 mediates peripheral actin polymerization and membrane protrusion via activation of a Crk-p130Cas-DOCK5 signaling cascade. This cascade is suppressed by GIT2, as depletion of GIT2 results in deregulated lamellipodial protrusions that depends on the activity of DOCK5 but not DOCK1. The non-redundant functions of DOCK1 and DOCK5 in epithelial invasion may result from an ability to regulate distinct actin structures. Although both DOCK1 and DOCK5 are present in peripheral ruffles, DOCK1 possesses a unique C-terminal motif that binds phosphatidic acid and mediates its recruitment to dorsal ruffles.31 As a result, loss of DOCK1, but not DOCK5, impairs dorsal ruffle formation.31 Dorsal ruffles seem important for migration through 3D environments.32 Therefore, assembly of distinct DOCK1- and DOCK5-dependent actin structures may be required for optimal cell invasiveness. DOCK1 and DOCK5 also form heterodimers, which appears essential for their in vivo biological function, offering an additional mechanism for non-redundancy in their function.33, 34, 35
Here, we establish that GIT2 represses the activity of DOCK5 but not DOCK1. GIT2 localizes to FAs via its association with the scaffolding protein paxillin.36 Our past and present observations indicate that active Rho, via a ROCK- and myosin II-dependent pathway, triggers the recruitment of GIT2 to nascent adhesions. This conclusion is supported by a comprehensive proteomic analysis of fibroblasts, which ranked GIT2 as among the top myosin II-dependent FA components.27 Our data suggest that GIT2 is rapidly targeted to nascent adhesions in response to Rho-initiated cytoskeletal tension, where it promotes transition from nascent DOCK5- and Rac-dependent protrusive adhesions to mature Rho-dependent contractile adhesions. In this context, we identify GIT2 as a novel mediator of Rho-Rac antagonism in control of motile and invasive capacities.
Our results suggest that oncogenic signaling, which inhibits GIT2 or activates DOCK5, will promote metastasis. Others have designed DOCK inhibitors, although none effectively distinguish between individual DOCK-A group members.13, 37 As DOCK5 is largely dispensable for gross embryonic development, our results indicate that DOCK5 is specifically required for wound repair and cell invasion. As such, DOCK5 may be part of a tumor-specific invasion pathway (Figure 9). It will be important for future studies to assess whether DOCK5 has a role in other integrin-dependent processes required for metastasis such as anchorage-independent growth, matrix degradation and cell extravasation.

Cartoon illustrating a novel signaling pathway for the coordinated regulation of Rho and Rac signaling in epithelial invasion. In this pathway, Rho-dependent activation of ROCK targets GIT2 to FAs, where GIT2 suppresses the activity of a Crk-p130Cas-DOCK5 signaling pathway required for activation of Rac and mammary epithelial cell invasion. The Crk-p130Cas-DOCK5 signaling pathway has non-redundant and essential role in epithelial invasion and metastasis, underscoring the need for tight regulation of this pathway by GIT2 in non-transformed cells.
Materials and methods
DOCK5 and DOCK1 mammalian expression vectors
Full-length human DOCK5 complementary DNA (cDNA; identical to NM_024940) was assembled in pCMV-Myc (Clontech/Takara, Mountain View, CA, USA) from partial cDNAs provided by NITE (www.nbrc.nite.go.jp) and RZPD (www.imagenes-bio.re). This DOCK5 cDNA was transferred to pLenti-puro (Addgene, Cambridge, MA, USA) for lentiviral transduction. DOCK1 expression vector was kindly provided by Kodi Ravichandran. An expression vector encoding the DOCK5 CBD (amino acids 1712–1871) was generated in pCMV-Myc.
Generation of DOCK5-specific antibody
DOCK5 cDNA encoding amino acids 1712–1871 and DOCK1 cDNA encoding amino acids 1691–1866 were inserted into pGEX-4T-1 (GE Healthcare Life Sciences, Pittsburgh, PA, USA). Recombinant GST-DOCK5-CT was used to generate antiserum reactive with DOCK5. Antibodies cross-reactive with DOCK1 or GST were removed by incubation with GST-DOCK1-CT coupled to glutathione sepharose followed by centrifugation. The supernatant containing DOCK5-specific antibody was used for experimentation.
Vector-based shRNA
To generate shRNAs for DOCK5, complimentary 80–87 oligonucleotides were annealed and cloned into pReSI-Puro, pReSI-Hygro or pReSI-GFP retroviral vectors as previously described.19 The target sequence of validated GIT2 shRNA was previously described.19 The target sequence of the primary DOCK5 shRNA (sense–antisense) was the following: 5′-GATCCCGAGCAGATCTTAAACTACTACGCCCGGGCCGCCCTGTAGTAGTTTAAGATCTGCTCTTTTTTGGAACTCGAGA-3′.
Additional DOCK5 shRNA sequences used in Supplementary Figure S2 were as follows: shRNA-1: 5′-GATCCCGAAGTACCTTCCTAGCATAATGCCCGGGCCGCCCTATTATGCTAGGAAGGTACTTCTTTTTTGGAACTCGAGA-3′; shRNA-2: 5′-GATCCCGGACCTGATTGGAAAGAATGTGCCCGGGCCGCCCTACATTCTTTCCAATCAGGTCCTTTTTTGGAACTCGAGA-3′; shRNA-3: 5′-GATCCCGAGAAGCTGTATCAAGAAATCGCCCGGGCCGCCCTGATTTCTTGATACAGCTTCTCTTTTTTGGAACTCGAGA-3′; shRNA-4: 5′-GATCCCGAATGTCTATGCCAAAGATTGGCCCGGGCCGCCCTCAATCTTTGGCATAGACATTCTTTTTTGGAACTCGAGA-3′.
Cell spreading and adhesion assay
For stable knockdown of DOCK5 in HeLa cells (Supplementary Figure S2), retrovirus was used to transduce cells with pReSI-Hygro vector encoding four distinct shRNA-targeting sequences and selected in the presence of hygromycin. For transient shRNA-mediated knockdown, HeLa cells were transfected with shRNA expression constructs as previously detailed.19 Next, cells were plated onto collagen-coated coverslips. To determine the requirement for DOCK1 and DOCK5 in GIT2-regulated cell spreading (Supplementary Figure S1), HeLa cells were transfected with 50 nM of siRNA duplexes targeting DOCK1 (Ambion ID# 145822; GE Healthcare Life Sciences) or DOCK5 (Ambion ID# 130801) using LipofectAmine 2000.
To quantify effects on cell adhesion, non-enzymatically harvested control or DOCK5 shRNA-expressing cell populations were resuspended in Dulbecco’s modified Eagle’s medium (supplemented with 10 mM HEPES pH 7.4 and 0.5% bovine serum albumin) and plated onto collagen-coated 96-well plates (50 000 cells per well). After 90-min incubation at 37 °C, wells were rinsed in phosphate-buffered saline+ and adherent cells quantified using the CyQuant cell proliferation assay kit (Molecular Probes; Thermo Fisher Scientific, Waltham, MA, USA).
Live cell imaging of HeLa cells expressing paxillin-GFP
A paxillin-GFP expression construct was generated in pLenti-Puro. HeLa cells expressing paxillin-GFP were selected with puromycin. Paxillin-GFP expressing cells were grown on 15 mm2 glass coverslips and GIT2 and/or DOCK5 expression was depleted as detailed above. The coverslips were mounted in a custom-imaging chamber maintained at 37 °C and pH 7.4 throughout the observation period. Analysis of FA assembly, disassembly and turnover rates was performed essentially as described by Webb et al.24
Immunofluorescence microscopy, immunoprecipitation and western blotting
Cells were fixed in methanol/acetone (1:1) with exception of samples processed for Rac-GTP labeling, which were fixed in 2% formalin and permeabilized with 0.1% Triton X-100. Processing of samples was carried out as described previously.19 Antibodies used for immunofluorescence were: rabbit anti-GIT2;19 rabbit anti-DOCK5 (described above); rabbit anti-DOCK1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA, clone H-70); mouse anti-paxillin (BD Biosciences, San Jose, CA, USA, clone 349), mouse anti-ARP3 (Sigma, St Louis, MO, USA, clone FMS338); mouse anti-Rac-GTP (NewEast Biosciences, Malvern, PA, USA, clone 26903); and mouse anti-Myc epitope (Cell Signaling Technologies, Danvers, MA, USA, clone 9B11). Confocal images were acquired using a spinning disk confocal microscope.
Antibodies used for immunoprecipitation and western blotting were: mouse anti-Crk (BD Biosciences; clone 22); rabbit anti-Crk (Santa Cruz Biotechnology; C-20), mouse anti-DOCK1 (Santa Cruz Biotechnology; clone H-4), mouse anti-p130Cas (BD Biosciences; clone 21), mouse anti-Rac1 (BD Biosciences; clone 102), rabbit anti-Rac3 (Abcam; clone EPR6680), rabbit anti-FAK (Santa Cruz Biotechnology; clone A-17), rabbit anti-phospho-p130Cas(Tyr410), rabbit anti-phospho-paxillin(Tyr118) and rabbit anti-phospho-FAK(Tyr397). All phospho-specific antibodies were from Cell Signaling Technologies.
MCF10A cell invasion assay
To generate cell lines with inducible knockdown of GIT2, MCF10A-TetR cells were retrovirally transduced with pReSI-Puro or pReSI-Puro-GIT2 shRNA and selected in puromycin. A clone exhibiting inducible GIT2 knockdown was subsequently transduced with pReSI-Hygro-DOCK5 and selected in hygromycin. To induce knockdown of GIT2 and/or DOCK5, cells were treated with 4 μg/ml of doxycyline for 72 h. Next, 1 × 105 cells were plated into the top chamber of a Matrigel invasion chamber (BD Biosciences). Cells were allowed to migrate overnight using 10 ng/ml of EGF in Dulbecco’s modified Eagle’s medium/F12 as chemoattractant. Migrated cells were quantified using CyQuant (see above).
MDA-MB-231 cell invasion assay
MDA-MB-231 were transfected with SMARTpool siRNA (siGeNOME, Dharmacon, Lafayette, CO, USA), targeting DOCK1 or DOCK5 (final concentration 50 nM), using Lipofectamine RNAiMax (Invitrogen, Carlsbad, CA, USA). Additional siRNA duplexes targeting DOCK5 with sequences as follows were custom ordered from Dharmacon: siRNA-1: 5′-GAGGAUGACUGCAAAGAAAUU-3′; siRNA-2: 5′-GGAGAGAACUCAAGCUGUAUU-3′; siRNA-3: 5′-UUUCAAACAUGGACAGUUAUU-3′.
Seventy-two hours post-transfection, cells were trypsinized and 1 × 105 cells were plated into the top well of a Matrigel-coated invasion chambers and incubated for 12 h. Assays were conducted using Dulbecco’s modified Eagle’s medium supplemented with either 10% serum or 10 ng/ml of EGF as a chemoattractant. Migrated cells were fixed in 4% formaldehyde, stained with crystal violet and counted.
Tail vein injection of MDA-MB-231 cells
Animal modeling of the metastatic potential of control and DOCK5-knockdown MDA-MB-231 cell lines was carried out by Pipeline Biotech A/S (Trige, Denmark). Pipeline Biotech was blinded to the identity of the individual cell lines. All animal experiments were performed according to standards set by the Animal Experiments Inspectorate in Denmark. Briefly, CB-17 female SCID mice were injected with 100 μl phosphate-buffered saline containing 1 × 106 cells in the tail vein. The injected mice were maintained under identical conditions and were monitored regularly. End point assays were conducted when significant morbidity required that the mouse be euthanized (see Supplementary Table S1 for details). Following killing, the thorax and abdomen of mice were opened and internal organs inspected for gross pathological analysis and lungs were evacuated and fixed by immersion in 4% formalin for 24 h followed by paraffin embedding and processed for histology as described below.
Histology
Paraffin-embedded lungs were cut into 1.5 mm slabs from apex to base. The slabs were mounted side-by-side and cut into 5 μm section that were deparaffinized and rehydrated before hematoxylin and eosin staining or histochemistry. Detection of human cytokeratin was carried out using AE1/AE3 clone M3515 (Dako, Glostrup, Denmark) by standard procedures. A NanoZoomer-2.0HT (Hamamatsu, Hamamatsu City, Shizuoka Pref., Japan) was used to scan entire sections. Lung metastasis volume was quantified blinded using a computer-assisted stereological technique using the Visiopharm stereology software package and the newCAST stereology module (Visiopharm, Hørsholm, Denmark).38
References
Guilluy C, Garcia-Mata R, Burridge K . Rho protein crosstalk: another social network? Trends Cell Biol 2011; 21: 718–726.
Laurin M, Cote JF . Insights into the biological functions of Dock family guanine nucleotide exchange factors. Genes Dev 2014; 28: 533–547.
Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, Tosello-Trampont AC et al. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol 2002; 4: 574–582.
Cote JF, Vuori K . In vitro guanine nucleotide exchange activity of DHR-2/DOCKER/CZH2 domains. Methods Enzymol 2006; 406: 41–57.
Hasegawa H, Kiyokawa E, Tanaka S, Nagashima K, Gotoh N, Shibuya M et al. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol 1996; 16: 1770–1776.
Kiyokawa E, Hashimoto Y, Kobayashi S, Sugimura H, Kurata T, Matsuda M . Activation of Rac1 by a Crk SH3-binding protein, DOCK180. Genes Dev 1998; 12: 3331–3336.
Kiyokawa E, Hashimoto Y, Kurata T, Sugimura H, Matsuda M . Evidence that DOCK180 up-regulates signals from the CrkII-p130(Cas) complex. J Biol Chem 1998; 273: 24479–24484.
Feng H, Hu B, Liu KW, Li Y, Lu X, Cheng T et al. Activation of Rac1 by Src-dependent phosphorylation of Dock180(Y1811) mediates PDGFRalpha-stimulated glioma tumorigenesis in mice and humans. J Clin Invest 2011; 121: 4670–4684.
Feng H, Hu B, Jarzynka MJ, Li Y, Keezer S, Johns TG et al. Phosphorylation of dedicator of cytokinesis 1 (Dock180) at tyrosine residue Y722 by Src family kinases mediates EGFRvIII-driven glioblastoma tumorigenesis. Proc Natl Acad Sci USA 2012; 109: 3018–3023.
Nishikimi A, Kukimoto-Niino M, Yokoyama S, Fukui Y . Immune regulatory functions of DOCK family proteins in health and disease. Exp Cell Res 2013; 319: 2343–2349.
Laurin M, Fradet N, Blangy A, Hall A, Vuori K, Cote JF . The atypical Rac activator Dock180 (Dock1) regulates myoblast fusion in vivo. Proc Natl Acad Sci USA 2008; 105: 15446–15451.
Sanders MA, Ampasala D, Basson MD . DOCK5 and DOCK1 regulate Caco-2 intestinal epithelial cell spreading and migration on collagen IV. J Biol Chem 2009; 284: 27–35.
Vives V, Laurin M, Cres G, Larrousse P, Morichaud Z, Noel D et al. The Rac1 exchange factor Dock5 is essential for bone resorption by osteoclasts. J Bone Miner Res 2011; 26: 1099–1110.
Ogawa K, Tanaka Y, Uruno T, Duan X, Harada Y, Sanematsu F et al. DOCK5 functions as a key signaling adaptor that links FcepsilonRI signals to microtubule dynamics during mast cell degranulation. J Exp Med 2014; 211: 1407–1419.
Watanabe M, Terasawa M, Miyano K, Yanagihara T, Uruno T, Sanematsu F et al. DOCK2 and DOCK5 act additively in neutrophils to regulate chemotaxis, superoxide production, and extracellular trap formation. J Immunol 2014; 193: 5660–5667.
Frank SR, Hansen SH . The PIX-GIT complex: a G protein signaling cassette in control of cell shape. Semin Cell Dev Biol 2008; 19: 234–244.
Brown MC, Cary LA, Jamieson JS, Cooper JA, Turner CE . Src and FAK kinases cooperate to phosphorylate paxillin kinase linker, stimulate its focal adhesion localization, and regulate cell spreading and protrusiveness. Mol Biol Cell 2005; 16: 4316–4328.
Nishiya N, Kiosses WB, Han J, Ginsberg MH . An alpha4 integrin-paxillin-Arf-GAP complex restricts Rac activation to the leading edge of migrating cells. Nat Cell Biol 2005; 7: 343–352.
Frank SR, Adelstein MR, Hansen SH . GIT2 represses Crk- and Rac1-regulated cell spreading and Cdc42-mediated focal adhesion turnover. EMBO J 2006; 25: 1848–1859.
Yu JA, Deakin NO, Turner CE . Paxillin-kinase-linker tyrosine phosphorylation regulates directional cell migration. Mol Biol Cell 2009; 20: 4706–4719.
Phee H, Dzhagalov I, Mollenauer M, Wang Y, Irvine DJ, Robey E et al. Regulation of thymocyte positive selection and motility by GIT2. Nat Immunol 2010; 11: 503–511.
Schaller MD, Parsons JT . pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol 1995; 15: 2635–2645.
Richardson A, Malik RK, Hildebrand JD, Parsons JT . Inhibition of cell spreading by expression of the C-terminal domain of focal adhesion kinase (FAK) is rescued by coexpression of Src or catalytically inactive FAK: a role for paxillin tyrosine phosphorylation. Mol Cell Biol 1997; 17: 6906–6914.
Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT et al. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol 2004; 6: 154–161.
Friedland JC, Lee MH, Boettiger D . Mechanically activated integrin switch controls alpha5beta1 function. Science 2009; 323: 642–644.
Pasapera AM, Schneider IC, Rericha E, Schlaepfer DD, Waterman CM . Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J Cell Biol 2010; 188: 877–890.
Kuo JC, Han X, Hsiao CT, Yates JR 3rd, Waterman CM . Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for beta-Pix in negative regulation of focal adhesion maturation. Nat Cell Biol 2011; 13: 383–393.
Brown MC, West KA, Turner CE . Paxillin-dependent paxillin kinase linker and p21-activated kinase localization to focal adhesions involves a multistep activation pathway. Mol Biol Cell 2002; 13: 1550–1565.
Frank SR, Bell JH, Frodin M, Hansen SH . A betaPIX-PAK2 complex confers protection against Scrib-dependent and cadherin-mediated apoptosis. Curr Biol 2012; 22: 1747–1754.
Laurin M, Huber J, Pelletier A, Houalla T, Park M, Fukui Y et al. Rac-specific guanine nucleotide exchange factor DOCK1 is a critical regulator of HER2-mediated breast cancer metastasis. Proc Natl Acad Sci USA 2013; 110: 7434–7439.
Sanematsu F, Nishikimi A, Watanabe M, Hongu T, Tanaka Y, Kanaho Y et al. Phosphatidic acid-dependent recruitment and function of the Rac activator DOCK1 during dorsal ruffle formation. J Biol Chem 2013; 288: 8092–8100.
Suetsugu S, Yamazaki D, Kurisu S, Takenawa T . Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Dev Cell 2003; 5: 595–609.
Patel M, Pelletier A, Cote JF . Opening up on ELMO regulation: new insights into the control of Rac signaling by the DOCK180/ELMO complex. Small GTPases 2011; 2: 268–275.
Premkumar L, Bobkov AA, Patel M, Jaroszewski L, Bankston LA, Stec B et al. Structural basis of membrane targeting by the Dock180 family of Rho family guanine exchange factors (Rho-GEFs). J Biol Chem 2010; 285: 13211–13222.
Terasawa M, Uruno T, Mori S, Kukimoto-Niino M, Nishikimi A, Sanematsu F et al. Dimerization of DOCK2 is essential for DOCK2-mediated Rac activation and lymphocyte migration. PLoS One 2012; 7: e46277.
West KA, Zhang H, Brown MC, Nikolopoulos SN, Riedy MC, Horwitz AF et al. The LD4 motif of paxillin regulates cell spreading and motility through an interaction with paxillin kinase linker (PKL). J Cell Biol 2001; 154: 161–176.
Nishikimi A, Uruno T, Duan X, Cao Q, Okamura Y, Saitoh T et al. Blockade of inflammatory responses by a small-molecule inhibitor of the Rac activator DOCK2. Chem Biol 2012; 19: 488–497.
Nielsen BS, Lund LR, Christensen IJ, Johnsen M, Usher PA, Wulf-Andersen L et al. A precise and efficient stereological method for determining murine lung metastasis volumes. Am J Pathol 2001; 158: 1997–2003.
Acknowledgements
This work was funded by NIH R01 CA142647 to SHH, as well as grants from the Danish Cancer Society R40-A1936-11-S2 to SHH and R2-A132-02-S2 to MF. SHH is a recipient of an Endowed Investigatorship from the Roy and Lynne Frank Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Frank, S., Köllmann, C., van Lidth de Jeude, J. et al. The focal adhesion-associated proteins DOCK5 and GIT2 comprise a rheostat in control of epithelial invasion. Oncogene 36, 1816–1828 (2017). https://doi.org/10.1038/onc.2016.345
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.345
