
Abstract
Wnt signaling is one of the key cascades regulating development and stemness, and has also been tightly associated with cancer. The role of Wnt signaling in carcinogenesis has most prominently been described for colorectal cancer, but aberrant Wnt signaling is observed in many more cancer entities. Here, we review current insights into novel components of Wnt pathways and describe their impact on cancer development. Furthermore, we highlight expanding functions of Wnt signaling for both solid and liquid tumors. We also describe current findings how Wnt signaling affects maintenance of cancer stem cells, metastasis and immune control. Finally, we provide an overview of current strategies to antagonize Wnt signaling in cancer and challenges that are associated with such approaches.
Similar content being viewed by others
Introduction
More than 40 years ago, the wingless gene was discovered in a mutagenesis screen for visual phenotypes, affecting various developmental patterning processes in Drosophila melanogaster.1 Subsequently, further genetic screens identified many components of the Wnt family of signaling proteins as key mediators of patterning decisions during embryonic development.2 A connection of the Wnt pathway to cancer was implicated by the discovery that activation of int1 (Wnt1), which, either by proviral insertion into the Wnt1 locus or transgenic overexpression in mice, resulted in mammary hyperplasia and tumors.3, 4, 5 It was also shown that the Drosophila gene wingless and the murine proto-oncogene Wnt1 are orthologous.6 Furthermore, injection of murine Wnt1 mRNA into embryos of Xenopus could induce axis duplication.7 These observations suggested that genes involved in Wnt signaling are highly conserved through evolution. In 1991, mutations of the adenomatous polyposis coli (APC) gene were discovered as the underlying cause of the hereditary colon cancer syndrome termed familial adenomatous polyposis.8, 9 The APC gene was found to interact with β-catenin10, 11 and loss of function of APC resulted in overactive T-cell factor (TCF)4/β-catenin signaling.12 These findings established a direct link between Wnt signaling and human colorectal cancer.
In the past years, many genetic and biochemical studies have sought to identify novel Wnt pathway components and their functions. Identified components and processes include the Wnt secretory machinery, Wnt co-receptors, components of the β-catenin destruction complex and nuclear co-factors. With the advance in sequencing technology and the comprehensive structural characterization of cancer genomes,13, 14 it became evident that mutations in the Wnt pathway occur frequently in human cancers.15, 16, 17, 18 Despite the fact that major pathway components have been characterized, the function of Wnt signaling within the context of cancer biology is intriguingly complex and remains only partially understood.
In this review we focus on novel insights into Wnt signaling in cancer, gained from studies published within the past 5 years. We describe recently discovered Wnt pathway components and novel functions of the Wnt pathway for cancer stemness, metastasis and immune surveillance. Furthermore, we review the current progress on targeting the Wnt pathway.
Canonical and non-canonical Wnt signaling
The Wnt pathway is commonly divided into β-catenin dependent (canonical) and independent (non-canonical) signaling. Both the canonical and non-canonical pathway are outlined in detail in Figure 1.

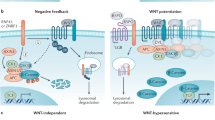
Overview of canonical and non-canonical Wnt signaling. (a) In canonical Wnt signaling, absence of Wnt ligands (Wnt signaling inactive state, left) leads to phosphorylation of β-catenin by the destruction complex, which contains the scaffold protein Axin, APC and the kinases GSK3β and casein kinase (CK1α). In this state, β-catenin is phosphorylated by GSK3β, ubiquitinated by β-TrCP200 and targeted for proteasomal degradation. In the absence of nuclear β-catenin, a repressive complex containing TCF/LEF and transducing-like enhancer protein (TLE/Groucho) recruits HDACs to repress target genes. The canonical pathway is activated upon binding of secreted Wnt ligands (for example, Wnt3a and Wnt1) to Fzd receptors and LRP co-receptors (Wnt signaling active, right). LRP receptors are then phosphorylated by CK1α and GSK3β, which recruits Dishevelled (Dvl) proteins to the plasma membrane where they polymerize and are activated.201 The Dvl polymers inactivate the destruction complex, for example, by sequestration in multivesicular bodies. This results in stabilization and accumulation of β-catenin which then translocates into the nucleus. There, β-catenin forms an active complex with LEF (lymphoid enhancer factor) and TCF (T-cell factor) proteins by displacing TLE/Groucho complexes and recruitment of histone modifying co-activators such as CBP/p300, BRG1, BCL9 and Pygo (reviewed in Lien and Fuchs48). This transcriptional switch leads to a change of multiple cellular processes.49, 202 (b) Non-canonical Wnt signaling is defined by β-catenin-independent mechanisms of signal transduction. During Wnt/PCP signaling, Wnt ligands bind to the ROR-Frizzled receptor complex to recruit and activate Dvl.203 Dvl binds to the small GTPase Rho by de-inhibition of the cytoplasmic protein DAAM1 (Dvl associated activator of morphogenesis 1).204 The small GTPase Rac1 and Rho together trigger ROCK (Rho kinase) and JNK. This leads to rearrangements of the cytoskeleton and/or transcriptional responses via for example, ATF2 (activating transcription factor 2).205 Next to Dvl, Vangl, a key member of Wnt/PCP signaling is activated by phosphorylation in a Wnt5a-dependent manner.206 Wnt/Ca2+ signaling is initiated by G-protein triggered phospholipase C activity207 leading to intracellular calcium fluxes and downstream calcium dependent cytoskeletal and/or transcriptional responses.208
In recent years, novel insights into multiple levels of canonical Wnt signaling were obtained, refining the model of how the pathway is regulated. Production of Wnt ligands in secreting cells is an important and surprisingly complex step in Wnt signaling. The ER resident acyl-transferase Porcupine is required for the attachment of palmitoleic acid to Wnt ligands.19 Thereafter, lipid-modified Wnt ligands bind to the transmembrane protein Evi/Wls and are shuttled to the plasma membrane via the Golgi apparatus.20, 21, 22 The transport of Wnts from the ER to the Golgi is assisted by p24 proteins.23, 24 After secretion of Wnt ligands, Evi/Wls is undergoing clathrin based endocytosis and is recycled to the Golgi apparatus by the retromer complex.25, 26 Finally, Evi/Wls is transported back to the ER to re-engage in Wnt secretion.22
Wnt proteins can either be tethered to the plasma membrane or exit the cell via multiple routes, including direct release from the plasma membrane by solubilization,27 the formation of exosomes28 or on lipid protein particles.29 The variety of mechanisms by which Wnt ligands are released may correspond to their diverse roles during development and organismal maintenance. For example, although membrane-bound Wnt3 ligands retain a short range, but high level of Wnt signaling in intestinal organoids,30, 31 exosome-bound Wnt2b in the epididymal lumen ensures long-range effects needed for sperm maturation.32 It is unclear which release mechanism of Wnt ligands is most prevalent in cancer. However, the presence of exosome-based Wnt signaling in the breast cancer microenviroment33 as well as short range Wnt signaling in RNF43/ZNRF3 double mutant intestinal organoids31 suggest that tissue-specific mechanisms exist.
Beyond secreted Wnts, members of the R-spondin ligand family were discovered as positive effectors of Wnt signaling.34, 35, 36 R-spondins bind to leucine-rich repeat containing G-protein-coupled receptors (Lgr) 4-6.37 In the absence of R-spondin binding, the two homologues E3 ubiquitin ligases ZNRF3/RNF43 target the Frizzled (Fzd) receptor for lysosomal degradation.37, 38 Binding of R-spondins to Lgr4-6 inhibits the activity of ZNRF3/RNF43 and leads to the accumulation of Fzd receptors on the cell surface.36, 39 Being transcriptional targets of Wnt signaling, ZNRF3 and RNF43 function as negative feedback regulators in Lgr5-positive cells.37, 38 The interaction of ZNRF3 and RNF43 with the Fzd receptor was found to be dependent on Dishevelled (Dsh).40 The important role of the R-spondin/Lgr5/RNF43 module in cancer has been demonstrated in several tumor subtypes of colorectal cancer (CRC), pancreatic ductal adenocarcinoma (PDAC) and endometrial cancer which all harbor inactivating mutations of RNF4341, 42 (Figure 2).

Mutation rates of Wnt pathway components in selected cancer entities. Percentage of cancer patients with mutations of selected canonical Wnt pathway related genes. Information was retrieved from the ICGC data portal (accessed 5/2016). The frequency of exonic mutations was determined based on cases with single nucleotide variant data in the MELA-AU, SKCA-BR, SKCM-US, PACA-US, PACA-CA, COAD-US, COCA-CN, READ-US, BRCA-UK and BRCA-US studies.
At the level of the destruction complex, YAP/TAZ, two transcriptional regulators of the Hippo pathway were recently identified as novel Wnt regulators.43 Complementing previous evidence for a negative effect of YAP/TAZ on Wnt signaling,44, 45, 46 Azzolin et al. proposed a double role for these factors: in the absence of Wnt ligands, YAP/TAZ are part of the destruction complex and recruit β-TrCP, thereby acting as negative regulators of Wnt signaling. However, in an activated pathway state, YAP/TAZ and bound β-TrCP are displaced from Axin1 by LRP during sequestration of the destruction complex. Free YAP/TAZ can subsequently act as positive transcriptional effectors of Wnt signaling.43, 47 The dual role of YAP/TAZ in the Wnt pathway underlines the close connection of Wnt and Hippo signaling.
The transcriptional response to Wnt signaling activation is orchestrated by a complex network of β-catenin binding factors in the nucleus (reviewed in Lien and Fuchs48 and MacDonald et al.49), of which novel cancer related components were recently identified. LATS2 kinase, a repressor of YAP/TAZ activity, was found to downregulate Wnt signaling by competing with BCL9,50 a co-activator of the TCF/LEF transcriptional complex which is highly expressed in human tumors.51 Furthermore, the DNA-repair gene PAF (PCNA-associated factor) was found to be specifically overexpressed in colon cancer and intestinal stem cells.52 Overexpression of PAF induced intestinal neoplasia in a mouse model. Mechanistically, PAF enhances Wnt signaling by recruiting the histone methyltransferase EZH2 to the TCF transcriptional complex (Figure 1).
Beyond the transcriptional response of canonical Wnt signaling, Wnt-dependent stabilization of proteins (Wnt/STOP) has been introduced as a novel β-catenin-independent mechanism. Canonical Wnt signaling, which peaks during G2/M phase due to priming of LRP6 by Cyclin Y/CDK14,53 was shown to promote β-catenin-independent stabilization of proteins.54 Activation of this β-catenin-independent Wnt cascade leads to inhibition of GSK3β and the subsequent blockade of poly-phosphorylation and poly-ubiquitination of target proteins, which were predicted to comprise 20% of the proteome.55, 56 These targets include prominent oncogenes such as c-Myc,54 which is degraded by the E3 ubiquitin ligase FBXW7 (Figure 1). Wnt/STOP has also been proposed to affect chromosomal stability, cell division and endolysosomal biogenesis.57, 58, 59 It remains to be further elucidated whether tissue- and time-dependent preferences for Wnt/STOP or transcriptional Wnt responses exist and can be exploited for cancer therapy.
Although the canonical pathway is comparatively well understood, the non-canonical Wnt pathways are more diverse and less well characterized (reviewed in Anastas and Moon60). The Wnt ligands Wnt5a and Wnt11 can bind to a panel of different receptors to preferentially activate non-canonical Wnt signaling, including receptors of the Fzd family and other receptors such as ROR2, ROR1 or RYK (reviewed in Wang61 and Katoh62). Binding of these non-canonical Wnt ligands can activate multiple intracellular pathways, of which the planar cell polarity (PCP) and calcium signaling pathways are most extensively studied (Figure 1b). The PCP pathway is implicated in cell orientation during development, but also has a role in metastasis formation, whereas the calcium signaling pathway controls intracellular influx of calcium which can activate various downstream kinases including PKC and CaM kinase II (reviewed in De63 and Yang and Mlodzik;64 see Figure 2c). The most prominent ligand receptor pair in non-canonical Wnt signaling is the Wnt5a-ROR2 module. Binding of Wnt5a leads to the formation of the ROR2 receptor complex (Figure 1b). In addition to Wnt/PCP and Wnt/Casignaling, further cascades can be triggered by ROR2. By binding of Filamin A to its prolin-rich domain (PRD), ROR2 can directly cause the formation of filopodia and subsequent cell migration.65 Of note, although related, ROR2 and ROR1 show a relatively low homology in their PRD, indicating differing functions of these receptors in cell migration.66 Although non-canonical Wnt signaling plays an undisputed—but understudied—role in most cancer types, our review will primarily focus on canonical Wnt signaling.
Emerging roles of Wnt in cancer
Gastrointestinal cancers
The impact of Wnt signaling on carcinogenesis of colorectal cancer is well-studied (reviewed in Polakis67). Loss of APC is the main driver of Wnt signaling in colorectal cancer and its important role was further highlighted by several recent studies. By genome editing of APC using the CRISPR/Cas9 technology, the carcinogenesis of CRC could be modeled ex vivo in human intestinal organoids.68, 69 Furthermore, studies of human CRC samples and tumors from mouse models revealed that different mutations of APC result in distinct levels of canonical Wnt pathway activity and are associated with characteristic tumor locations within the large intestine.70, 71 Using a mouse model with reversible knockdown of APC via shRNA, it was demonstrated that adenomas could regress to normal tissue if APC function is restored, underlining the importance of continuous Wnt signaling for tumor maintenance.72 Moreover, it was also shown that in spite of truncated APC, Wnt pathway activity can still be modulated by interference with Wnt secretion.73 Interestingly, a molecular classification of colorectal cancers based on expression and mutation data demonstrates that despite comparable frequencies of APC mutations between subtypes, Wnt target genes can be differentially expressed. Molecular subtypes with high expression levels of Wnt target genes were associated with better overall survival rate after relapse compared to subtypes with low expression levels of the respective genes.74
Besides APC, mutations in the R-spondin/Lgr5/RNF43 module were implicated as drivers of Wnt-dependent tumor growth. Deleterious RNF43 mutations have been described in ~19% of CRC cases and are mutually exclusive to APC mutations.41 In addition, R-spondin3 mutations and fusion proteins expressed at a high level have been described in 10% of CRC cases.75 RNF43 mutant CRC are strongly dependent on Wnt secretion, rendering them highly susceptible to Wnt secretion targeted therapy.76
Chromosomal instability (CIN) is frequently observed in CRC and associated with poor prognosis. Loss of function of Wnt pathway components, particularly APC, have been linked to CIN by multiple mechanisms.77, 78, 79 Although direct interactions of APC with the cytoskeleton and the transcriptional Wnt response via β-catenin are known routes to CIN, Wnt/STOP was introduced as new mechanism: Ertych et al. recently showed that loss of basal Wnt/STOP leads to increased microtubule assembly rates and subsequent CIN in HCT116 cells,57, 80 while reconstitution of normal assembly rates could reverse the CIN phenotype.57
Development of PDAC is mainly driven by oncogenic Ras signaling and the impact of Wnt signaling has not been fully understood. Unlike CRC, mutations of key Wnt pathway components are rare in PDAC (Figure 2), but nuclear localization of β-catenin is regularly found.81 Results from mouse models indicate that Wnt signaling can initiate tumor formation when activated at distinct tumor stages (reviewed in White et al.82). However, stabilized β-catenin can also inhibit reprogramming of acini into preneoplastic lesions in the presence of mutated KRAS,83 indicating a more complex role of Wnt signaling during tumor development. Recent studies show that PDAC relies on Wnt ligand stimulation, as PDAC cell lines carrying a mutation in RNF43 are particularly sensitive towards treatment with the Porcupine inhibitor LGK974.42 Furthermore, induction of the Wnt antagonist DKK1 as well as treatment with the anti-Fzd antibody OMP18R5 delays PDAC formation.84, 83 Autocrine Wnt7b was found to increase Wnt signaling in pancreatic cell lines and to promote anchorage-independent cell growth.85
Wnt signaling is also activated in cholangiocarcinoma, but genomic alternations of major Wnt pathway components are rare, with the exception of RNF43.76 Pharmacological inhibition of Wnt signaling, both at the level of β-catenin and Wnt secretion, reduces proliferation of cholangiocarcinoma cells in a mouse model.86 Interestingly, Wnt secreting inflammatory macrophages in the microenvironment are required to maintain high Wnt signaling in the tumor.86, 87 Furthermore, secreted inhibitors of Wnt signaling such as SFRP2 are frequently silenced by hypermethylation in cholangiocarcinoma.88, 89 Taken together, novel findings reinforce the view that dependence on Wnt signaling of gastrointestinal cancers can be mediated by different, tissue-specific routes.
Leukemia
In recent years, knowledge about the role of Wnt signaling in hematopoiesis and leukemia has increased.90 Normal hematopoietic stem cells (HSC) depend on a finely controlled level of Wnt signaling for long-term maintenance, whereas Wnt activity is substantially increased in most leukemias.91 Acute myelogenous leukemia (AML) is the most common type of acute leukemia in adults and characterized by frequent chromosomal translocations. In MLL-fusion positive AML mouse models, leukemia initiating cells (LIC) can arise from HSC as well as myeloid progenitor cells after progression through a pre-LIC state.92, 93 β-catenin appears to be essential for the progression of pre-LICs to the LIC state and for LIC self-renewal.94, 95 Frequent translocation products found in AML, such as AML1-ETO, MLL-AF9 and PML-RARα positively affect canonical Wnt signaling in patient samples and derived cell lines.93, 96, 97
The most common leukemia in childhood is acute lymphoblastic leukemia (ALL). The majority of LICs in T-cell ALL (T-ALL) harbor activating mutations of the Notch signal pathway (reviewed in Ferrando98). However, canonical Wnt signaling in HSC and thymocytes synergizes with PTEN loss and c-Myc amplification to generate a β-catenin dependent and Notch independent T-ALL subset in mouse studies and human T-ALL patients.99, 100 Besides the driving role of canonical Wnt signaling during tumorigenesis of specific T-ALL subsets, active β-catenin appears to play an important role during LIC self-renewal in a broader context. Giambra and colleagues showed that LICs in bulk NOTCH1 driven T-ALL mouse models are marked by high Wnt activity.101 Inactivation of β-catenin in these tumors eliminated LICs without affecting the short-term viability of the bulk tumor.
Chronic lymphocytic leukemia (CLL) is the most prevalent form of adult leukemia in western countries. Canonical Wnt signaling is active in CLL cells and its inhibition increases apoptosis in vitro.102 Next to frequent silencing of Wnt inhibiting factors such as DKK1/2,103 somatic mutations in Wnt pathway related genes (for example, FZD5, BCL9) were found in 14% of studied cases.104 Knockdown of mutated Wnt pathway members reduced cell viability in CLL cells carrying the targeted Wnt pathway alteration, while those without Wnt pathway mutations remained unaffected.104 These findings demonstrate that a subset of CLL is dependent on active Wnt signaling for survival. In summary, canonical Wnt signaling is able to drive tumor development in major leukemia subtypes and is required for maintenance of leukemia initiating cells.
Melanoma
About 25% of melanomas arise from benign nevi (commonly known as moles) which typically consist of quiescent BRAFV600E or NRASQ61K mutant melanocytes that have undergone a process of oncogene induced senescence105, 106 (reviewed in Jones and Cichowski107). Canonical Wnt signaling has been found to delay the onset of oncogene induced senescence in both BRAFV600E or NRASQ61K expressing primary melanocytes and thereby increase the chance of tumor development.108, 109, 110
Although canonical Wnt signaling appears to contribute to melanoma development, its role in disease progression is controversial.111, 112 Several clinical and translational studies have shown an increased overall survival rate of patients carrying melanoma with elevated nuclear β-catenin levels.113, 114, 115 However, in a mouse model with a mutant PTEN and BRAF genoype,116 activated β-catenin lead to accelerated melanoma development and promotion of metastasis. The impact of Wnt signaling on response towards BRAF inhibitors in melanoma is also unclear. Active Wnt signaling was shown to cooperate with BRAFV600E inhibition to induce apoptosis in melanoma cell lines.117 However, clinical studies demonstrated that patients with lower nuclear β-catenin levels had a better prognosis under BRAF inhibitor treatment.118
The role of non-canonical Wnt signaling in melanoma progression has been investigated extensively. Activity of Wnt5a/ROR2 leads to increased cell motility as well as a pseudo-senescent phenotype, which is induced by external stresses.119, 120, 121, 122 In this reversible senescence-like state, melanoma cells are increasingly chemo- and radioresistant and show a senescence associated secretory phenotype120 marked by the secretion of pro-angiogenic and pro-inflammatory cytokines, for example, IL-6.123 Interestingly, stimulation with IL-6 has been show to induce the expression of Wnt5a in melanoma cells itself,124 thereby forming a positive feedback loop. Thus, Wnt5a mediated non-canonical Wnt signaling leads to self-promoting invasive and resistant phenotypes in melanomas. It is important to note that Wnt5a ligand binding to alternative co-receptors, such as LRP6, can result in a canonical Wnt signaling response in a subset of melanoma cells.125, 126 Consequently, the distinction between canonical and non-canonical signaling effects is not dichotomous and might explain controversial findings.
The progression of melanoma has been described by a phenotype switching model with melanoma cells changing between proliferative and invasive states.127, 128 These prevailing phenotypes are determined in part by the balance between canonical and non-canonical Wnt signaling. Recently, ROR1 and ROR2 co-receptor abundance was linked to the two different melanoma phenotypes. Wnt5a treatment of proliferative ROR1 positive melanoma cells led to ROR1 degradation, a high ROR2 expression and increased invasiveness of melanoma cells in vivo.122 Hypoxic culture conditions were identified as a trigger for changing the cellular Wnt signaling response eventually leading to phenotypic switching.122
Breast cancer
Wnt signaling is activated in over 50% of breast cancer patients and linked to reduced overall survival.129 The role of canonical Wnt signaling in triple negative breast cancer development and progression has been studied intensively.130, 131, 132 However, high levels of nuclear β-catenin were also found in other breast cancer subtypes.133 Only a small fraction of tumors harbor somatic mutations of key pathway regulators such as β-catenin130 (Figure 2), but canonical Wnt ligands and receptors are often overexpressed in breast cancers134, 135, 136 whereas secreted antagonists are silenced.137 In mice, MMTV-Wnt induced tumors are dependent on continuous Wnt signaling,138 which leads to progenitor-like signatures in tumor cells.139 Overexpression of R-spondin2 alone was shown to initiate mammary tumors in mouse models.140
Recently the model of canonical Wnt signaling in mammary tumors has been refined by analysis of clonal heterogeneity within the tumor. Cleary et al.141 identified two tumor cell-lineages of luminal and basal descent in a MMTV-Wnt model. The luminal subclone was characterized by secretion of canonical Wnt ligands, which was necessary for tumor growth of the basal-like recipient cells.141, 142 These findings indicate that mammary tumors can consist of polyclonal cell populations that cooperate to generate distinct, subpopulation specific Wnt activity levels.
Wnt signaling and cancer stem cells
The self-renewal potential of cancer cells is described by the cancer stemness model and has been used to explain many malignant phenotypes.143 Although the concept of cancer stemness is still controversially discussed, the vital role of the Wnt pathway for the function of normal and cancer stem cells is commonly accepted (reviewed in Reya and Clevers144). One of the hallmarks of stem cells is their ability to maintain long telomeres by function of the TERT gene. TERT expression was found to be directly enhanced by binding of β-catenin to its promoter region and thereby links telomerase activity to Wnt signaling.145 The R-spondin receptor Lgr5 is a marker of intestinal stem cells and can fuel tumor growth when APC is deleted in these cells.146 Lineage tracing experiments demonstrate that single Lgr5-positive cells can give rise to additional Lgr5-positive cells and other cell types in colon adenoma, indicating that Lgr5 is a potential cancer stem cell marker.147 Myant et al.148 show that RAC1 is required for expansion of the Lgr5 population after APC loss. RAC1 activation drives ROS production and thereby activates NFκB signaling, which then enhances Wnt signaling. These findings are supported by another study showing that co-activation of the NFκB and Wnt pathway can induce dedifferentiation of normal intestinal cells into stem cells and thereby initiate tumor development in a mouse model.149 The important role of the tumor environment for maintenance of cancer stemness is highlighted by several studies. Hepatocyte growth factor secreted by myofibroblasts in the tumor microenvironment can increase Wnt activity and induce stemness features in colorectal cancer cells.150 Malanchi et al.151 demonstrated that breast tumor cells induce the stromal expression of the extracellular matrix protein periostin in order to form a metastatic niche. Periostin interacts with Wnt1 and Wnt3a, thereby inducing Wnt signaling and sustaining a CSC phenotype.151 In another study, MMP3 secreted by mammary epithelial cells was found to stimulate canonical Wnt signaling in mammary stem cells by sequestration of Wnt5b, thereby counteracting the inhibitory effect of the non-canonical Wnt pathway.152 CD44v6 was described as another CSC marker in colorectal cancer and its expression is promoted by canonical Wnt signaling and cytokines secreted from tumor-associated cells, resulting in increased metastatic capacity.153
Recently, several studies uncovered potential links between non-coding RNAs and Wnt signaling in cancer stem cells. microRNA-146a was shown to stabilize β-catenin by repression of Numb, leading to maintenance of Wnt signaling and symmetric division of colorectal cancer stem cells.154 In mammary stem cells, miR-142 recruits the APC mRNA for degradation and thereby increases canonical Wnt signaling.155 The long non-coding RNA lncTCF7, which is highly expressed in hepatocellular carcinoma and liver stem cells, was found to activate expression of TCF7 by recruitment of the SWI/SNF complex to the promoter of TCF7. This activation of canonical Wnt signaling is associated with an increased self-renewal capacity of liver CSC.156 In non-small cellular lung cancer, overexpressed miR-582-3p maintains stemness features by targeting negative the regulators of Wnt signaling Axin2, DKK3 and SRP1 for degradation, thereby increasing β-catenin mediated Wnt activity.157
Expression signatures of intestinal stemness in tumor samples were correlated with disease prognosis in colorectal cancer.158, 159 Although both the intestinal stem cell and cancer stem cell signature could readily identify patients with poor prognosis, Melo et al. show that the expression of accepted Wnt target/stemness genes such as Axin2 and Lgr5 were lower in patients with poor prognosis, due to promoter methylation of those genes. No correlation was found between expression level of Wnt target/stemness genes and the number of CD133-positive stem cells or nuclear β-catenin levels. Thus, stem cell signatures likely reflect the general differentiation state of the tumor tissue rather than the number of Wnt driven cancer stem cells.
Wnt signaling in metastasis
Metastasis is a hallmark of late stage cancer and a major challenge to therapy. A main adaptive change of tumors during therapy is an epithelial to mesenchymal transition (EMT, reviewed in Scheel and Weinberg160). EMT describes the process by which polarized epithelial cells transform into migratory mesenchymal cells with invasive properties.161, 162 Transcriptional factors that are responsible for EMT include, among others, SNAI2. Cytoplasmic SNAI2 concentration is kept in check by GSK3β phosphorylation and subsequent ubiquitinylation by β-TrCP. Activation of canonical Wnt signaling stabilizes SNAI2 by inhibiting GSK3β kinase activity and initiates EMT transcriptional programs in breast cancer cells.163 Another candidate gene that regulates EMT is ASPP2, a protein that binds to a β-catenin/E-cadherin complex and inhibits N-terminal phosphorylation of β-catenin, leading to its stabilization. Reduced expression of ASPP2 leads to EMT and is associated with poor survival in hepatocellular and breast cancer.164 In colon cancer cells with hyperactivated canonical Wnt signaling, pharmacological inhibition of the PI3K-Akt signaling leads to a nuclear accumulation of β-catenin and FOXO3a which results in increased cell scattering and metastasis.165 These results show that both active and non-active canonical Wnt signaling can enhance EMT, depending on the tissue type. An involvement of the non-canonical Wnt pathway in EMT was implied by high co-expression of Fzd2, its ligands Wnt5a/b and EMT markers. It was shown that Fzd2 expression enhances EMT and cell migration via Fyn and Stat3. Targeting of Fzd2 by a specific antibody reduces tumor growth and metastasis in a xenograft mouse model of colon cancer.166
Recently, exosomes were found to be potential mechanism by which tumors prime their metastatic niche.167 Exosomes are small vesicles secreted by cells and function in intercellular communication. It was shown that they can be vehicles for the transport of active Wnt ligands28 or incorporate β-catenin.168 Exosomes secreted from fibroblast in the tumor microenvironment can enhance motility and protrusive activity of breast cancer cells via the Wnt/PCP pathway.169 Co-injection of breast cancer cells with fibroblast in orthotopic mouse models was shown to promote metastasis. Mechanistically, this results from a tethering of Wnt11 to fibroblast-derived exosomes.169 Another route by which distant metastasis is proposed to spread is via circulating tumor cells (CTCs).170 Single-cell RNA sequencing of CTCs was performed for prostate and pancreatic cancer and both studies identified a role for Wnt signaling. In CTCs of pancreatic cancer, Wnt2 expression increased anchorage-independent sphere formation and their metastatic propensity.171 In another study, the non-canonical Wnt signaling pathway was found to be upregulated in CTCs of prostate cancer cells that are resistant to androgen receptor inhibition.172 Taken together, there is increasing evidence that both canonical and non-canonical Wnt signaling can support tumor metastasis in a highly tissue-specific manner.
Wnt signaling and anti-tumor immunity
Overcoming immune evasion by cancer cells is a promising therapeutic approach and immune checkpoint blockade was shown to be highly effective in the treatment of melanoma173 and other tumor types.174, 175 Wnt signaling controls proliferation, maturation and differentiation of T-cells and dendritic cells,176 but a role of tumor intrinsic Wnt signaling in immune evasion has only recently emerged. Spranger et al. show that a Wnt signature in cutaneous melanoma samples correlates with T-cell exclusion. Using a mouse model of melanoma with Braf/PTEN mutant background and constitutively high β-catenin activity, the authors show that T-cell priming against tumor antigens is failing due to defective recruitment of CD103+ dendritic cells.177 β-catenin signaling downregulates the chemokine CCL4, which negatively affects the recruitment of dendritic cells to the tumor. Restoration of intra-tumoral dendritic cells by injection could furthermore increase the efficiency of anti-CTLA4 and anti-PD-L1 therapy. Moreover, upregulation of IL-12 production in melanoma by increased β-catenin signaling can also lead to impaired dendritic cell maturation and induction of regulatory dendritic cells.178 A different mechanism of immune evasion was recently demonstrated in lung and breast cancer. It was shown that latency competent cancers self-impose a slow-cycling state by autocrine inhibition of Wnt signaling by DKK1, thereby evading innate immune response.179 As therapy against immune checkpoint inhibitors are showing promising results also in other tumor entities such as colorectal cancer,180 further studies investigating the interplay between tumor intrinsic Wnt signaling and immune response are expected.
Pharmacological inhibitors and clinical trials
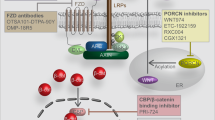
The concept that inhibition of Wnt signaling is an universal strategy for the treatment of cancer has been controversially discussed in the past.60 As clinical data suggests, elevated Wnt signaling is only linked to a worse outcome for a subset of human cancers.181 Therefore, current strategies aim at targeting Wnt signaling in distinct tumor subclasses or with specific mutational backgrounds. An overview of small molecular inhibitors and antibodies that are currently in clinical testing is presented in Figure 3 and Table 1.

Currently tested pharmaceuticals targeting the Wnt pathway in cancer. Schematic representation of the canonical Wnt signaling pathway with pharmaceutical modulators. All depicted drugs are currently undergoing testing in Phase 1/2 against various types of cancer (see also Table 1).
In recent years, the knowledge on the role of Wnt secretion for carcinogenesis has advanced considerably and unveiled novel therapeutic targets. Small molecule inhibitors, including IWPs182 and LGK974, were shown to selectively inhibit the acyl-transferase Porcupine and thus Wnt secretion, leading to a size reduction of MMTV-Wnt1-driven tumors and head and neck cancer xenotransplants. Furthermore, Jiang et al. showed that mutations of RNF43 results in dependency of pancreatic adenocarcinomas on Wnt ligands.42 Mutations of RNF43 and R-spondin fusion proteins, which occur mutually exclusive with APC mutations in colorectal cancer,41 were subsequently presented as a predictors for an effective therapy targeting Wnt secretion.75, 183 Based on these results, a phase I/II trial of LGK974 was initiated for patients with metastatic colorectal cancer harboring mutations of RNF43 or R-spondin fusions. In addition, a novel orally available Porcupine inhibitor ETC-159184 that was found to prevent growth of R-spondin-fusion positive CRC, is undergoing clinical testing since July 2015.185 Although anti-Wnt secretion therapeutics appear promising, the number and impact of potential side effects are currently unclear.
Besides the intracellular perturbation of Wnt secretion, an array of drugs targeting extracellular Wnt ligands and their receptors are under development. OMP-54F28 is a fusion protein consisting of a Fzd8 and a human IgG1 Fc domain. This decoy receptor for Wnt ligands reduces the size of tumor xenografts and overall tumor initiating cell number in mouse models of hepatocellular carcinoma and ovarian cancer.186 Currently, the substance is undergoing three phase 1b trials in liver, ovarian and pancreatic cancer in combination with established therapeutics. Furthermore, a phase I clinical trial testing the safety of OMP131R10, a RSPO3-binding antibody, has been initiated in June 2015 for patients with solid tumors and metastasized colorectal cancer.
OMP18R5 is a monoclonal antibody targeting five of ten human Fzd receptors. It was shown to inhibit the growth of human tumor xenografts and to synergize with standard-of-care therapeutic agents,187 but first data from clinical studies suggested adverse effects on skeletal constitution.188 This finding was considered as an on-target effect of Wnt inhibition since Wnt signaling has a key role in bone development and disease.189 Currently, OMP18R5 is studied in phase I clinical trials alone or in combination with taxanes in breast and pancreatic cancer, as well as non-small cell lung cancer. Besides targeting key functional members of the Wnt signaling cascade, other pharmaceuticals exploit the specific expression patterns of Fzd receptors by cancer cells. For example, Fzd10 is almost exclusively overexpressed in a variety of defined cancer types.190 OTSA101 is a radioactive anti-Fzd10 antibody which is currently in a phase I trial for the treatment of advanced synovial sarcoma.191
In addition to approaches targeting Wnt secretion and ligands, an inhibitor of the downstream Wnt pathway is currently undergoing clinical trials. PRI-724 and the closely related compound ICG-001 specifically target the complex formation of β-catenin and CBP while enhancing the formation of β-catenin/p300 complexes in ALL cells.192 Treatment with PRI-724 therefore inhibits the self-renewing downstream effects of β-catenin-CBP activity and leads to reduction of tumor burden.193 Following promising results from phase I trials,194 a new phase II trial of PRI-724 in combination with bevacizumab therapy in metastatic colorectal carcinoma patients is planned.195 Tankyrase inhibitors such as XAV939, which stabilize Axin by blocking its PARsylation, have shown promising results as Wnt inhibitors.196 Subsequently, additional compounds targeting this enzyme have been developed.197 However, no tankyrase inhibitor is currently undergoing clinical testing, which may be linked to their toxicity in preclinical models.198
A better understanding of non-canonical Wnt signaling in tumor metastasis and growth has led to novel therapeutic approaches. Two Wnt5a analog small peptides, Foxy-5 and Box-5 have been developed to either activate or inhibit Wnt5a-dependent signaling, thereby reducing metastasis in selected tumors. First results from a phase I clinical trial of Foxy-5 suggest a good tolerability.199
The introduction of both Wnt5a agonists and antagonists, as well as the identification of anti-secretion therapy responsive tumor subsets further guide the clinical practice into the direction of personalized treatments. However, as data from selected clinical trials suggests, the possibility of serious on-target side effects across stem cell niches in the organism needs to be considered in future drug safety evaluations.
Outlook
In recent years, a multitude of studies have contributed to a deeper understanding of canonical and non-canonical Wnt signaling on a mechanistic level. The effect of aberrant canonical Wnt signaling is not only restricted to cancer cells, but dynamically interacts with the microenvironment and immune system. It also became clear that the function of non-canonical Wnt signaling is similar in development and cancer. While non-canonical Wnt signaling regulates convergent extension and tissue mobility during development, it can also mediate motility of cancer cells during metastasis. Moreover, there is a better understanding of how canonical and non-canonical Wnt signaling interact. The balance between both pathways is maintained by mechanisms that are distinct for different tissue types and their corresponding tumors. This knowledge is currently translated into a refined approach of targeting the Wnt pathway in cancer, taking into account both the functional and mutational status of canonical and non-canonical Wnt pathways in different cancer types.
References
Sharma R . Wingless, a new mutant in D. melanogaster. Drosoph Inf Serv 1973; 50: 134.
Nüsslein-Volhard C, Wieschaus E . Mutations affecting segment number and polarity in Drosophila. Nature 1980; 287: 795–801.
Tsukamoto AS, Grosschedl R, Guzman RC, Parslow T, Varmus HE . Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell 1988; 55: 619–625.
Nusse R, van Ooyen A, Cox D, Fung YK, Varmus H . Mode of proviral activation of a putative mammary oncogene (int-1) on mouse chromosome 15. Nature 307: 131–136.
Nusse R, Varmus HE . Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982; 31: 99–109.
Rijsewijk F, Schuermann M, Wagenaar E, Parren P, Weigel D, Nusse R . The Drosophila homolog of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell 1987; 50: 649–657.
McMahon AP, Moon RT . Ectopic expression of the proto-oncogene int-1 in Xenopus embryos leads to duplication of the embryonic axis. Cell 1989; 58: 1075–1084.
Kinzler K, Nilbert M, Su L, Vogelstein B, Bryan T, Levy D et al. Identification of FAP locus genes from chromosome 5q21. Science 1991; 253: 661–665.
Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991; 253: 665–669.
Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain S, Masiarz F et al. Association of the APC gene product with beta-catenin. Science 1993; 262: 1731–1734.
Su L, Vogelstein B, Kinzler K . Association of the APC tumor suppressor protein with catenins. Science 1993; 262: 1734–1737.
Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 1997; 275: 1784–1787.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW . Cancer genome landscapes. Science 2013; 339: 1546–1558.
Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010; 463: 191–196.
Clements WM, Wang J, Sarnaik A, Kim OJ, MacDonald J, Fenoglio-Preiser C et al. {beta}-Catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res 2002; 62: 3503–3506.
Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet 2000; 24: 245–250.
Dahmen RP, Koch A, Denkhaus D, Tonn JC, Sorensen N, Berthold F et al. Deletions of AXIN1, a component of the WNT/wingless pathway, in poradic Medulloblastomas. Cancer Res 2001; 61: 7039–7043.
Segditsas S, Tomlinson I . Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006; 25: 7531–7537.
Kadowaki T, Wilder E, Klingensmith J, Zachary K, Perrimon N . The segment polarity gene porcupine encodes a putative multitransmembrane protein involved in Wingless processing. Genes Dev 1996; 10: 3116–3128.
Bartscherer K, Pelte N, Ingelfinger D, Boutros M . Secretion of Wnt ligands requires Evi, a conserved transmembrane protein. Cell 2006; 125: 523–533.
Bänziger C, Soldini D, Schütt C, Zipperlen P, Hausmann G, Basler K . Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell 2006; 125: 509–522.
Yu J, Chia J, Canning CA, Jones CM, Bard FA, Virshup DM . WLS Retrograde transport to the endoplasmic reticulum during Wnt secretion. Dev Cell 2014; 29: 277–291.
Buechling T, Chaudhary V, Spirohn K, Weiss M, Boutros M . p24 proteins are required for secretion of Wnt ligands. EMBO Rep 2011; 12: 1265–1272.
Port F, Hausmann G, Basler K . A genome-wide RNA interference screen uncovers two p24 proteins as regulators of Wingless secretion. EMBO Rep 2011; 12: 1144–1152.
Coudreuse DYM, Roël G, Betist MC, Destrée O, Korswagen HC . Wnt gradient formation requires retromer function in Wnt-producing cells. Science 2006; 312: 921–924.
Gasnereau I, Herr P, Chia PZC, Basler K, Gleeson PA . Identification of an endocytosis motif in an intracellular loop of Wntless protein, essential for its recycling and the control of Wnt protein signaling. J Biol Chem 2011; 286: 43324–43333.
Mulligan KA, Fuerer C, Ching W, Fish M, Willert K, Nusse R . Inaugural Article: Secreted Wingless-interacting molecule (Swim) promotes long-range signaling by maintaining Wingless solubility. Proc Natl Acad Sci USA 2012; 109: 370–377.
Gross JC, Chaudhary V, Bartscherer K, Boutros M . Active Wnt proteins are secreted on exosomes. Nat Cell Biol 2012; 14: 1036–1045.
Neumann S, Coudreuse DYM, van der Westhuyzen DR, Eckhardt ERM, Korswagen HC, Schmitz G et al. Mammalian Wnt3a is released on lipoprotein particles. Traffic 2009; 10: 334–343.
Boutros M, Niehrs C . Sticking around: short-range activity of Wnt ligands. Dev Cell 2016; 36: 485–486.
Farin HF, Jordens I, Mosa MH, Basak O, Korving J, Tauriello DVF et al. Visualization of a short-range Wnt gradient in the intestinal stem-cell niche. Nature 2016; 530: 340–343.
Koch S, Acebron SP, Herbst J, Hatiboglu G, Niehrs C . Post-transcriptional Wnt signaling governs epididymal sperm maturation. Cell 2015; 163: 1225–1236.
Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012; 151: 1542–1556.
Kazanskaya O, Glinka A, del Barco Barrantes I, Stannek P, Niehrs C, Wu W . R-Spondin2 is a secreted activator of Wnt/b-catenin signaling and is required for Xenopus myogenesis. Dev Cell 2004; 7: 525–534.
Glinka A, Dolde C, Kirsch N, Huang Y-L, Kazanskaya O, Ingelfinger D et al. LGR4 and LGR5 are R-spondin receptors mediating Wnt/β-catenin and Wnt/PCP signalling. EMBO Rep 2011; 12: 1055–1061.
Hao H-X, Xie Y, Zhang Y, Charlat O, Oster E, Avello M et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012; 485: 195–200.
de Lau W, Barker N, Low TY, Koo B-K, Li VSW, Teunissen H et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011; 476: 293–297.
De Lau W, Peng WC, Gros P, Clevers H . The R-spondin / Lgr5 / Rnf43 module: regulator of Wnt signal strength the R-spondin / Lgr5 / Rnf43 module: regulator of Wnt signal strength. Genes Dev 2014; 28: 305–316.
Koo B-K, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012; 488: 665–669.
Jiang X, Charlat O, Zamponi R, Yang Y, Cong F . Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol Cell 2015; 58: 522–533.
Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet 2014; 46: 1264–1266.
Jiang X, Hao H-X, Growney JD, Woolfenden S, Bottiglio C, Ng N et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci USA 2013; 110: 12649–12654.
Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 2014; 158: 157–170.
Barry ER, Morikawa T, Butler BL, Shrestha K, de la Rosa R, Yan KS et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 2012; 493: 106–110.
Varelas X, Miller BW, Sopko R, Song S, Gregorieff A, Fellouse FA et al. The Hippo pathway regulates Wnt/β-catenin signaling. Dev Cell 2010; 18: 579–591.
Heallen T, Zhang M, Wang J, Bonilla-claudio M, Klysik E, Randy L et al. NIH public access. Science 2012; 332: 458–461.
Rosenbluh J, Nijhawan D, Cox AG, Li X, James T, Schafer EJ et al. NIH public access. Cell 2013; 151: 1457–1473.
Lien WH, Fuchs E . Wnt some lose some: transcriptional governance of stem cells by Wnt/β-catenin signaling. Genes Dev 2014; 28: 1517–1532.
MacDonald BT, Tamai K, He X . Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17: 9–26.
Li J, Chen X, Ding X, Cheng Y, Zhao B, Lai ZC et al. LATS2 suppresses oncogenic Wnt signaling by disrupting β-Catenin/BCL9 interaction. Cell Rep 2013; 5: 1650–1663.
Takada K, Zhu D, Bird GH, Sukhdeo K, Zhao J-J, Mani M et al. Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling. Sci Transl Med 2012; 4: 148ra117.
Jung H-Y, Jun S, Lee M, Kim H-C, Wang X, Ji H et al. PAF and EZH2 induce Wnt/β-catenin signaling hyperactivation. Mol Cell 2013; 52: 193–205.
Davidson G, Shen J, Huang Y-L, Su Y, Karaulanov E, Bartscherer K et al. Cell cycle control of wnt receptor activation. Dev Cell 2009; 17: 788–799.
Acebron SP, Karaulanov E, Berger BS, Huang Y-L, Niehrs C . Mitotic Wnt signaling promotes protein stabilization and regulates cell size. Mol Cell 2014; 54: 663–674.
Taelman VF, Dobrowolski R, Plouhinec J-L, Fuentealba LC, Vorwald PP, Gumper I et al. Wnt Signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 2010; 143: 1136–1148.
Xu C, Kim NG, Gumbiner BM . Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 2009; 8: 4032–4039.
Stolz A, Neufeld K, Ertych N, Bastians H . Wnt-mediated protein stabilization ensures proper mitotic microtubule assembly and chromosome segregation. EMBO Rep 2015; 16: 490–499.
Huang Y-L, Anvarian Z, Döderlein G, Acebron SP, Niehrs C . Maternal Wnt/STOP signaling promotes cell division during early Xenopus embryogenesis. Proc Natl Acad Sci USA 2015; 112: 5732–5737.
Ploper D, Taelman VF, Robert L, Perez BS, Titz B, Chen H-W et al. MITF drives endolysosomal biogenesis and potentiates Wnt signaling in melanoma cells. Proc Natl Acad Sci USA 2015; 112: E420–E429.
Anastas JN, Moon RT . WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 2013; 13: 11–26.
Wang Y . Wnt/Planar cell polarity signaling: a new paradigm for cancer therapy. Mol Cancer Ther 2009; 8: 2103–2109.
Katoh M . WNT/PCP signaling pathway and human cancer (Review). Oncol Rep 2005; 14: 1583–1588.
De A . Wnt/Ca2+ signaling pathway: a brief overview. Acta Biochim Biophys Sin (Shanghai) 2011; 43: 745–756.
Yang Y, Mlodzik M . Wnt-frizzled/planar cell polarity signaling: cellular orientation by facing the wind (Wnt). Annu Rev Cell Dev Biol 2015; 31: 623–646.
Nomachi A, Nishita M, Inaba D, Enomoto M, Hamasaki M, Minami Y . Receptor tyrosine kinase Ror2 mediates Wnt5a-induced polarized cell migration by activating c-Jun N-terminal kinase via actin-binding protein filamin A. J Biol Chem 2008; 283: 27973–27981.
Endo M, Nishita M, Fujii M, Minami Y . Insight into the role of Wnt5a-induced signaling in normal and cancer cells. Int Rev Cell Mol Biol 2015; 314: 117–148.
Polakis P . Wnt signaling in cancer. Cold Spring Harb Perspect Biol 2012; 4: 9.
Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y et al. Modeling colorectal cancer using CRISPR-Cas9–mediated engineering of human intestinal organoids. Nat Med 2015; 21: 256–262.
Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015; 521: 43–47.
Christie M, Jorissen RN, Mouradov D, Sakthianandeswaren A, Li S, Day F et al. Different APC genotypes in proximal and distal sporadic colorectal cancers suggest distinct WNT/β-catenin signalling thresholds for tumourigenesis. Oncogene 2013; 32: 4675–4682.
Buchert M, Athineos D, Abud HE, Burke ZD, Faux MC, Samuel MS et al. Genetic dissection of differential signaling threshold requirements for the Wnt/beta-catenin pathway in vivo. PLoS Genet 2010; 6: e1000816.
Dow LE, O’Rourke KP, Simon J, Tschaharganeh DF, van Es JH, Clevers H et al. Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell 2015; 161: 1539–1552.
Voloshanenko O, Erdmann G, Dubash TD, Augustin I, Metzig M, Moffa G et al. Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat Commun 2013; 4: 2610.
Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015; 21: 1350–1356.
Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB et al. Recurrent R-spondin fusions in colon cancer. Nature 2012; 488: 660–664.
van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A et al. Prospective derivation of a living organoid Biobank of colorectal cancer patients. Cell 2015; 161: 933–945.
Rusan NM, Peifer M . Original CIN: reviewing roles for APC in chromosome instability. J Cell Biol 2008; 181: 719–726.
Caldwell CM, Green RA, Kaplan KB . APC mutations lead to cytokinetic failures in vitro and tetraploid genotypes in Min mice. J Cell Biol 2007; 178: 1109–1120.
Aoki K, Aoki M, Sugai M, Harada N, Miyoshi H, Tsukamoto T et al. Chromosomal instability by beta-catenin/TCF transcription in APC or beta-catenin mutant cells. Oncogene 2007; 26: 3511–3520.
Ertych N, Stolz A, Stenzinger A, Weichert W, Kaulfuß S, Burfeind P et al. Increased microtubule assembly rates influence chromosomal instability in colorectal cancer cells. Nat Cell Biol 2014; 16: 779–791.
Zeng G, Germinaro M, Micsenyi A, Monga NK, Bell A, Sood A et al. Aberrant Wnt/beta-catenin signaling in pancreatic adenocarcinoma. Neoplasia 2006; 8: 279–289.
White BD, Chien AJ, Dawson DW . Dysregulation of Wnt/β-catenin signaling in gastrointestinal cancers. Gastroenterology 2012; 142: 219–232.
Morris JP, Cano DA, Sekine S, Wang SC, Hebrok M . Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest 2010; 120: 508–520.
Zhang Y, Morris JP, Yan W, Schofield HK, Gurney A, Simeone DM et al. Canonical Wnt signaling is required for pancreatic carcinogenesis. Cancer Res 2013; 73: 4909–4922.
Dougherty DM, Marsh-richard DM, Hatzis ES, Nouvion SO, Mathias CW . WNT7B mediates autocrine Wnt/β-catenin signaling and anchorage-independent growth in pancreatic adenocarcinoma. Oncogene 2014; 96: 111–120.
Boulter L, Guest R V, Kendall TJ, Wilson DH, Wojtacha D, Robson AJ et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J Clin Invest 2015; 125: 1269–1285.
Loilome W, Bungkanjana P, Techasen A, Namwat N, Yongvanit P, Puapairoj A et al. Activated macrophages promote Wnt/β-catenin signaling in cholangiocarcinoma cells. Tumour Biol 2014; 35: 5357–5367.
Chan-On W, Nairismägi M-L, Ong CK, Lim WK, Dima S, Pairojkul C et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet 2013; 45: 1474–1478.
Goeppert B, Konermann C, Schmidt CR, Bogatyrova O, Geiselhart L, Ernst C et al. Global alterations of DNA methylation in cholangiocarcinoma target the Wnt signaling pathway. Hepatology 2014; 59: 544–554.
Luis TC, Ichii M, Brugman MH, Kincade P, Staal FJT . Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia 2012; 26: 414–421.
Lento W, Congdon K, Voermans C, Kritzik M, Reya T . Wnt signaling in normal and malignant hematopoiesis. Cold Spring Harb Perspect Biol e-pub ahead of print 1 February 2013 doi:10.1101/cshperspect.a008011.
Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D et al. β-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 2010; 18: 606–618.
Lane SW, Wang YJ, Lo Celso C, Ragu C, Bullinger L, Stephen M et al. Differential niche and Wnt requirements during acute myeloid leukemia progression. Blood 2011; 118: 2849–2856.
Wang Y, Krivtsov A, Sinha A, North T . The Wnt/β-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010; 327: 1650–1653.
Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D et al. Beta-catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 2010; 18: 606–618.
Cheng CK, Li L, Cheng SH, Lau KM, Chan NPH, Wong RSM et al. Transcriptional repression of the RUNX3/AML2 gene by the t(8;21) and inv(16) fusion proteins in acute myeloid leukemia. Blood 2008; 112: 3391–3402.
Müller-Tidow C, Steffen B, Cauvet T, Tickenbrock L, Ji P, Diederichs S et al. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol Cell Biol 2004; 24: 2890–2904.
Ferrando AA . The role of NOTCH1 signaling in T-ALL. Hematology Am Soc Hematol Educ Program 2009; 2009: 353–361.
Guo W, Lasky JL, Chang C-J, Mosessian S, Lewis X, Xiao Y et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 2008; 453: 529–533.
Kaveri D, Kastner P, Dembélé D, Nerlov C, Chan S, Kirstetter P . β-Catenin activation synergizes with Pten loss and Myc overexpression in Notch-independent T-ALL. Blood 2013; 122: 694–704.
Giambra V, Jenkins CE, Lam SH, Hoofd C, Belmonte M, Wang X et al. Leukemia stem cells in T-ALL require active Hif1α and Wnt signaling. Blood 2015; 125: 3917–3927.
Lu D, Zhao Y, Tawatao R, Cottam HB, Sen M, Leoni LM et al. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc Natl Acad Sci USA 2004; 101: 3118–3123.
Moskalev EA, Luckert K, Vorobjev IA, Mastitsky SE, Gladkikh AA, Stephan A et al. Concurrent epigenetic silencing of wnt / β -catenin pathway inhibitor genes in B cell chronic lymphocytic leukaemia. BMC Cancer 2012; 12: 1.
Wang L, Shalek AK, Lawrence M, Ding R, Gaublomme JT, Pochet N et al. Somatic mutation as a mechanism of Wnt/beta-catenin pathway activation in CLL. Blood 2014; 124: 1089–1098.
Stolz W, Schmoeckel C, Landthaler M, Braun-Falco O . Association of early malignant melanoma with nevocytic nevi. Cancer 1989; 63: 550–555.
Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell 2009; 15: 294–303.
Jones SL, Cichowski K . Many roads lead to oncogene-induced senescence. Oncogene 2008; 27: 2801–2809.
Delmas V, Beermann F, Martinozzi S, Carreira S, Ackermann J, Kumasaka M et al. Beta-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes Dev 2007; 21: 2923–2935.
Juan J, Muraguchi T, Iezza G, Sears RC, McMahon M . Diminished WNT ->beta -catenin->c-MYC signaling is a barrier for malignant progression of BRAFV600E-induced lung tumors. Genes Dev 2014; 28: 561–575.
Pawlikowski JS, McBryan T, van Tuyn J, Drotar ME, Hewitt RN, Maier AB et al. Wnt signaling potentiates nevogenesis. Proc Natl Acad Sci USA 2013; 110: 16009–16014.
Lim X, Nusse R . Wnt signaling in skin development, homeostasis, and disease. Cold Spring Harb Perspect Biol e-pub ahead of print 1 February 2013 doi:10.1101/cshperspect.a008029.
Webster MR, Weeraratna AT . A Wnt-er migration: the confusing role of β-catenin in melanoma metastasis. Sci Signal 2013; 6: pe11.
Kageshita T, Hamby CV, Ishihara T, Matsumoto K, Saida T, Ono T . Loss of beta-catenin expression associated with disease progression in malignant melanoma. Br J Dermatol 2001; 145: 210–216.
Bachmann IM . Importance of P-Cadherin, -Catenin, and Wnt5a/Frizzled for progression of melanocytic tumors and prognosis in cutaneous melanoma. Clin Cancer Res 2005; 11: 8606–8614.
Chien AJ, Moore EC, Lonsdorf AS, Kulikauskas RM, Rothberg BG, Berger AJ et al. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc Natl Acad Sci USA 2009; 106: 1193–1198.
Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE et al. β-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell 2011; 20: 741–754.
Biechele TL, Kulikauskas RM, Toroni R a, Lucero OM, Swift RD, James RG et al. Wnt/β-catenin signaling and AXIN1 regulate apoptosis triggered by inhibition of the mutant kinase BRAFV600E in human melanoma. Sci Signal 2012; 5: ra3.
Chien AJ, Haydu LE, Biechele TL, Kulikauskas RM, Rizos H, Kefford RF et al. Targeted BRAF inhibition impacts survival in melanoma patients with high levels of Wnt/β-catenin signaling. PLoS One 2014; 9: e94748.
O’Connell MP, Fiori JL, Baugher KM, Indig FE, French AD, Camilli TC et al. Wnt5A activates the calpain-mediated cleavage of filamin A. J Invest Dermatol 2009; 129: 1782–1789.
Webster MR, Xu M, Kinzler KA, Kaur A, Appleton J, Connell MPO et al. Wnt5A promotes an adaptive, senescent-like stress response, while continuing to drive invasion in melanoma cells. Pigment Cell Melanoma Res 2015; 28: 184–195.
Anastas JN, Kulikauskas RM, Tamir T, Rizos H, Long G V, von Euw EM et al. WNT5A enhances resistance of melanoma cells to targeted BRAF inhibitors. J Clin Invest 2014; 124: 2877–2890.
O’Connell MP, Marchbank K, Webster MR, Valiga AA, Kaur A, Vultur A et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov 2013; 3: 1378–1393.
Ekström EJ, Bergenfelz C, von Bülow V, Serifler F, Carlemalm E, Jönsson G et al. WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol Cancer 2014; 13: 1–15.
Linnskog R, Jönsson G, Axelsson L, Prasad CP, Andersson T . Interleukin-6 drives melanoma cell motility through p38α-MAPK-dependent up-regulation of WNT5A expression. Mol Oncol 2014; 8: 1365–1378.
Grossmann AH, Yoo JH, Clancy J, Sorensen LK, Sedgwick A, Tong Z et al. The small GTPase ARF6 stimulates β-catenin transcriptional activity during WNT5A-mediated melanoma invasion and metastasis. Sci Signal 2013; 6: ra14.
Mikels AJ, Nusse R . Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol 2006; 4: e115.
Hoek KS, Schlegel NC, Brafford P, Sucker A, Ugurel S, Kumar R et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res 2006; 19: 290–302.
Hoek KS, Eichhoff OM, Schlegel NC, Döbbeling U, Kobert N, Schaerer L et al. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res 2008; 68: 650–656.
Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y et al. Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci USA 2000; 97: 4262–4266.
Geyer FC, Lacroix-Triki M, Savage K, Arnedos M, Lambros MB, MacKay A et al. β-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod Pathol 2011; 24: 209–231.
Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH . Wnt/{beta}-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am J Pathol 2010; 176: 2911–2920.
Xu J, Prosperi JR, Choudhury N, Olopade OI, Goss KH . B-catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS One 2015; 10: 1–11.
Li S, Li S, Sun Y, Li L . The expression of β-catenin in different subtypes of breast cancer and its clinical significance. Tumour Biol 2014; 35: 7693–7698.
Howe LR, Brown AMC . Wnt signaling and breast cancer. Cancer Biol Ther 2004; 3: 36–41.
Liu C-C, Prior J, Piwnica-Worms D, Bu G . LRP6 overexpression defines a class of breast cancer subtype and is a target for therapy. Proc Natl Acad Sci USA 2010; 107: 5136–5141.
Yang L, Wu X, Wang Y, Zhang K, Wu J, Yuan Y-C et al. FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene 2011; 30: 4437–4446.
Klarmann GJ, Decker A, Farrar WL . Epigenetic gene silencing in the Wnt pathway in breast cancer. Epigenetics 2008; 3: 59–63.
Gunther EJ, Moody SE, Belka GK, Hahn KT, Innocent N, Dugan KD et al. Impact of p53 loss on reversal and recurrence of conditional Wnt-induced tumorigenesis. Genes Dev 2003; 17: 488–501.
Li Y, Welm B, Podsypanina K, Huang S, Chamorro M, Zhang X et al. Evidence that transgenes encoding components of the Wnt signaling pathway preferentially induce mammary cancers from progenitor cells. Proc Natl Acad Sci USA 2003; 100: 15853–15858.
Klauzinska M, Baljinnyam B, Raafat A, Rodriguez-Canales J, Strizzi L, Endo Greer Y et al. Rspo2/Int7 regulates invasiveness and tumorigenic properties of mammary epithelial cells. J Cell Physiol 2012; 227: 1960–1971.
Cleary AS, Leonard TL, Gestl SA, Gunther EJ . Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature 2014; 508: 113–117.
Kim S, Goel S, Alexander CM . Differentiation generates paracrine cell pairs that maintain basaloid mouse mammary tumors: proof of concept. PLoS One 2011; 6: e19310.
Beck B, Blanpain C . Unravelling cancer stem cell potential. Nat Rev Cancer 2013; 13: 727–738.
Reya T, Clevers H . Wnt signalling in stem cells and cancer. Nature 2005; 434: 843–850.
Park J-I, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009; 460: 66–72.
Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2008; 457: 608–611.
Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 2012; 337: 730–735.
Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ, Cordero JB et al. ROS production and NF-κB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell 2013; 12: 761–773.
Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Göktuna SI, Ziegler PK et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013; 152: 25–38.
Vermeulen L, De Sousa E, Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 2010; 12: 468–476.
Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr H-A, Delaloye J-F et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012; 481: 85–89.
Kessenbrock K, Dijkgraaf GJP, Lawson DA, Littlepage LE, Shahi P, Pieper U et al. A role for matrix metalloproteinases in regulating mammary stem cell function via the Wnt signaling pathway. Cell Stem Cell 2013; 13: 300–313.
Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014; 14: 342–356.
Hwang W-L, Jiang J-K, Yang S-H, Huang T-S, Lan H-Y, Teng H-W et al. MicroRNA-146a directs the symmetric division of Snail-dominant colorectal cancer stem cells. Nat Cell Biol 2014; 16: 268–280.
Isobe T, Hisamori S, Hogan DJ, Zabala M, Hendrickson DG, Dalerba P et al. miR-142 regulates the tumorigenicity of human breast cancer stem cells through the canonical WNT signaling pathway. Elife 2014; 3: 1–23.
Wang Y, He L, Du Y, Zhu P, Huang G, Luo J et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell 2015; 16: 413–425.
Fang L, Cai J, Chen B, Wu S, Li R, Xu X et al. Aberrantly expressed miR-582-3p maintains lung cancer stem cell-like traits by activating Wnt/β-catenin signalling. Nat Commun 2015; 6: 8640.
Merlos-Suárez A, Barriga FM, Jung P, Iglesias M, Céspedes MV, Rossell D et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 2011; 8: 511–524.
de Sousa E, Melo F, Colak S, Buikhuisen J, Koster J, Cameron K, de Jong JH et al. Methylation of cancer-stem-cell-associated Wnt target genes predicts poor prognosis in colorectal cancer patients. Cell Stem Cell 2011; 9: 476–485.
Scheel C, Weinberg RA . Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol 2012; 22: 396–403.
Kalluri R . EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest 2009; 119: 1417–1419.
De Craene B, Berx G . Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 2013; 13: 97–110.
Wu Z-Q, Li X-Y, Hu CY, Ford M, Kleer CG, Weiss SJ . Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc Natl Acad Sci USA 2012; 109: 16654–16659.
Wang Y, Bu F, Royer C, Serres S, Larkin JR, Soto MS et al. ASPP2 controls epithelial plasticity and inhibits metastasis through β-catenin-dependent regulation of ZEB1. Nat Cell Biol 2014; 16: 1092–1104.
Tenbaum SP, Ordóñez-Morán P, Puig I, Chicote I, Arqués O, Landolfi S et al. β-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat Med 2012; 18: 892–901.
Gujral TS, Chan M, Peshkin L, Sorger PK, Kirschner MW, MacBeath G . A Noncanonical frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014; 159: 844–856.
Kahlert C, Kalluri R . Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl) 2013; 91: 431–437.
Chairoungdua A, Smith DL, Pochard P, Hull M, Caplan MJ . Exosome release of β-catenin: a novel mechanism that antagonizes Wnt signaling. J Cell Biol 2010; 190: 1079–1091.
Luga V, Zhang L, Viloria-Petit AM, Ogunjimi A a, Inanlou MR, Chiu E et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012; 151: 1542–1556.
Maheswaran S, Haber DA . Circulating tumor cells: a window into cancer biology and metastasis. Curr Opin Genet Dev 2010; 20: 96–99.
Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature 2012; 487: 510–513.
Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015; 349: 1351–1356.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015; 373: 23–34.
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med 2015; 373: 1627–1639.
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015; 373: 1803–1813.
Staal FJT, Luis TC, Tiemessen MM . WNT signalling in the immune system: WNT is spreading its wings. Nat Rev Immunol 2008; 8: 581–593.
Spranger S, Bao R, Gajewski TF . Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015; 523: 231–235.
Yaguchi T, Goto Y, Kido K, Mochimaru H, Sakurai T, Tsukamoto N et al. Immune suppression and resistance mediated by constitutive activation of Wnt/β-catenin signaling in human melanoma cells. J Immunol 2012; 189: 2110–2117.
Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 2016; 165: 45–60.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015; 372: 2509–2520.
Kahn M . Can we safely target the WNT pathway? Nat Rev Drug Discov 2014; 13: 513–532.
Dodge ME, Moon J, Tuladhar R, Lu J, Jacob LS, Zhang L et al. Diverse chemical scaffolds support direct inhibition of the membrane-bound O-acyltransferase porcupine. J Biol Chem 2012; 287: 23246–23254.
van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015; 161: 933–945.
Madan B, Ke Z, Harmston N, Ho SY, Frois AO, Alam J et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2016; 35: 2197–2207.
D3 (Drug Product and Development) BSI. A Study to Evaluate the Safety and Tolerability of ETC-1922159 in Advanced Solid Tumours. ClinicalTrials.gov, 2015, p NCT02521844.
Yeung P, Beviglia L, Cancilla B, Dee-Hoskins C, Evans JW, Fischer MM et al. Abstract 1907: Wnt pathway antagonist OMP-54F28 (FZD8-Fc) inhibits tumor growth and reduces tumor-initiating cell frequency in patient-derived hepatocellular carcinoma and ovarian cancer xenograft models. Cancer Res 2014; 74: 1907–1907.
Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci USA 2012; 109: 11717–11722.
Smith DC, Rosen LS, Chugh R, Goldman JW, Xu L, Kapoun A et al. General Poster Session (Board # 1H), Mon, 8: 00 AM-11: 45 AM First-in-human evaluation of the human monoclonal antibody vantictumab (OMP- 18R5; anti-Frizzled) targeting the WNT pathway in a phase I study for patients with advanced solid tumors. ASCO Annu Meet Abstr 2013; J Clin Onc: 1345201.
Baron R, Kneissel M . WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med 2013; 19: 179–192.
Nagayama S, Yamada E, Kohno Y, Aoyama T, Fukukawa C, Kubo H et al. Inverse correlation of the up-regulation of FZD10 expression and the activation of beta-catenin in synchronous colorectal tumors. Cancer Sci 2009; 100: 405–412.
Giraudet A-L, Badel J-N, Cassier P, Desuzinges C, Kriza D, Perol D et al. SYNFRIZZ-A phase Ia/Ib of a radiolabelled monoclonal AB for the treatment of relapsing synovial sarcoma. J Nucl Med 2014; 55: 223.
Gang EJ, Hsieh Y-T, Pham J, Zhao Y, Nguyen C, Huantes S et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene 2014; 33: 2169–2178.
Rebel VI, Kung AL, Tanner EA, Yang H, Bronson RT, Livingston DM . Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc Natl Acad Sci USA 2002; 99: 14789–14794.
Robert R, McWilliams, Andrew H, Ko E, Gabriela Chiorean, Eunice Lee Kwak et al. A phase Ib dose-escalation study of PRI-724, a CBP/beta-catenin modulator, plus gemcitabine (GEM) in patients with advanced pancreatic adenocarcinoma (APC) as second-line therapy after FOLFIRINOX or FOLFOX. J Clin Oncol 2015; 33: abstr e15270.
University of Southern California. Combination Chemotherapy and Bevacizumab With or Without PRI-724 in Treating Patients With Newly Diagnosed Metastatic Colorectal Cancer (PRIMIER). In: Clinicaltrials.Gov. 2015, p NCT02413853.
Huang S-MA, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009; 461: 614–620.
Riffell JL, Lord CJ, Ashworth A . Tankyrase-targeted therapeutics: expanding opportunities in the PARP family. Nat Rev Drug Discov 2012; 11: 923–936.
Lau T, Chan E, Callow M, Waaler J, Boggs J, Blake RA et al. A novel Tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res 2013; 73: 3132–3144.
Andersson T, Axelsson L, Mohapatra P, Prasad C, Soerensen PG, Mau-Soerensen M et al. Abstract A116: targeting the Wnt-5a signaling pathway as a novel anti-metastatic therapy. Mol Cancer Ther 2015; 14: A116–A116.
Latres E, Chiaur DS, Pagano M . The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene 1999; 18: 849–854.
Metcalfe C, Mendoza-Topaz C, Mieszczanek J, Bienz M . Stability elements in the LRP6 cytoplasmic tail confer efficient signalling upon DIX-dependent polymerization. J Cell Sci 2010; 123: 1588–1599.
Clevers H . Wnt/Beta-catenin signaling in development and disease. Cell 2006; 127: 469–480.
Tree DRP, Shulman JM, Scott MP, Gubb D, Axelrod JD . Prickle mediates feedback amplification to generate asymmetric planar cell polarity signaling. Cell 2002; 109: 371–381.
Habas R, Kato Y, He X . Wnt / frizzled activation of Rho regulates vertebrate gastrulation and requires a novel formin homology protein Daam1. Cell 2001; 107: 843–854.
Kikuchi A, Yamamoto H, Sato A, Matsumoto S . New insights into the mechanism of Wnt signaling pathway activation. Int Rev Cell Mol Biol 2011; 291: 21–71.
Gao B, Song H, Bishop K, Elliot G, Garrett L, English MA et al. Wnt signaling gradients establish planar cell polarity by inducing Vangl2 phosphorylation through Ror2. Dev Cell 2011; 20: 163–176.
Gao C, Chen Y . Dishevelled: the hub of Wnt signaling. Cell Signal 2010; 22: 717–727.
Sheldahl LC, Slusarski DC, Pandur P, Miller JR, Kuhl M, Moon RT . Dishevelled activates Ca2+ flux, PKC, and CamKII in vertebrate embryos. J Cell Biol 2003; 161: 769–777.
Zhang C, Cattaruzza F, Yeung P, Yen W-C, Fischer M, Brunner A et al. Abstract A30: Predictive and pharmacodynamic biomarkers of vantictumab (OMP-18R5; anti-Frizzled) in non-small cell lung cancer. Mol Cancer Ther 2015; 14: A30–A30.
Acknowledgements
We thank Iris Augustin, Fillip Port, Katharina Kober and Lucie Wolf for carefully reading the manuscript and contributing valuable ideas. T.Z. was supported by a DKFZ Postdoctoral Fellowship. We apologize to all colleagues whose work was not cited due to space constrains.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Zhan, T., Rindtorff, N. & Boutros, M. Wnt signaling in cancer. Oncogene 36, 1461–1473 (2017). https://doi.org/10.1038/onc.2016.304
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.304
This article is cited by
-
The SEMA3F-NRP1/NRP2 axis is a key factor in the acquisition of invasive traits in in situ breast ductal carcinoma
Breast Cancer Research (2024)
-
Cytosolic Cadherin 4 promotes angiogenesis and metastasis in papillary thyroid cancer by suppressing the ubiquitination/degradation of β-catenin
Journal of Translational Medicine (2024)
-
Wnt signaling regulates chemokine production and cell migration of circulating human monocytes
Cell Communication and Signaling (2024)
-
Intracellular domain of epithelial cell adhesion molecule induces Wnt receptor transcription to promote colorectal cancer progression
Journal of Biomedical Science (2024)
-
Cancer, metastasis, and the epigenome
Molecular Cancer (2024)
