
Abstract
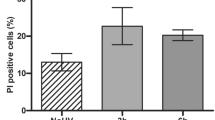
The role of UVA-radiation—the major fraction in sunlight—in human skin carcinogenesis is still elusive. We here report that different UVA exposure regime (4 × 5 J/cm2 per week or 1 × 20 J/cm2 per week) caused tumorigenic conversion (tumors in nude mice) of the HaCaT skin keratinocytes. While tumorigenicity was not associated with general telomere shortening, we found new chromosomal changes characteristic for each recultivated tumor. Since this suggested a nontelomere-dependent relationship between UVA irradiation and chromosomal aberrations, we investigated for alternate mechanisms of UVA-dependent genomic instability. Using the alkaline and neutral comet assay as well as γ-H2AX foci formation on irradiated HaCaT cells (20–60 J/cm2), we show a dose-dependent and long lasting induction of DNA single and double (ds) strand breaks. Extending this to normal human skin keratinocytes, we demonstrate a comparable damage response and, additionally, a significant induction and maintenance of micronuclei (MN) with more acentric fragments (indicative of ds breaks) than entire chromosomes particularly 5 days post irradiation. Thus, physiologically relevant UVA doses cause long-lasting DNA strand breaks, a prerequisite for chromosomal aberration that most likely contribute to tumorigenic conversion of the HaCaT cells. Since normal keratinocytes responded similarly, UVA may likewise contribute to the complex karyotype characteristic for human skin carcinomas.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

References
Armitage P . (1955). Test for linear trends in proportions and frequencies. Biometrics 11: 375–386.
Armstrong BK, Kricker A . (2001). The epidemiology of UV induced skin cancer. J Photochem Photobiol B 63: 8–18.
Bleuel K, Popp S, Fusenig NE, Stanbridge EJ, Boukamp P . (1999). Tumor suppression in human skin carcinoma cells by chromosome 15 transfer or thrombospondin-1 overexpression through halted tumor vascularization. Proc Natl Acad Sci USA 96: 2065–2070.
Boukamp P . (2005b). UV-induced skin cancer: similarities—variations. J Dtsch Dermatol Ges 3: 493–503.
Boukamp P, Breitkreutz D, Stark HJ, Fusenig NE . (1990a). Mesenchyme-mediated and endogenous regulation of growth and differentiation of human skin keratinocytes derived from different body sites. Differentiation 44: 150–161.
Boukamp P, Peter W, Pascheberg U, Altmeier S, Fasching C, Stanbridge EJ et al. (1995). Step-wise progression in human skin carcinogenesis in vitro involves mutational inactivation of p53, rasH oncogene activation and additional chromosome loss. Oncogene 11: 961–969.
Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE . (1988). Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 106: 761–771.
Boukamp P, Popp S, Altmeyer S, Hulsen A, Fasching C, Cremer T et al. (1997). Sustained nontumorigenic phenotype correlates with a largely stable chromosome content during long-term culture of the human keratinocyte line HaCaT. Genes Chromosomes Cancer 19: 201–214.
Boukamp P, Popp S, Bleuel K, Tomakidi E, Burkle A, Fusenig NE . (1999). Tumorigenic conversion of immortal human skin keratinocytes (HaCaT) by elevated temperature. Oncogene 18: 5638–5645.
Boukamp P, Stanbridge EJ, Foo DY, Cerutti PA, Fusenig NE . (1990b). c-Ha-ras oncogene expression in immortalized human keratinocytes (HaCaT) alters growth potential in vivo but lacks correlation with malignancy. Cancer Res 50: 2840–2847.
Burnworth B, Popp S, Stark HJ, Steinkraus V, Brocker EB, Hartschuh W et al. (2006). Gain of 11q/cyclin D1 overexpression is an essential early step in skin cancer development and causes abnormal tissue organization and differentiation. Oncogene 25: 4399–4412.
Cadet J, Sage E, Douki T . (2005). Ultraviolet radiation-mediated damage to cellular DNA. Mutat Res 571: 3–17.
Cochran WG . (1954). Some methods for strengthening the common chi square test. Biometrics 11: 417–451.
Cohen J . (1960). A coefficient of agreement for nominal scales. Educ Psychol Meas 20: 37–46.
de Gruijl FR . (2002). Photocarcinogenesis: UVA vs. UVB radiation. Skin Pharmacol Appl Skin Physiol 15: 316–320.
Emri G, Wenczl E, Van EP, Jans J, Roza L, Horkay I et al. (2000). Low doses of UVB or UVA induce chromosomal aberrations in cultured human skin cells. J Invest Dermatol 115: 435–440.
Federal Office for Radiation Protection (2006). Continuous Solar UV-Monitoring in Germany. Federal Office for Radiation Protection: Munich, Germany, www.bfs.de.
Fenech M . (2000). The in vitro micronucleus technique. Mutat Res 455: 81–95.
Fenech M . (2002). Biomarkers of genetic damage for cancer epidemiology. Toxicology 181–182: 411–416.
Figueroa R, Lindenmaier H, Hergenhahn M, Nielsen KV, Boukamp P . (2000). Telomere erosion varies during in vitro aging of normal human fibroblasts from young and adult donors. Cancer Res 60: 2770–2774.
Folkman J, Hanahan D . (1991). Switch to the angiogenic phenotype during tumorigenesis. Princess Takamatsu Symp 22: 339–347.
Garinis GA, Mitchell JR, Moorhouse MJ, Hanada K, de Waard H, Vandeputte D et al. (2005). Transcriptome analysis reveals cyclobutane pyrimidine dimers as a major source of UV-induced DNA breaks. EMBO J 24: 3952–3962.
He YY, Pi J, Huang JL, Diwan BA, Waalkes MP, Chignell CF . (2006). Chronic UVA irradiation of human HaCaT keratinocytes induces malignant transformation associated with acquired apoptotic resistance. Oncogene 25: 3680–3688.
Ikehata H, Kudo H, Masuda T, Ono T . (2003). UVA induces C → T transitions at methyl-CpG-associated dipyrimidine sites in mouse skin epidermis more frequently than UVB. Mutagenesis 18: 511–519.
Ikehata H, Nakamura S, Asamura T, Ono T . (2004). Mutation spectrum in sunlight-exposed mouse skin epidermis: small but appreciable contribution of oxidative stress-mediated mutagenesis. Mutat Res 556: 11–24.
Ikehata H, Ono T . (2007). Significance of CpG methylation for solar UV-induced mutagenesis and carcinogenesis in skin. Photochem Photobiol 83: 196–204.
Kielbassa C, Roza L, Epe B . (1997). Wavelength dependence of oxidative DNA damage induced by UV and visible light. Carcinogenesis 18: 811–816.
Krude T . (1999). Mimosine arrests proliferating human cells before onset of DNA replication in a dose-dependent manner. Exp Cell Res 247: 148–159.
Lehman TA, Modali R, Boukamp P, Stanek J, Bennett WP, Welsh JA et al. (1993). p53 mutations in human immortalized epithelial cell lines. Carcinogenesis 14: 833–839.
Limoli CK, Giedzinski E, Bonner WM, Cleaver JE . (2002). UV-induced replication arrest in the xeroderma pigmentosum variant leads to DNA double-strand breaks, γ-H2AX formation, and Mre11 relocalization. Proc Natl Acad Sci USA 99: 233–238.
Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE . (2006). H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci USA 103: 9891–9896.
Murnane JP, Sabatier L . (2004). Chromosome rearrangements resulting from telomere dysfunction and their role in cancer. Bioessays 26: 1164–1174.
Mutzhas MF, Holzle E, Hofmann C, Plewig G . (1981). A new apparatus with high radiation energy between 320–460 nm: physical description and dermatological applications. J Invest Dermatol 76: 42–47.
Obermueller E, Vosseler S, Fusenig NE, Mueller MM . (2004). Cooperative autocrine and paracrine functions of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor in the progression of skin carcinoma cells. Cancer Res 64: 7801–7812.
Oikawa S, Kawanishi S . (1999). Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett 453: 365–368.
Olive PL . (1999). DNA damage and repair in individual cells: applications of the comet assay in radiobiology. Int J Radiat Biol 75: 395–405.
Olive PL, Frazer G, Banath JP . (1993). Radiation-induced apoptosis measured in TK6 human B lymphoblast cells using the comet assay. Radiat Res 136: 130–136.
Parangi S, O’Reilly M, Christofori G, Holmgren L, Grosfeld J, Folkman J et al. (1996). Antiangiogenic therapy of transgenic mice impairs de novo tumor growth. Proc Natl Acad Sci USA 93: 2002–2007.
Phillipson RP, Tobi SE, Morris JA, McMillan TJ . (2002). UV-A induces persistent genomic instability in human keratinocytes through an oxidative stress mechanism. Free Radic Biol Med 32: 474–480.
Popp S, Waltering S, Herbst C, Moll I, Boukamp P . (2002). UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int J Cancer 99: 352–360.
Popp S, Waltering S, Holtgreve-Grez H, Jauch A, Proby C, Leigh IM et al. (2000). Genetic characterization of a human skin carcinoma progression model: from primary tumor to metastasis. J Invest Dermatol 115: 1095–1103.
Rapp A, Bock C, Dittmar H, Greulich KO . (2000). UV-A breakage sensitivity of human chromosomes as measured by COMET-FISH depends on gene density and not on the chromosome size. J Photochem Photobiol B 56: 109–117.
Rapp A, Greulich KO . (2004). After double-strand break induction by UV-A, homologous recombination and nonhomologous end joining cooperate at the same DSB if both systems are available. J Cell Sci 117: 4935–4945.
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM . (1998). DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273: 5858–5868.
Rothkamm K, Lobrich M . (2003). Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA 100: 5057–5062.
Sedelnikova OA, Rogakou EP, Panyutin IG, Bonner WM . (2002). Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat Res 158: 486–492.
Skobe M, Fusenig NE . (1998). Tumorigenic conversion of immortal human keratinocytes through stromal cell activation. Proc Natl Acad Sci USA 95: 1050–1055.
Tefferi A, Dewald GW, Litzow ML, Cortes J, Mauro MJ, Talpaz M et al. (2005). Chronic myeloid leukemia: current application of cytogenetics and molecular testing for diagnosis and treatment. Mayo Clin Proc 80: 390–402.
Thompson LH, Limoli CL . (2003). Origin, recognition, signaling and repair of DNA double-strand breaks in mammalian cells (Chapter 6). In: Caldecott KW (ed). Eukaryotic DNA Damage Surveillance and Repair. Kluwer Academic/Plenum Press, pp 107–145.
von Zglinicki T, Pilger R, Sitte N . (2000). Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic Biol Med 28: 64–74.
Wischermann K, Boukamp P, Schmezer P . (2007). Improved alkaline comet assay protocol for adherent HaCaT keratinocytes to study UVA-induced DNA Damage. Mutat Res 630: 122–128.
Wondrak GT, Jacobson MK, Jacobson EL . (2006). Endogenous UVA-photosensitizers: mediators of skin photodamage and novel targets for skin photoprotection. Photochem Photobiol Sci 5: 215–237.
Acknowledgements
We thank Cees Guikers for the first experiments with daily exposures, Heinrich Steinbauer for performing the tumor experiments, Heidi Holtgreve for her excellent technical assistance in M-FISH hybridization and Stefan Henning for comparing the different UVA-sources. This work was supported in part from the Sander Stiftung, Deutsche Krebshilfe e.V. and EU (LSHC-CT-2004-502943) (both to PB), QLK4-1999-01084 (to PB and KSK) as well as the German Research Foundation (SCHA 411/10-1,-2) and the European Community Marie Curie Program MEIF-CT-2005-023821 (to AR).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
Rights and permissions
About this article
Cite this article
Wischermann, K., Popp, S., Moshir, S. et al. UVA radiation causes DNA strand breaks, chromosomal aberrations and tumorigenic transformation in HaCaT skin keratinocytes. Oncogene 27, 4269–4280 (2008). https://doi.org/10.1038/onc.2008.70
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2008.70
Keywords
This article is cited by
-
Probing photoprotection properties of lipophilic chain conjugated thiourea-aryl group molecules to attenuate ultraviolet-A induced cellular and DNA damages
Scientific Reports (2022)
-
Development and characterisation of an irradiation device for biomedical studies covering the solar spectrum with individual regulated spectral bands
Photochemical & Photobiological Sciences (2022)
-
Unravelling the molecular mechanism of mutagenic factors impacting human health
Environmental Science and Pollution Research (2022)
-
Sunscreens and their usefulness: have we made any progress in the last two decades?
Photochemical & Photobiological Sciences (2021)
-
Antiapoptotic effects of scutellarin on ultraviolet A-irradiated HaCaT human keratinocytes
Biomedical Dermatology (2018)
