
Abstract
To date more than 90 loci that show an association with body mass index (BMI) and other obesity-related traits, have been discovered through genome-wide association studies. These findings have been widely replicated, mostly in European and Asian populations, but systematic investigation in African cohorts is still lacking. Therefore, the aim of our study was to replicate the association of six single-nucleotide polymorphisms (SNPs) previously linked to BMI, in a South African black adolescent cohort. The SNPs were in or near GNPDA2 (rs10938397), MTCH2 (rs10838738), NEGR1 (rs2568958), SH2B1 (rs7498665), STK33 (rs10769908) and TMEM18 (rs6548238). The SNPs were genotyped in 990 adolescents from the Birth to Twenty study, using an Illumina VeraCode assay, and association with BMI statistically assesed by using PLINK. Three of the SNPs tested were associated with BMI in this African cohort, and showed a consistent (albeit smaller) directional effect to that observed in non-African cohorts. We identified significant association between BMI and rs10938397 (effect allele-G) near GNPDA2 (Padj=0.003), rs7498665 (effect allele-G) in SH2B1 (Padj=0.014) and rs6548238 (effect allele-C) near TMEM18 (Padj=0.030). This data suggests that common genetic variants potentially contributes to obesity risk in diverse population groups.
Similar content being viewed by others
Introduction
Recent studies show that the mean prevalence of overweight and obesity (combined) in South African children and adolescents is ~15%.1 The prevention of childhood obesity is a key global health priority as obesity is a major contributor to increased mortality in adulthood,2 and is increasing in prevalence in both developed and developing nations, such as South Africa.
The risk of developing obesity is modulated by both heritable and environmental factors.3 Heritability studies of body mass index (BMI) demonstrated that a significant proportion of the variance in BMI (40–70%) is because of genetics4 with genome-wide association studies of obesity-related traits identifying more than 90 risk loci thus far.5, 6, 7 An explicit deficit of African-centric genome-wide association studies data for body composition and obesity exist, especially for sub-Saharan African populations. In addition, most published genetic association studies of BMI have primarily focussed on the association with adult BMI. Identifying loci that predispose to obesity early in life could provide a better understanding of the early determinants of adult obesity and may also uncover potential new targets for the therapeutic prevention of obesity.
Previously, our group investigated the role of gene variants in appetite regulating genes with BMI in an adolescent cohort,8 replicating single-nucleotide polymorphism (SNP) associations in FTO and MC4R, as well as establishing a novel association with variants in the LEP gene. Here, we describe the replication within the same cohort of six variants, selected based on previous evidence of robust association with BMI in non-African populations in large meta-analyses undertaken by the GIANT consortium.9, 10, 11 These include SNPs in or near GNPDA2, MTCH2, NEGR1, SH2B1, STK33 and TMEM18.
Subjects and methods
We used data and samples from participants of the Birth to Twenty (Bt20) study that have been described in detail elsewhere.12 Written assent was obtained from all adolescents in conjunction with written consent from caregivers, prior to a blood sample collection. This study was approved by the Human Research Ethics Committee of the University of the Witwatersrand (certificate number M010556). The participants in the Bt20 cohort are South Africans who self-identified as Sotho speakers (that is, of southeastern Bantu-speaking descent), thus belonging to the Niger-Kordofanian ethno-linguistic group. A subset of individuals (43%, n=990) from the Bt20 cohort were randomly selected for this study, and consisted of 524 (53%) female and 466 (47%) male adolescents, with a mean (±s.d.) age of 13.7±0.2 years. Anthropometric measurements were obtained by using standard methods13 and pubertal stage was assessed by using a validated self-assessment method.14
SNPs previously associated with BMI9, 10, 11 were selected for analysis, and include rs2568958 near NEGR1, rs6548238 near TMEM18, rs10938397 near GNPDA2, rs10769908 in STK33, rs10838738 in MTCH2 and rs7498665 in SH2B1. Genotyping was performed using the GoldenGate VeraCode assay (Illumina, San Diego, CA, USA). Internal quality control was performed on all raw genotype data according to the supplier’s specifications using the genotyping module of BeadStudio (Framework version 3.1.3.0; Illumina). Further quality control filters based on minor allele frequency (MAF<0.01) and Hardy–Weinberg equilibrium (HWE<0.05) were used as exclusion criteria. Ancestry information markers previously genotyped in this cohort show no significant population structure within the cohort.8
PLINK v.1.9 software was used for all statistical analysis, unless stated otherwise.15 The distribution of BMI was skewed and therefore was log-transformed to normality for all analyses. Linear regression was used to assess the association of the selected SNPs with BMI. As BMI correlated significantly with gender, pubertal stage and age, analyses were adjusted for these variables by including them in the linear model as covariates (Padj). Given the strong prior information about the correlation of the SNPs tested here with BMI, we considered this a replication study, and therefore P-values below 0.05 were considered significant. Given the minor allele frequencies of the SNPs tested, the study achieves 80% power to detect differences among the means of BMI, with a standard F-test for linear regression, as previously calculated.8 To assess the combined impact of risk alleles on BMI, we calculated a risk allele score by summing the number of BMI-increasing alleles per individual. This score was calculated including the risk alleles described here, as well as the four previously identified BMI-associated variants in this cohort: FTO (rs17817449), LEP (rs10954174 and rs6966536) and MC4R (rs17782313).8
Results
The study group consisted of 524 (53%) female and 466 (47%) male adolescents, with a mean (±s.d.) age of 13.7±0.2 years. Summary statistics and trends related to BMI in this subset were described previously,8 and based on these data all analyses were adjusted for gender, gender-specific pubertal stage and age.
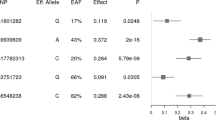
Table 1 reports the association of the SNPs with (log)BMI. Of the six SNPs investigated, associations with BMI were replicated for three SNPs in this African cohort, and showed a similar (albeit smaller) directional effect to that observed in the discovery studies. We identified significant correlations between BMI and rs10938397 (effect allele-G) near GNPDA2 (Padj=0.003), rs7498665 (effect allele-G) in SH2B1 (Padj=0.014), and with rs6548238 (effect allele-C) near TMEM18 (Padj=0.030).
To assess the combined impact of risk alleles on BMI, we calculated a risk alllele score (Figure 1), in which we included the three risk alleles described here, as well as four previously identified in the same cohort.8 The difference in average BMI between individuals with a high genetic-susceptibility score (defined as having ⩾10 BMI-increasing alleles) and those with a low genetic-susceptibility score (⩽4 BMI-increasing alleles) was 3.90 kg m−2, signifying a 21.7% increase in average BMI between these two groups. In comparison, if only the three SNPs from this paper is assesed, the increase in average BMI (comparing the lowest with the highest risk score) is 2.06 log kg m−2, signifying a 10.5% increase in average BMI between these two groups. This implies that both set of risk SNPs contribute to the effect that we see on BMI.

Combined impact of risk alleles on average BMI in the Bt20 cohort. Risk alleles were summed for each individual. The number of individuals in each risk allele category is shown along the x axis.
Discussion
Several common genetic variants have been robustly associated with adult obesity risk. This study provides confirmation that three of these variants are also associated with BMI in a South African cohort of adolescents. The G-allele of both rs7498665 (SH2B1) and rs10938397 (GNPDA2), and the C-allele of rs6548238 (TMEM18) were shown to be associated with an increase in BMI. A similar, albeit smaller, directional effect was observed to that seen in the discovery studies in non-African cohorts. These results demonstrate that genetic variants for adult BMI are also associated with BMI earlier in life, which may provide insights into the genetic aetiology of obesity within an indigenous African population.
GNPDA2, TMEM18 and SH2B1 are all highly plausable biological candidates for adiposity—all three are expressed in the brain, with evidence of a role in appetite regulation or affecting adipose tissue biology.16 GNPDA2 is expressed in the hypothalamus, alluding to a neuronal influence on energy balance, and has been associated with BMI in both pediatric17 and adult cohorts, including a replication in an African-American cohort.18 SH2B1’s link to metabolic function is well established19 and deletions in this gene are associated with severe early-onset obesity.20 Furthermore, rs7498665 in SH2B1 is a missense variant and results in a substitution of alanine with threonine, which likely affects protein activity and expression. TMEM18 is ubiquitously expressed, and although a direct link to obesity is still elusive, early evidence suggests a likely role through transcriptional regulation of critical targets.16
We acknowledge a number of limitations in our study. The sample size is moderate and therefore not powered to detect small effects on BMI, suggesting that the potential contribution of MTCH2 and STK33 warrants follow-up investigation in a larger cohort. The significant differences in the genomic structure between Africans and non-Africans genomes, could lead to a situation where SNPs shown to be associated with a trait in European populations may be weak predictors for causal variants in African populations, due to difference in linkage disequilibrium.21 Another consideration is that, although BMI is an established obesity index, it is not the best indicator of adiposity22 and the use of more suitable measures may help to elucidate the role of genetics in adiposity.
Finally, the data in this study are derived from a cohort of adolescents in the midst of puberty. It is therefore possible that the effects on weight of some polymorphisms may have been masked by puberty-associated changes in body fat mass. Furthermore, the effects of some polymorphisms on BMI are conceivably only observed later in life. Elucidating the genetic component of obesity in children is important because it may uncover factors that have a stronger phenotypic effect than those gene variants that only become apparent after years of exposure to an obesogenic environment. Also, gene variants than give rise to childhood obesity may provide important information on important metabolic or neurological pathways that could be therapeutically manipulated to reduce adipose tissue accumulation. The discovery of such polymorphisms may also help identify individuals with a high risk of obesity and hence allow early lifestyle interventions.
In conclusion, our study has replicated associations for increased BMI with SNPs present in or near TMEM18, SH2B1 and GNPDA2 in an African adolescent population. These observations suggest that variants in these genes or neighbouring loci may be important in body weight regulation in divergent populations.
References
Shishana O, Labadarios D, Rehle T, Simbayi L, Zuma K, Dhansay A et al The South African National Health and Nutrition Examination Survey, 2012: SANHANES-1: the health and nutritional status of the nation. HSRC Press, South Africa, 2014, p 424.
Reilly JJ, Kelly J . Long-term impact of overweight and obesity in childhood and adolescence on morbidity and premature mortality in adulthood: systematic review. Int J Obes (Lond) 2010; 35: 891–898.
Wardle J, Carnell S, Haworth CM, Plomin R . Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr 2008; 87: 398–404.
Bodurtha JN, Mosteller M, Hewitt JK, Nance WE, Eaves LJ, Moskowitz WB et al. Genetic analysis of anthropometric measures in 11-year-old twins: the Medical College of Virginia Twin Study. Pediatr Res 1990; 28: 1–4.
Fall T, Ingelsson E . Genome-wide association studies of obesity and metabolic syndrome. Mol Cell Endocrinol 2014; 382: 740–757.
Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015; 518: 197–206.
Loos RJ . Genetic determinants of common obesity and their value in prediction. Best Pract Res Clin Endocrinol Metab 2012; 26: 211–226.
Lombard Z, Crowther NJ, van der Merwe L, Pitamber P, Norris SA, Ramsay M . Appetite regulation genes are associated with body mass index in black South African adolescents: a genetic association study. BMJ Open 2012; 2: e000873.
Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010; 42: 937–948.
Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet 2009; 41: 25–34.
Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Helgadottir A et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet 2009; 41: 18–24.
Richter L, Norris S, Pettifor J, Yach D, Cameron N . Cohort Profile: Mandela's children: the 1990 Birth to Twenty study in South Africa. Int J Epidemiol 2007; 36: 504–511.
Cameron N, De Wet T, Ellison GT, Bogin B . Growth in height and weight of South African urban infants from birth to five years: the Birth to Ten Study. Am J Hum Biol 1998; 10: 495–504.
Norris SA, Richter L . Usefulness and reliability of tanner pubertal self‐rating to urban black adolescents in South Africa. J Res Adolesc 2005; 15: 609–624.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Speakman JR . Functional analysis of seven genes linked to body mass index and adiposity by genome-wide association studies: a review. Hum Hered 2013; 75: 57–79.
Zhao J, Bradfield JP, Li M, Wang K, Zhang H, Kim CE et al. The role of obesity-associated loci identified in genome-wide association studies in the determination of pediatric BMI. Obesity (Silver Spring) 2009; 17: 2254–2257.
Gong J, Schumacher F, Lim U, Hindorff LA, Haessler J, Buyske S et al. Fine mapping and identification of BMI Loci in African Americans. Am J Hum Genet 2013; 93: 661–671.
Ren D, Zhou Y, Morris D, Li M, Li Z, Rui L . Neuronal SH2B1 is essential for controlling energy and glucose homeostasis. J Clin Invest 2007; 117: 397–406.
Bochukova EG, Huang N, Keogh J, Henning E, Purmann C, Blaszczyk K et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature 2010; 463: 666–670.
Teo YY, Small KS, Kwiatkowski DP . Methodological challenges of genome-wide association analysis in Africa. Nat Rev Genet 2010; 11: 149–160.
Wells JC . Commentary: the paradox of body mass index in obesity assessment: not a good index of adiposity, but not a bad index of cardio-metabolic risk. Int J Epidemiol 2014; 43: 672–674.
Acknowledgements
We thank the participants in the Birth to Twenty study and Punita Pitamber for assistance with the laboratory work. This work is based on the research supported in part by the Wellcome Trust (grant number 080535/Z/06/Z to SAN); the Fogarty International Centre of the National Institutes of Health (Award number D43 TW008330); the South African Medical Research Council, the National Research Foundation of South Africa (grant number TTK20110713000020761 to ZL) and the University of the Witwatersrand for the collection of the samples for the Bt20 cohort and genotyping. In addition genotyping was funded by the South African National Research Foundation (grant number IFR2011062100004 to MR). The content is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health. Any opinion, finding and conclusion or recommendation expressed in this material is that of the authors and the NRF does not accept any liability in this regard.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pillay, V., Crowther, N., Ramsay, M. et al. Exploring genetic markers of adult obesity risk in black adolescent South Africans—the Birth to Twenty Cohort. Nutr & Diabetes 5, e157 (2015). https://doi.org/10.1038/nutd.2015.7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nutd.2015.7
