
Abstract
Obesity has drastically increased over the last few decades. Obesity is associated with elevated insulin levels, which can gain access to the brain, including into dopamine neurons of the ventral tegmental area (VTA), a brain region critical for mediating reward-seeking behavior. Synaptic plasticity of VTA dopamine neurons is associated with altered motivation to obtain reinforcing substances such as food and drugs of abuse. Under physiological circumstances, insulin in the VTA can suppress excitatory synaptic transmission onto VTA dopamine neurons and reduce aspects of palatable feeding behavior. However, it is unknown how insulin modulates excitatory synaptic transmission in pathological circumstances such as hyperinsulinemia. Using patch-clamp electrophysiology, we demonstrate that, in a hyperinsulinemic mouse model, insulin has reduced capacity to cause a synaptic depression of VTA dopamine neurons, although both low-frequency stimulation-induced long-term depression and cannabinoid-induced depression were normal. These results suggest that insulin action in the VTA during pathological hyperinsulinemia is disrupted and may lead to increased feeding behavior.
Similar content being viewed by others
Introduction
In recent times, hyperinsulinemia has been proposed not only to be associated with obesity but also to be a cause of obesity (reviewed in the study by Shanik et al.1). Hyperinsulinemia can result in insulin receptor insensitivity leading to type 2 diabetes.1 Dopamine neurons of the ventral tegmental area (VTA) have been implicated in the incentive, reinforcing and motivational aspects of food intake.2 We have recently demonstrated that insulin either applied exogenously or elevated by a sweetened high-fat meal can induce long-term depression (LTD) at excitatory synapses onto dopamine neurons.3 This reduction of synaptic efficacy in the VTA is linked to a reduced anticipatory activity or preference for palatable food. Thus, under physiological circumstances, insulin action in the VTA has a vital role in regulating food intake by decreasing salience for food-related cues after a meal. However, it is unknown how insulin regulates dopamine neurons of the VTA under pathological circumstances such as hyperinsulinemia. BTBR T+ Itpr3tf/J (BTBR) mice constitute a hyperinsulinemic mouse strain that is predisposed to obesity.4, 5 These mice have a mutation in the Itpr3 gene at the Tufted locus, which gives rise to tufted hair in older mice as well as to different taste perceptions due to alterations in the taste receptor Inositol 1,4,5-trisphosphate receptor, type 3.6 Furthermore, the BTBR mouse strain has elevated plasma insulin levels relative to the C57BL/6 J strain and has a higher fat mass than many other inbred mouse strains.4, 5 Therefore, we hypothesized that insulin-induced LTD of VTA neurons of BTBR mice would be disrupted.
Materials and Methods
Animals
All protocols were in accordance with the ethical guidelines established by the Canadian Council for Animal Care and were approved by the University of British Columbia or University of Calgary Animal Care Committees. C57BL/6 J mice were obtained from the University of British Columbia breeding facility or Jackson Laboratories (Sacramento, CA, USA). BTBR mice were obtained from the Jackson Laboratory and bred in the UBC facility. Both strains were fed chow ad libitum (Teklad 2918).
Electrophysiology
All electrophysiological recordings were performed in male mice ranging from P19 to P30 as per.3 Slices in the recording chamber were superfused with bicarbonate-buffered solution (artificial cerebrospinal fluid) saturated with 95% O2/5% CO2 and containing (in mM) 126 NaCl, 1.6 KCl, 1.1 NaH2PO4, 1.4 MgCl2, 2.4 CaCl2, 26 NaHCO3 and 11 glucose as well as picrotoxin (100 μM) (at 32–34 °C). Electrodes (3–4.5 MΩ) contained (in mM) 117 cesium methansulfonate, 20 HEPES, 0.4 EGTA, 2.8 NaCl, 5 TEA-Cl, 2.5 MgATP and 0.25 NaGTP (pH 7.2–7.3, 270–285 mOsm). Series resistance (10–25 MΩ) and input resistance were monitored online with a 10 mV depolarizing step (400 ms) given before every afferent stimulus. Dopamine neurons were identified by the presence of a hyperpolarizing cation current (Ih), which is a good predictor of tyrosine hydroxylase (TH)+ neurons in mice.7 A bipolar stimulating electrode was placed 100–300 μm rostral to the recording electrode and was used to stimulate excitatory afferents at 0.1 Hz.
Immunohistochemistry
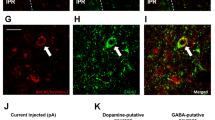
Coronal brain sections (30 μm) were fixed in 4% paraformaldehyde overnight, blocked with 5% goat serum/0.3% Triton X-100/0.2% bovine serum albumin in phosphate-buffered saline (pH 7.4) for 2 h at room temperature and then incubated for 48 h at 4 °C with mouse TH (1:1000) and rabbit insulin receptor (1:100) monoclonal antibodies. The sections were then washed and incubated for 2 h at room temperature with donkey anti-mouse Texas Red (1:50) and goat anti-rabbit FITC (1:50) secondary antibodies. Slices were washed and mounted onto slides and coverslipped (Fluromount, Sigma, Oakville, ON, Canada). Immunofluorescent images were captured using an FV10i Olympus confocal laser scanning microscope with a × 60 phase-contrast oil-immersion objective/NA 1.35. Immunoflurescence was quantified after background subtraction using Image J software.
Results
Consistent with previous reports,4, 5 fasted plasma insulin concentrations (measured as in3 were significantly higher in BTBR mice than in C57BL/6J mice (BTBR: 2.9±0.6 ng ml−1, n=8; C57BL/6 J: 1.3±0.3 ng ml−1, n=7; P<0.05, t-test, t=2.357, d.f.=13). To test the effects of insulin on excitatory synaptic transmission, we recorded 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl) propanoic acid receptor (AMPAR)-mediated excitatory postsynaptic currents (EPSCs) evoked in VTA dopamine neurons, voltage clamped at −60 mV in midbrain slices acutely obtained from BTBR or C57BL/6 mice. Insulin induced an LTD to a maximum of 65% of baseline 30 min after a 10 min insulin application to VTA brain slices of C57BL/6J mice (Figures 1a and b; n=7, P<0.05, paired t-test baseline vs 30 min after insulin application t=6.15, d.f.=6). In contrast, although insulin caused a modest transient depression of AMPAR EPSCs (82±9% 14 min after insulin), it did not induce a significant LTD (104±8% of baseline 30 min post insulin) in dopamine neurons of BTBR mice (Figures 1c and d; n=7 ). To determine whether the disrupted insulin-induced LTD was due to differential insulin receptor expression in the VTA between the strains, we used immunohistochemical analysis to test the colocalization of insulin receptors on TH-containing neurons within the VTA. Insulin receptor expression on TH+ neurons was similar for BTBR and C57BL/6J mice (relative intensity: 345±22, n=20 vs 371±58, n=20; P>0.05, t-test, t=0.42, d.f.=38; Figure 1e). To determine whether insulin-induced LTD in BTBR mice was deficient because of the inability to induce a synaptic depression in VTA dopamine neurons, we tested whether synapses could undergo LTD induced by low-frequency stimulation (−40 mV, 6 min, 1 Hz stimulation3, 8). Low-frequency stimulation induced a significant LTD at excitatory synapses of VTA dopamine neurons in BTBR mice (66±5% of baseline at 30 min, Figures 2a and b, P<0.05, paired t-test, t=4.79, d.f.=5), which was not different from that observed in C57BL/6J mice (63±4%; Figure 2a, P>0.05, t-test, t=0.40, d.f.=7). Insulin-induced LTD required synthesis of endocannabinoids that act retrogradely at cannabinoid-1 receptors to depress glutamate release onto VTA dopamine neurons.3 To determine whether the inability of insulin to induce LTD in the VTA of hyperinsulinemic mice was due to insufficient CB1R-dependent synaptic depression, we bath applied the CB1R agonist WIN55232-2 (1 μM) for 5 min to VTA slices. WIN55232-2 significantly decreased AMPAR EPSCs (55±4%; Figures 2c and d; n=5, P<0.05, paired t-test, t=10.13, d.f.=4) to a similar maximum as C57BL/6J mice (62±6%; Figure 2c, n=4, P>0.05, t-test, t=0. 98, d.f.=7). Taken together, these data suggest that, whereas regulation of synaptic efficacy at VTA synapses of hyperinsulinemic BTBR mice is normal, insulin-induced LTD is deficient.

Insulin-induced LTD is disrupted in hyperinsulinemic BTBR mice. AMPAR EPSCs recorded at −60 mV were evoked using a bipolar stimulating electrode placed 100–300 μM rostrally to the recorded neuron. (a) Bath application of insulin (500 nM, 10 min) to VTA slices of C57BL/6 J mice induced an LTD (n=7). (b) Example time course of AMPAR EPSC amplitude in a single dopamine neuron from a C57BL6 mouse. Example recordings of AMPAR EPSCs at 5 (black) and 40 (gray) min are shown above the time course. Scale bars, 5 ms and 50 pA. (c) Bath application of insulin (500 nM, 10 min) to VTA slices of BTBR mice did not induce LTD (n=6). (d) Example time course of AMPAR EPSC amplitude in a single dopamine neuron from a BTBR mouse. Example recordings of AMPAR EPSCs at 5 (black) and 40 (gray) min are shown above the time course. Scale bars, 5 ms and 50 pA. (e) Immunostaining of insulin receptors labeled with anti-IR β subunit and FITC (left) and tyrosine hydroxylase (TH) labeled with anti-TH antibodies and Texas red (middle) taken from VTA coronal slices from C57BL/6 J (top) or BTBR (bottom) mice. Scale bars represent 20 μm. Insets represent a single TH+ neuron at higher magnification (scale bars=5 μm).

Excitatory synaptic transmission is normal in BTBR mice. (a) WIN 55212-2 (1 μM, 5 min) induced equivalent LTD of AMPAR EPSCs in BTBR (n=6, filled circles) or C57BL6/J mice (n=5, open circles). Inset, example traces of AMPAR EPSCs at 5 (black) and 40 (gray) min for BTBR (filled circle) or C57BL6/J (open circle) mice. Scale bars, 5 ms and 50 pA. (b) Example time course of WIN 55212-2-induced LTD. (c) Low–frequency-stimulation-induced LTD in VTA dopamine neurons of BTBR (n=5, filled circles) or C67BL6/J mice (n=4, open circles). Inset, example traces of AMPAR EPSCs at 5 (black) and 40 (gray) min from BTBR (filled circle) or C57BL6/J mice (open circle). Scale bars, 5 ms and 50 pA. Stimulus artifacts have been removed for clarity. (d) Example time course of low-frequency stimulation-induced LTD in BTBR mice. Error bars represent s.e.m.
Discussion
Obesity-associated insulin resistance has been reported in the brain.9 Several possible explanations exist for the inability of insulin to induce an LTD at excitatory synapses of dopamine neurons. In physiological situations, insulin gains access to the brain by active transport across the blood–brain barrier 10 and can mediate its effects by signaling through insulin receptors expressed throughout the brain.11 Here, we observed a similar expression pattern of insulin receptors in TH+ neurons in BTBR mice as in C57BL/6J mice, suggesting that disrupted insulin-induced LTD in BTBR mice is not due to reduced insulin receptor expression. BTBR mice have higher fat mass than C57BL/6 J mice5 likely resulting in higher circulating leptin. As leptin decreases glutamatergic release onto dopamine neurons,12 it is possible that insulin-induced endocannabinoids acting presynaptically at CB1 receptors to inhibit glutamate release have a blunted effect because of high leptin levels lowering the release probability. However, this is unlikely as a CB1 receptor agonist induced a similar LTD in BTBR or C57BL/6 J mice. Alternatively, disrupted insulin-induced LTD may be due to insulin resistance because of impaired signaling at the level of the insulin receptor or its downstream effectors, due to genetic differences in insulin receptors, cognate signaling pathways or hyperinsulinemia. Indeed, others have demonstrated that diet-induced hyperinsulinemia can induce impaired insulin signaling in hippocampal neurons.13, 14 Hyperinsulinemia can disrupt aspects of synaptic transmission in the hippocampus. For example, LTD was not present15 and LTP was reduced along with a reduction in spine density16 in hippocampal neurons from high-calorie-diet-fed mice compared with controls. In contrast, others have observed that hyperinsulinemia caused no change in plasticity at the hippocampal synapses but a reduction in the ability of insulin to induce LTD in CA1 neurons.13 Consistent with this, we found that hyperinsulinemia did not alter the plasticity of excitatory synapses onto VTA neurons but reduced insulin-induced LTD.
Our results imply that reduced insulin receptor efficacy in the VTA may promote obesity. Insulin action in the VTA decreases opioid-stimulated food intake,17 food anticipatory behavior, preference for food3 and palatable food intake when sated.18 Enhanced synaptic transmission in the VTA has been associated with learning about cues that predict food delivery.19 Therefore, one may speculate that a suppression of excitatory synaptic transmission in the VTA by post-ingestive insulin release makes food-predicting cues less salient. An inability of insulin to dampen salience to food-predicting cues may lead to increased caloric intake, even when sated. In summary, our results suggest that, in hyperinsulinemic mice, insulin in the VTA does not cause an LTD of excitatory inputs to dopamine neurons, and thus information relayed to the VTA about cues predicting food is not suppressed after feeding when brain insulin levels are normally elevated.
References
Shanik MH, Xu Y, Skrha J, Dankner R, Zick Y, Roth J . Insulin resistance and hyperinsulinema: is hyperinsulinemia the cart or the horse? Diabetes Care 2008; 31: S262–S268.
Palmiter RD . Is dopamine a physiologically relevant mediator of feeding behavior? Trends Neurosci 2007; 30: 375–381.
Labouèbe G, Liu S, Dias C, Zou HY, Wong CYJ, Karunakaran S et al. Insulin mediates long term depression of VTA dopamine neurons via an endocannabinoid mediated mechanism. Nat Neurosci 2013; 16: 300–308.
Stoehr JP, Nadler ST, Schueler KL, Rabaglia ME, Yandell BS, Metz SA et al. Genetic obesity unmasks nonlinear interactions between murine type 2 diabetes susceptibility loci. Diabetes 2000; 49: 1946–1954.
Flowers JB, Oler AT, Nadler ST, Choi Y, Schueler KL, Yandell BS et al. Abdominal obesity in BTBR male mice is associated with peripheral but not hepatic insulin resistance. Am J Physiol Endocrinol Metab 2007; 292: E936–E945.
Ellis HT, Tordoff MG, Parker MR . Itpr3 is responsible for the mouse tufted (tf) locus. J Hered 2013; 104: 295–297.
Wanat MJ, Hopf FW, Stuber GD, Phillips PE, Bonci A . Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol 2008; 586: 2157–2170.
Bonci A, Malenka RC . Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J Neurosci 1999; 19: 3723–3730.
Gerozissis K . Brain insulin, energy and glucose homeostasis; genes, environment and metabolic pathologies. Eur J Pharmacol 2008; 585: 38–49.
Woods SC, Seeley RJ, Baskin DG, Schwartz MW . Insulin and the blood-brain barrier. Curr Pharm Des 2003; 9: 795–800.
Woods SC, D'Alessio DA . Central control of body weight and appetite. J Clin Endocrinol Metab 2008; 93: S37–S50.
Thompson JT, Borgland SL . Presynaptic leptin action suppresses excitatory synaptic transmission onto ventral tegmental area dopamine neurons. Biol Psych 2013; 73: 860–868.
Mielke JG, Taghibiglou C, Liu L, Zhang Y, Jia Z, Adeli K et al. A biochemical and functional characterization of diet-induced brain insulin resistance. J Neurochem 2005; 93: 1568–1578.
Battú CE, Rieger D, Loureiro S, Furtado GV, Bock H, Saraiva-Pereira ML et al. Alterations of PI3K and Akt signaling pathways in the hippocampus and hypothalamus of Wistar rats treated with highly palatable food. Nutr Neurosci 2012; 15: 10–17.
Hwang LL, Wang CH, Li TL, Chang SD, Lin LC, Chen CP et al. Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity 2010; 18: 463–469.
Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM et al. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus 2008; 18: 1085–1088.
Sipols AJ, Bayer J, Bennett R, Figlewicz DP . Intraventricular insulin decreases kappa opioid-mediated sucrose intake in rats. Peptides 2002; 23: 2181–2187.
Mebel DM, Wong JCY, Dong JY, Borgland SL . Insulin in the ventral tegmental area modulates somatodendritic dopamine release and hedonic feeding. Eur J Neurosci 2012; 36: 2336–2346.
Stuber GD, Klanker M, de Ridder B, Bowers MS, Joosten RN, Feenstra MG et al. Reward-predictive cues enhance excitatory synaptic strength onto midbrain dopamine neurons. Science 2008; 321: 1690–1692.
Acknowledgements
We thank Katie Lee for performing the insulin measurements. This research was supported by a CIHR operating grant and a CIHR new investigator award to SLB. GL was supported by a Swiss National Science Foundation postdoctoral fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Liu, S., Labouèbe, G., Karunakaran, S. et al. Effect of insulin on excitatory synaptic transmission onto dopamine neurons of the ventral tegmental area in a mouse model of hyperinsulinemia. Nutr & Diabetes 3, e97 (2013). https://doi.org/10.1038/nutd.2013.38
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nutd.2013.38
Keywords
This article is cited by
-
Genome-wide association study of psychiatric and substance use comorbidity in Mexican individuals
Scientific Reports (2021)
-
Neuroprotective Actions of Glucagon-Like Peptide-1 (GLP-1) Analogues in Alzheimer’s and Parkinson’s Diseases
CNS Drugs (2019)
-
Impact of Brain Insulin Signaling on Dopamine Function, Food Intake, Reward, and Emotional Behavior
Current Nutrition Reports (2019)
-
Central insulin modulates food valuation via mesolimbic pathways
Nature Communications (2017)
