
Abstract
Objective:
Initiation and intensification of insulin therapy commonly causes weight gain, a barrier to therapy. A contrasting body of evidence indicates that insulin functions as an adiposity negative feedback signal and reduces food intake, weight gain and adiposity via action in the central nervous system. Basal insulin analogs, detemir (Det) and glargine (Glar), have been associated with less hypoglycemia compared with neutral protamine hagedorn insulin, and Det with less weight gain, especially in patients with higher body mass index (BMI). We sought to determine whether insulin therapy per se causes body weight and fat mass gain when delivered via a clinically relevant subcutaneous (SC) route in the absence of hypoglycemia and glycosuria in non-diabetic lean and diet-induced obese rats.
Materials and methods:
Rats were exposed to either a low-fat diet (LFD; 13.5% fat) or high-fat diet (HFD; 60% fat), and received Det (0.5 U kg−1), Glar (0.2 U kg−1) or vehicle (Veh) SC once daily for 4 weeks. These dosages of insulin were equipotent in rats with respect to blood–glucose concentration and did not induce hypoglycemia.
Results:
As predicted by current models of energy homeostasis, neither insulin Det nor Glar therapy affected food intake and weight gain in LFD rats. Det treatment significantly attenuated food intake, body weight gain and fat mass gain relative to the Glar and Veh in high-fat fed animals, mirroring observations in humans.
Conclusions:
That neither insulin group gained excess weight, suggests weight gain with SC basal insulin therapy may not be inevitable. Our data further suggest that Det possesses a unique property to attenuate the development of obesity associated with a HFD.
Similar content being viewed by others

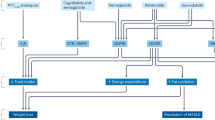

Introduction
Together with obesity, diabetes is epidemic in the US and worldwide.1 Weight gain is both a risk factor for diabetes and a consequence of initiation and intensification of insulin therapy, as illustrated in landmark diabetes control trials.2, 3, 4 Weight gain associated with increased adiposity can worsen insulin resistance leading to higher insulin requirements, and thus perpetuates a vicious cycle. Weight gain additionally generates further risk for macrovascular outcomes such as cardiovascular disease5, 6 and generates negative ‘biofeedback’ to the patient and physician struggling to achieve glycemic control.
Mechanisms involved in weight gain associated with insulin therapy are incompletely understood. Hypoglycemia is a potent stimulus to feed,7 improving glycemic control reverses the negative energy balance associated with glycosuria, and insulin potently stimulates lipogenesis. Indeed insulin is an anabolic hormone in peripheral tissues.8 Although weight gain on insulin initiation or intensification is common, there are reports in randomized, controlled human clinical trials of attenuation of this effect associated with the use of a basal insulin analog, detemir (Det), compared with other insulins used in subjects with type 1 and type 2 diabetes9 (as reviewed in Hermansen and Davies10). Intriguingly, the observed weight sparing effects of Det appear to be amplified in the most obese subjects,9 who are predicted to be more insulin resistant. Det has also been shown to have enhanced effects on central nervous system (CNS) function in both experimental animals11 and human subjects.12, 13 So-called basal insulins are long-acting insulin formulations that maintain euglycemia between meals and overnight (that is, fasting) primarily by limiting hepatic glucose production.14 Frequently, type 2 diabetes insulin therapy is initiated with basal insulin.
In contrast to ample evidence associating insulin therapy with increased adiposity and weight gain, a distinct body of evidence indicates that insulin functions as an adiposity negative feedback signal to the CNS.15 Insulin (and leptin) is secreted in proportion to body fat stores,16 and regulates the function of key homeostatic neurons found in the mediobasal hypothalamus17 and elsewhere,18 which in turn, control feeding, energy expenditure and other aspects of neuroendocrine physiology (reviewed in Niswender et al.17 and Plum et al.19). Although a potent anabolic stimulus in peripheral tissues, insulin action in the CNS is catabolic in the sense that it reduces food intake, weight gain and adiposity. Thus, a paradox emerges when one considers that insulin therapy is associated with weight gain, whereas in experimental situations insulin functions to limit weight gain.
A number of elegant basic science approaches have clarified the role of insulin in energy homeostasis. Insulin administration directly into the brain reduces food intake20, 21, 22 and targeted mutation of insulin signaling components in neurons leads to weight gain.23, 24 Here, we sought to engage a more clinically relevant approach to the question of whether it is insulin therapy per se (rather than hypoglycemia, reversal of negative energy balance and so on) that causes weight gain in a well-controlled preclinical model. Herein we tested the chronic effects of modern basal insulin analogs, Det and glargine (Glar). As basal insulins, Det and Glar, are used clinically to control between meal and overnight hyperglycemia,14 and were chosen here because they could be delivered by the clinically relevant subcutaneous (SC) route without causing hypoglycemia due to their pharmacodynamic profiles.25 We have controlled for confounding variables such as glycosuria, negative energy balance and hypoglycemia by studying non-diabetic, but high-fat (HF) fed diet-induced obese rats compared with low-fat (LF) fed rats. Thus, we used long-acting basal insulin analogs to replace some percentage of endogenous basal insulin (that rate needed to maintain euglycemia in the fasting state) allowing us to avoid hypoglycemia and thereby to test the effects of these insulins per se on feeding and weight gain. We hypothesized that the anabolic effects of insulin in peripheral tissues will be balanced by the catabolic effects in the CNS when confounders are controlled, and that Det might attenuate weight gain, especially on HF diet. Thus, in the absence of negative energy balance and hypoglycemia, we sought to ask whether chronic SC basal insulin therapy still leads to weight gain and increased adiposity and whether we can model the observed effects of Det to limit weight gain in the setting of obesity.
Materials and methods
Animals and dietary treatment
After an acclimation period, individually housed male Sprague–Dawley rats (250–274 g, Harlan, Indianapolis, IN, USA) were fed a LF diet (LFD; 5001, Lab diet, Richmond, IN, USA; 3.02 kcal g−1, 58% carbohydrate, 28.5% protein, 13.5% fat; n=15) or a HF diet (HFD; D12492, Research Diets, New Brunswick, NJ, USA; 5.24 kcal g−1, 20% carbohydrate, 20% protein, 60% fat; n=21) to induce obesity.26 Food intake and body weight were measured daily and adiposity measured three times weekly using an EchoMRI-700 nuclear magnetic resonance (NMR, Echo Medical Systems, Houston TX, USA). All experimental protocols were approved by the Vanderbilt Institutional Animal Care and Use Committee.
Insulin preparation
Det (Levemir; 100 U ml−1; Novo Nordisk, Bagsvaerd, Denmark) was freshly prepared daily in Det dilution media (140 mM NaCl, 70 p.p.m. Tween-20, 5 mM NaH2PO4, pH 7.4) and administered SC at 0.5 U kg−1 body weight in 250 μl. Similarly, Glar (Lantus; 100 U ml−1; Sanofi-Aventis, Paris, France) was freshly prepared daily in Glar dilution media (1 ml contains 30 μg zinc, 20 mg of 85% glycerol, 20 μg Tween-20, pH 4.0) dosed SC at 0.2 U kg−1 body weight in 250 μl. Modestly, insulin resistant humans might use between 0.2–0.8 U basal insulin per kg body weight per 24 h (KD Niswender, personal experience). All the insulin was purchased from the Vanderbilt Outpatient Pharmacy (Nashville, TN, USA). Vehicle (Veh) treated rats received either Det dilution media or Glar dilution media in the same volume as the insulin treated groups (250 μl). Outcomes of both Veh groups were not different and were combined for presentation.
Insulin dosing and equipotency
Different insulin formulations have varying potencies among species,27 and insulin is used clinically for glycemic control. Therefore, we sought to conduct our studies at dosages of Det and Glar that were equipotent in rats. After a baseline blood–glucose measurement, SC insulin injections were administered and blood samples were collected by tail prick at 30-min intervals up to 360 min. Blood–glucose concentration was measured using a glucometer (Cardinal Health, Dublin, OH, USA). Extensive insulin tolerance testing (ITT, data not shown) revealed that a dose of 0.5 U kg−1 body weight of Det was equal to 0.2 U kg−1 body weight of Glar (2.5:1 unit ratio, Det:Glar) for effects on glycemia. This relative potency in rats was the same as observed by other investigators using hyperinsulinemic–euglycemic clamp techniques in rats.27
Study design
Animals were randomized to receive Det (n=11), Glar (n=13) or Veh (n=12). Insulin or Veh was administered in the subscapular region daily at the onset of the dark cycle for the initial 4-week acclimation period of LF-feeding. Subsequent to acclimation, at day 0, a subset of animals from each treatment group were randomly assigned to HFD (n=21; 6 Det, 8 Glar and 7 Veh) or remained on LFD (n=15; 5 Det, 5 Glar, 5 Veh), and all animals continued to receive the same insulin or Veh treatments for an additional 4 weeks.
Indirect calorimetry
Indirect calorimetry was performed on LF- (n=7–8 per group) and HF-fed (n=6–8 per group) rats using an open-circuit indirect calorimeter (Oxymax; Columbus Instruments, Columbus, OH, USA). Average respiratory quotient (RQ) was calculated as the molar ratio of VCO2:VO2 in a 24-h period. Energy expenditure (kcal per day) was calculated as heat (kcal /h−1)=(3.815+1.232 × RQ) × VO2.28 Energy balance (kcal per day) was calculated from the energy expenditure (kcal per day) minus the energy intake (kcal per day). Rats received their respective insulin or Veh treatments before onset of the dark cycle period during calorimetry.
Study termination
Four hours after receiving SC injection of their respective treatments both LFD and HFD groups were euthanized. Plasma was collected, as were epididymal white adipose tissue (snap frozen on dry ice), and stored at −80 °C until assayed.
RNA quantification using real-time PCR
Epididymal white adipose tissue samples were homogenized and RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). From these RNA samples, complementary DNA (cDNA) was synthesized using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR was conducted using standard curves in each assay (six-point serial dilution of cDNA synthesized from XpressRef rat universal total RNA; SuperArray Bioscience, Frederick, MD, USA), IQ SYBR Green Supermix and performed in triplicate using the I-Cycler thermal cycler (Bio-Rad, Hercules, CA, USA) with 1 cycle of 95 °C for 3 min followed by 40 cycles of 95 °C for 30 s, optimal annealing temperature for 30 s and 72 °C for 30 s. Only experiments with >90% efficiency of amplification were used. Quantification results for each RNA of interest were normalized to the housekeeping gene, ribosomal protein L13α (RPL13α) and for comparative analysis, RNA ratios of HF and LF groups were normalized to the Veh-treated LF control group. The sequences and optimal annealing temperature of each primer pair used for RNA quantification are available on request.
Lipid assays
Lipids were assayed from plasma with enzymatic colorimetric assays using the following reagent kits; triglycerides and total cholesterol (Raichem, San Diego, CA, USA), non-esterified fatty acids (Wako Diagnostics, Richmond, VA, USA) and glycerol (Sigma-Aldrich, St Louis, MO, USA). The reported intra-assay coefficient of variation (CV) for all lipid assays is within 1%.
Plasma hormones and metabolites
Plasma levels of leptin (Rat Leptin RIA kit, Millipore, Billerica, MA, USA) and corticosterone (ImmuChem Double Antibody RIA kit, MP Biomedicals, Solon, OH, USA) were quantified using radioimmunoassay by the Vanderbilt Diabetes Center Hormone Assay and Analytical Services Core (Nashville, TN, USA). The reported CVs for leptin and corticosterone were 2–5% and 4%, respectively. Adiponectin was assessed by ELISA (RT Biosource, Invitrogen, Camarillo, CA, USA) with a reported CV of 2–4%.
Statistical analysis
Data are presented as the mean±s.e.m. Equipotency and lack of hypoglycemia in the ITT testing of Det vs Glar treatment were analyzed using Student's two-tailed, unpaired t-test. The main effect of diet and insulin therapy on food intake, body weight and adipose mass over the 4-week period were compared by two-way repeated measures analysis of variance (ANOVA) with Bonferroni's post-test analysis. The main effect of diet and insulin therapy on body weight gain and fat mass gain, energy homeostasis and metabolic parameters were compared using two-way ANOVA with Bonferroni's post-test analysis. All analysis was performed using Prism, version 5.02, 2008 (GraphPad Software, San Diego, CA, USA). Differences of P<0.05 were considered statistically significant.
Results
Insulin potency and dosing
We sought to study the effects of insulin Det and Glar on food intake and weight gain at doses that were equipotent with respect to blood–glucose concentration, since insulin is used clinically for glycemic management. In preliminary ITTs (data not shown), we determined that a dose of 0.5 U kg−1 body weight of Det was equipotent with that of 0.2 U kg−1 body weight of Glar with respect to changes in blood glucose, which is consistent with observations of others in rats.27 Because hypoglycemia is a potent hyperphagic stimulus,29 additional ITTs were conducted during this study in both HF- and LF-fed rats receiving Det (0.5 U kg−1) and Glar (0.2 U kg−1) to assess insulin-induced hypoglycemia (Figure 1). These doses of insulin did not induce hypoglycemia with blood glucose at 90 min in LF-fed rats (Figure 1a; Glar 5.1±0.1 mmol l−1 vs Det 5.2±0.1 mmol l−1, P=NS) and in HF-fed rats (Figure 1b; Glar 4.7±0.1 mmol l−1 vs Det 4.7±0.1 mmol l−1, P=NS). ITTs were also conducted in animals included in this study covering 24 h of insulin exposure to assess for a delayed onset of insulin-induced hypoglycemia and to assure that the doses of insulin remained equipotent. Twenty hours after insulin injection blood–glucose concentrations were not significantly different among the HFD (5.6±0.1 mmol l−1 for Glar and 5.3±0.1 mmol l−1 for Det, P=NS) and LFD (5.3±0.1 mmol l−1 for Glar vs 5.3±0.1 mmol l−1 for Det, P=NS) groups. In a separate study, we observed that higher doses of insulin did significantly lower glycemia and induce compensatory increases in feeding (data not shown).

Glycemic effects of Det and Glar in rats. (a, b) Blood glucose was determined at indicated time points after SC insulin administration of Det 0.5 U kg−1 (triangle) or Glar 0.2 U kg−1 (diamond) in LF-fed rats (a; n=11–13 per group) or HF-fed rats (b; n=6–8 per group). Data are presented as the mean±s.e.m. and were analyzed by Student's two-tailed, unpaired t-test; P=NS, Glar vs Det.
Food intake
Here, we sought to determine the effects of relatively low-dose basal insulin therapy on food intake in both LF- and HF-fed animals. We observed in the LFD groups that neither insulin Det or Glar increased nor decreased cumulative food intake (Figures 2a and c) compared with Veh. Conversely, Det reduced cumulative food intake in HF-fed rats by 7% relative to Veh and by 10% relative to Glar (whereas Glar had no effect compared with Veh) resulting in a lower 4-week cumulative food intake (Figures 2b and c; Veh 2296±55 kcal, Glar 2379±72 kcal, Det 2130±47 kcal; Veh vs Det P<0.05, Glar vs Det P<0.001, Veh vs Glar P=NS). Clearly, the effect of Det to reduce food intake was dependent on HFD intake (F(5, 30)=44.82; P<0.0001).

Effects of Det and Glar on food intake in LFD and HFD rats. Food intake was measured daily for each treatment group, Veh (circle), Det (triangle) and Glar (diamond); and presented as cumulative food intake in rats fed either LFD (a; n=5 per group) or HFD (b; n=6–8 per group). Total cumulative food intake for each treatment and diet group is shown (c). Data are presented as the mean±s.e.m. and were analyzed by two-way repeated measures ANOVA. Bonferroni's post-test shows that Det reduced cumulative food intake in HF-fed rats relative to Glar from 10 to 27 days and relative to Veh from 24 to 27 days. For all panels: *P<0.05 for Veh vs Det; #P<0.05, αP<0.01 and βP<0.001 for Glar vs Det comparisons as analyzed by Bonferroni's post-test.
Body weight and adipose mass
Because we observed significant attenuation of cumulative food intake in HF-fed rats receiving Det, the effect of basal insulin therapy on body weight and fat mass gain was of primary interest. Consistent with the food intake data in the LFD group neither Det nor Glar therapy had any effect on daily body weight and fat mass (Figures 3a and d) or body weight or fat mass gain (Figures 3c and f). In contrast, in HFD rats, Det trended to attenuate daily body weight (Figure 3b), but this did not reach statistical significance for any individual day over the 4-week period. Det did, however, reduce body weight gain relative to both Veh and Glar, whereas Glar had no effect compared with Veh (Figure 3c; Veh 72.0±4.8 g, Glar 72.5±4.0, Det 59.2±3.5; Veh vs Det P<0.05, Glar vs Det P<0.05, Veh vs Glar P=NS). The effect of Det to reduce body weight was clearly dependent on HFD intake (F(5, 30)=6.10; P<0.001).

Insulin Det attenuates weight and adipose gain in HFD, but not LFD rats. Body weight was measured daily in rats fed either LFD (a; n=5 per group) or HFD (b; n=6–8 per group) for each treatment group; Veh (circle), Det (triangle) and Glar (diamond). Body weight gain was determined for the 4-week study period for all groups (c). Fat mass was measured three times weekly by NMR in rats fed either LFD (d; n=5 per group) or HFD (e; n=6–8 per group) for each treatment group; Veh (circle), Det (triangle) and Glar (diamond). Fat mass gain was determined for the 4-week study period for all groups (f). Data are presented as the mean±s.e.m. and were analyzed by either two-way repeated measures ANOVA (for body weight and fat mass) or two-way ANOVA (for body weight and fat mass gain). Bonferroni's post-test shows that daily fat mass (e) was reduced by Det relative to Veh from 13 to 26 days and relative to Glar from 10 to 26 days. For all panels: *P<0.05, **P<0.01, and ***P<0.001 for Veh vs Det; #P<0.05, αP<0.01 and βP<0.001 for Glar vs Det comparisons as analyzed by Bonferroni's post-test.
Fat mass was significantly reduced by Det compared with both Veh and Glar on HFD (Figure 3e, P<0.05 from day 10 onward). Fat mass gain was significantly lower in the Det group relative to Veh and Glar, Glar had no effect compared with Veh (Figure 3f; Veh 36.6±2.9 g, Glar 39.2±3.3 g, Det 25.6±2.9 g; Veh vs Det P<0.05, Glar vs Det P<0.01, Veh vs Glar P=NS). The effect of Det to reduce fat mass was clearly dependent on HFD intake (F(5, 30)=8.15, P<0.0001).
Energy balance
We assessed energy expenditure in LF- and HF-fed rats by indirect calorimetry using an open-circuit indirect calorimeter. No significant differences in energy expenditure were noted (Table 1). HF-fed rats treated with Det had reduced calculated positive energy balance relative to Veh (Veh vs Det, P<0.05) and to Glar (Glar vs Det, P<0.01) and this was due entirely to a significant reduction in energy intake (Table 1; Veh vs Det, P<0.05 and Glar vs Det, P<0.001). Also as expected, lower RQ values were observed among the HFD groups in comparison to LFD groups indicating that the HF-fed animals were oxidizing fats to a greater extent as their primary fuel substrate (F(1, 38)=678.40; P<0.0001).
Plasma lipids, hormones and metabolites
We assessed several plasma lipid, hormonal and metabolite markers to determine whether insulin therapy in HF-fed rats alters the metabolic milieu. We observed that only Glar treatment in the LFD and not the HFD rats had significantly lower plasma triglycerides (TG) relative to Veh (Table 2; Veh vs Glar, P<0.05). Overall, we observed no effects of insulin Det or Glar therapy on total cholesterol (Chol), free fatty acids (FFA) or glycerol (Table 2). We measured adiponectin (ADN) levels as an index of adiposity and insulin sensitivity,30 and no differences were observed (Table 2). Corticosterone (Cort) levels were quantified as an index of potential counter-regulatory response to unrecognized insulin-induced hypoglycemia,31 and no differences were observed (Table 2). As might be predicted with lower fat mass in the HFD group, Det-treated animals had lower plasma leptin levels compared with Veh- and Glar-treated animals (Table 2). However, the effect of Det to reduce leptin in HF-fed rats did not reach statistical significance (F(2, 28)=1.65; P=0.21).
White adipose tissue markers of insulin action
Insulin is a potent stimulator of lipogenesis,32 thus we assessed the insulin-sensitive transcriptional markers,33 fatty acid synthase (FAS) and lipoprotein lipase (LPL), by real-time PCR on cDNA prepared from epididymal white adipose tissue (WAT) samples to determine whether adipose-specific effects could be identified. Glar treatment in HFD rats significantly increased FAS RNA levels relative to Veh and Det, whereas Det had no effect compared with Veh (Figure 4a; Veh 1.2±0.3, Glar 2.1±0.4, Det 1.1±0.1; Veh vs Glar P<0.05, Glar vs Det P<0.05, Veh vs Det P=NS). Clearly, the effect of Glar to increase FAS RNA expression was dependent on HFD intake (F(2, 30)=7.69; P<0.01). Glar-treated LFD rats had increased LPL RNA relative to Veh and Det, whereas Det had no effect relative to Veh (Figure 4b; Veh 1.0±0.1, Glar 2.3±0.3, Det 0.9±0.3; Veh vs Glar P<0.001, Glar vs Det P<0.001, Veh vs Det P=NS). The effect of Glar on LPL RNA was dependent on LFD intake (F(2, 32)=16.00; P<0.0001). It is well documented that HFD can induce adipose tissue inflammation, which has been correlated with insulin resistance and cardiovascular disease.34 On both LFD and HFD, basal insulin therapy had no significant effect on the expression of the proinflammatory cytokine, tumor necrosis factor-α (data not shown).

Effects of Det and Glar on RNA levels of white adipose tissue markers of lipogenesis. RNA isolated from epididymal fat of rats of all treatment groups (Veh, Det and Glar) after 4 weeks of diet consisting of either LFD (n=5 per group) or HFD (n=6–8 per group) was assessed by real-time PCR for changes in RNA levels of WAT lipogenic markers, fatty acid synthase (FAS) (a) and lipoprotein lipase (LPL) (b). RNAs of interest were normalized to RPL13α and for comparative analysis, RNA ratios were normalized to the Veh LF control. Data are presented as the mean±s.e.m. and were analyzed by two-way ANOVA. For all panels, analysis with Bonferroni's post-test shows: *P<0.05 and ***P<0.001 for Veh vs Det; #P<0.05, βP<0.001 for Glar vs Det comparisons.
Discussion
For clinicians, the concept that insulin functions as an adiposity negative feedback signal to the CNS and controls food intake and weight gain, first proposed by Porte and Woods in the late 1970s,35, 36 is quite at odds with clinical experience where insulinization rather uniformly leads to weight gain. Whereas ∼2 kg of weight gain is typically expected for each 1% reduction in HbA1C, insulin administered directly into the CNS of animals ranging from baboons20 to rodents22 will lower food intake and body weight. Genetic modifications that reduce insulin signaling in mice23, 24 as well as mutations in insulin signaling homologs in even more primitive organisms, such as flies and nematodes,17 lead to increased fat mass in these organisms.
Here we describe, for the first time to our knowledge, the robust effect of peripheral SC Det insulin administration to reduce food intake and limit weight and adipose gain. Explanations for weight gain on insulin therapy commonly include reversal of the negative energy balance associated with glycosuria, anabolic effects of insulin in peripheral tissues, particularly the potent effect to stimulate lipogenesis, and the fact that hypoglycemia is a potent stimulus to feed. Finally, it has been described that, with imperfect pharmacodynamic profiles, humans will learn to anticipate hypoglycemia and eat defensively.8 By controlling for these variables, we appear to have uncovered a unique effect of insulin Det, but not Glar, to modulate weight gain.
In these studies, we sought to carefully examine the role of insulin delivered ‘clinically’ (that is, given subcutaneously, once daily) on energy homeostasis. Of course, insulin delivered directly into the brain decreases feeding,20, 21, 22 but our primary objective was to specifically address whether it is the insulin per se that causes weight gain when given via a clinically relevant (SC) route. Here, we sought to bridge clinical experience with our basic science understanding of the role of insulin in energy homeostasis. We additionally controlled for confounders that may explain weight gain by studying non-glycosuric, non-diabetic animals without causing hypoglycemia. Furthermore, because potential weight effects might be amplified in a model of rapid weight gain (diet-induced obese) and because of the clinical observation that weight sparing effects of Det appear to be amplified in the most obese subjects, we also studied the effects in animals on HFD.
First, as current models of energy homeostasis predict, SC insulin neither increased nor decreased food intake and weight gain in LFD rats. The doses of insulin used here are within the clinically relevant dosage range for treatment of diabetes in humans with modest insulin resistance (∼0.2–0.8 U kg−1 per day, KD Niswender, personal experience).
Conversely, on HFD, a surprisingly robust effect of Det, but not Glar, to attenuate food intake, weight gain and adipose gain was observed. First, this implies an important interaction of diet with Det for effects on weight and is intriguingly consistent with observations in humans where those individuals with higher body mass index benefited most.9 Of course, in terms of fat content, the typical human diet much more closely resembles the HFD, rather than the LFD, which consists of only 13.5% calories from fat. Indeed, this food intake and weight attenuating effect is eliminated with increasing dose of insulin (not shown), suggesting, not surprisingly, a narrow effective ‘window.’ Because it is clinically very difficult in humans to remain precisely within such a window due to variability in (1) caloric and macronutrient intake, (2) in insulin administration, and (3) in insulin absorption it is perhaps not surprising that weight gain is common. Weight gain is clearly one of the major barriers to successful clinical use of insulin and our findings, albeit in a preclinical model, suggest that with careful use, insulin need not inevitably cause weight gain. This conclusion is similar to that drawn from a longitudinal study comparing weight gain in controlled diabetics on insulin vs oral agents.37 That the weight effects of Det were observed under HF dietary conditions that provoke weight gain, and that most experts agree contribute to human obesity and diabetes suggests that this model may hold promise for understanding pathophysiological and potentially therapeutic mechanisms at the interface of feeding, obesity and diabetes. Finally, that the effect is observed only on HFD implies that the increased molar ratio of Det to Glar required to obtain similar glycemic effects is not the sole explanation for the observed weight effects, as the same ratio was utilized in LF-fed rats where no effects were observed.
Several studies investigating the effect of chronic administration of insulin in normal-weight or obese non-diabetic rat models have largely reported findings that insulin treatment either had no effect38 or induced hyperphagia and weight gain.39, 40, 41, 42, 43, 44, 45 Notably, only a study by Vanderweele46 showed an 11% reduction of spontaneous daily food intake in normal-weight chow-fed rats administered 2 U per day of regular insulin via an osmotic minipump implanted into the peritoneal cavity. Thus, our findings with insulin Det appear to be fundamentally novel.
Although the primary objective of our study was to determine the effects of SC basal insulin regimens on energy homeostasis in a unique preclinical diet-induced obese model, we also took the opportunity to explore, in a preliminary way, insulin-sensitive markers of potential mechanisms underlying the phenotype. Others have hypothesized that relative tissue selectivity, particularly for the CNS, could explain such weight effects.47, 48 For example, a recent study in humans demonstrated effects of Det on brain function and to reduce spontaneous food intake in a test meal.12 Conversely, others have determined that Det has attenuated transport into the CNS,49 suggesting the possibility that findings of reduced food intake in humans and rodents may be indirect.
Given that insulin has potent lipogenic effects, and that we observed significant attenuation of fat mass gain in Det-treated HFD rats, we assessed expression of key insulin-sensitive target genes in adipose tissue. Here, Glar was noted to increase fatty acid synthase gene expression in both HFD and LFD groups, and to increase lipoprotein lipase gene expression in LFD rats, both effects that would promote storage of fat in adipose. Det showed no effects on adipose tissue gene expression among the groups. These findings are consistent with potency studies in vitro where Glar was noted to have roughly double the lipogenic potency of Det and ∼80-fold increased mitogenic potency.50 Coupling in vitro and in vivo findings such as these raises the possibility that future insulin analog development may enhance specific aspects of insulin's pleiotropic effects while minimizing others, including those on weight. This will clearly require an improved understanding of the molecular pharmacological mechanisms underlying such effects.
Insulin-associated weight gain contributes to clinical inertia, resulting in many patients with diabetes failing to achieve adequate glycemic control, and increasing risk for other metabolic comorbidities. We conclude here that insulin therapy may not necessarily and inevitably lead to weight gain and that it is not the insulin per se that is responsible when weight gain occurs. Next, we have clearly unmasked a unique property of Det in these studies to influence energy balance. Obesity is thought to arise from a very small mismatch in caloric intake vs expenditure; as little as a 1% mismatch can lead to significant weight gain. Here, we observed an overall 7–10% reduction in caloric intake, consistent with recent findings in human volunteers where Det significantly reduced food intake compared with regular human insulin.12 Recognizing significant differences in rat vs human physiology, if such a magnitude of effect were indeed achievable in humans it would have an enormous impact on diabetes progression and obesity related comorbidities. All too commonly, insulin therapy in humans is reserved for ‘rescue’ or ‘last-ditch’ therapy, after the failure of multiple oral medications to control blood–glucose concentration. Several compelling recent publications, however, demonstrated that early insulinization had potential beneficial effects on disease progression.51, 52 These were primarily glycemia-focused intervention studies, but it is intriguing to consider what a similar, but weight-focused approach might achieve.
References
Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP . The continuing epidemics of obesity and diabetes in the United States. JAMA 2001; 286: 1195–1200.
UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352: 837–853.
United Kingdom Prospective Diabetes Study Group. United Kingdom Prospective Diabetes Study 24: a 6-year, randomized, controlled trial comparing sulfonylurea, insulin, and metformin therapy in patients with newly diagnosed type 2 diabetes that could not be controlled with diet therapy. Ann Intern Med 1998; 128: 165–175.
The Diabetes Control and Complications Trial Research Group. Influence of intensive diabetes treatment on body weight and composition of adults with type 1 diabetes in the Diabetes Control and Complications Trial. Diabetes Care 2001; 24: 1711–1721.
Willett WC, Dietz WH, Colditz GA . Guidelines for healthy weight. N Engl J Med 1999; 341: 427–434.
Purnell JQ, Hokanson JE, Marcovina SM, Steffes MW, Cleary PA, Brunzell JD . Effect of excessive weight gain with intensive therapy of type 1 diabetes on lipid levels and blood pressure: results from the DCCT. Diabetes Control and Complications Trial. JAMA 1998; 280: 140–146.
Dunn-Meynell AA, Sanders NM, Compton D, Becker TC, Eiki J, Zhang BB et al. Relationship among brain and blood glucose levels and spontaneous and glucoprivic feeding. J Neurosci 2009; 29: 7015–7022.
Russell-Jones D, Khan R . Insulin-associated weight gain in diabetes—causes, effects and coping strategies. Diabetes Obes Metab 2007; 9: 799–812.
Hermansen K, Davies M, Derezinski T, Martinez Ravn G, Clauson P, Home P . A 26-week, randomized, parallel, treat-to-target trial comparing insulin detemir with NPH insulin as add-on therapy to oral glucose-lowering drugs in insulin-naive people with type 2 diabetes. Diabetes Care 2006; 29: 1269–1274.
Hermansen K, Davies M . Does insulin detemir have a role in reducing risk of insulin-associated weight gain? Diabetes, Obesity, and Metabolism 2007; 9: 209–217.
Hennige AM, Sartorius T, Tschritter O, Preissl H, Fritsche A, Ruth P et al. Tissue selectivity of insulin detemir action in vivo. Diabetologia 2006; 49: 1274–1282.
Hallschmid M, Jauch-Chara K, Korn O, Molle M, Rasch B, Born J et al. Euglycemic infusion of insulin detemir compared with human insulin appears to increase direct current brain potential response and reduces food intake while inducing similar systemic effects. Diabetes 2010; 59: 1101–1107.
Tschritter O, Hennige AM, Preissl H, Porubska K, Schafer SA, Lutzenberger W et al. Cerebrocortical beta activity in overweight humans responds to insulin detemir. PLoS One 2007; 2: e1196.
Owens DR, Bolli GB . Beyond the era of NPH insulin—long-acting insulin analogs: chemistry, comparative pharmacology, and clinical application. Diabetes Technol Ther 2008; 10: 333–349.
Woods SC, Seeley RJ . Insulin as an adiposity signal. Int J Obes Relat Metab Disord 2001; 25 (Suppl 5): S35–S38.
Bagdade JD, Bierman EL, Porte Jr D . The significance of basal insulin levels in the evaluation of the insulin response to glucose in diabetic and nondiabetic subjects. J Clin Invest 1967; 46: 1549–1557.
Niswender KD, Baskin DG, Schwartz MW . Insulin and its evolving partnership with leptin in the hypothalamic control of energy homeostasis. Trends Endocrinol Metab 2004; 15: 362–369.
Grill HJ, Kaplan JM . The neuroanatomical axis for control of energy balance. Front Neuroendocrinol 2002; 23: 2–40.
Plum L, Belgardt BF, Bruning JC . Central insulin action in energy and glucose homeostasis. J Clin Invest 2006; 116: 1761–1766.
Woods SC, Lotter EC, McKay LD, Porte Jr D . Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 1979; 282: 503–505.
Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers Jr MG et al. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes 2003; 52: 227–231.
Air EL, Benoit SC, Blake Smith KA, Clegg DJ, Woods SC . Acute third ventricular administration of insulin decreases food intake in two paradigms. Pharmacol Biochem Behav 2002; 72: 423–429.
Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC et al. Role of brain insulin receptor in control of body weight and reproduction. Science 2000; 289: 2122–2125.
Masaki T, Chiba S, Noguchi H, Yasuda T, Tobe K, Suzuki R et al. Obesity in insulin receptor substrate-2-deficient mice: disrupted control of arcuate nucleus neuropeptides. Obes Res 2004; 12: 878–885.
Klein O, Lynge J, Endahl L, Damholt B, Nosek L, Heise T . Albumin-bound basal insulin analogues (insulin detemir and NN344): comparable time-action profiles but less variability than insulin glargine in type 2 diabetes. Diabetes Obes Metab 2007; 9: 290–299.
Posey K, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab 2009; 296: E1003–E1012.
Sorensen AR, Stidsen CE, Ribel U, Nishimura E, Sturis J, Jonassen I et al. Insulin detemir is a fully efficacious, low affinity agonist at the insulin receptor. Diabetes Obes Metab 2010; 12: 665–673.
Ayala JE, Bracy DP, Julien BM, Rottman JN, Fueger PT, Wasserman DH . Chronic treatment with sildenafil improves energy balance and insulin action in high fat-fed conscious mice. Diabetes 2007; 56: 1025–1033.
Dryden S, Pickavance L, Henderson L, Williams G . Hyperphagia induced by hypoglycemia in rats is independent of leptin and hypothalamic neuropeptide Y (NPY). Peptides 1998; 19: 1549–1555.
Kadowaki T, Yamauchi T . Adiponectin and adiponectin receptors. Endocr Rev 2005; 26: 439–451.
Galassetti P, Davis SN . Effects of insulin per se on neuroendocrine and metabolic counter-regulatory responses to hypoglycaemia. Clin Sci (Lond) 2000; 99: 351–362.
Kersten S . Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep 2001; 2: 282–286.
O’Brien RM, Granner DK . Regulation of gene expression by insulin. Biochem J 1991; 278 (Part 3): 609–619.
Berg AH, Scherer PE . Adipose tissue, inflammation, and cardiovascular disease. Circ Res 2005; 96: 939–949.
Woods SC, Porte Jr D . Relationship between plasma and cerebrospinal fluid insulin levels of dogs. Am J Physiol 1977; 233: E331–E334.
Bernstein IL, Lotter EC, Kulkosky PJ, Porte Jr D, Woods SC . Effect of force-feeding upon basal insulin levels of rats. Proc Soc Exp Biol Med 1975; 150: 546–548.
Huizinga MM, Niswender KD, Gebretsadik T, Rothman RL, Shintani AK, Elasy TA . Insulin use and weight maintenance in well-controlled type 2 diabetes: a prospective cohort study. Obesity (Silver Spring) 2008; 16: 1933–1937.
Destefano MB, Stern JS, Castonguay TW . Effect of chronic insulin administration on food intake and body weight in rats. Physiol Behav 1991; 50: 801–806.
Panksepp J, Pollack A, Krost K, Meeker R, Ritter M . Feeding in response to repeated protamine zinc insulin injections. Physiol Behav 1974; 14: 487–493.
Woodward CJ, Emery PW . Energy balance in rats given chronic hormone treatment. 1. Effects of long-acting insulin. Br J Nutr 1989; 61: 437–444.
MacKay EM, Callaway JW, Barnes RH . Hyperalimentation in normal animals produced by protamine insulin. J Nutr 1940; 20: 59–66.
Kanarek RB, Marks-Kaufman R, Lipeles BJ . Increased carbohydrate intake as a function of insulin administration in rats. Physiol Behav 1980; 25: 779–782.
Larue-Achagiotis C, Goubern M, Laury MC . Concomitant food intake and adipose tissue responses under chronc insulin infusion in rats. Physiol Behav 1988; 44: 95–100.
Brandes JS . Insulin induced overeating in the rat. Physiol Behav 1977; 18: 1095–1102.
Lotter EC, Woods SC . Injections of insulin and changes of body weight. Physiol Behav 1977; 18: 293–297.
VanderWeele DA, Haraczkiewicz E, Van Itallie TB . Elevated insulin and satiety in obese and normal-weight rats. Appetite 1982; 3: 99–109.
Fritsche A, Haring H . At last, a weight neutral insulin? Int J Obes Relat Metab Disord 2004; 28 (Suppl 2): S41–S46.
Tibaldi J . Actions of insulin beyond glycemic control: a perspective on insulin detemir. Adv Ther 2007; 24: 868–882.
Banks WA, Morley JE, Lynch JL, Lynch KM, Mooradian AD . Insulin detemir is not transported across the blood-brain barrier. Peptides 2010; 31: 2284–2288.
Kurtzhals P, Schäffer L, Sørensen A, Kristensen C, Jonassen I, Schmid C et al. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes 2000; 49: 999–1005.
Weng J, Li Y, Xu W, Shi L, Zhang Q, Zhu D et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multicentre randomised parallel-group trial. Lancet 2008; 371: 1753–1760.
Pfutzner A, Lorra B, Abdollahnia MR, Kann PH, Mathieu D, Pehnert C et al. The switch from sulfonylurea to preprandial short-acting insulin analog substitution has an immediate and comprehensive beta-cell protective effect in patients with type 2 diabetes mellitus. Diabetes Technol Ther 2006; 8: 375–384.
Acknowledgements
We thank Maxine Turney, Leena George and Sanaz Saadat for providing superb technical support. This investigator-initiated, peer-reviewed study was funded by Novo Nordisk. JMR was supported by the Vanderbilt Molecular Endocrinology Training Program (5T 32 DK07563). The Vanderbilt Diabetes Center (DK20593) Hormone Assay and Analytical Services Core performed several of the described assays.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Dr KD Niswender has received investigator-initiated, peer-reviewed research funds from Novo Nordisk, has participated in Scientific Advisory Boards for Novo Nordisk, and is a site PI for a weight loss study sponsored by Novo Nordisk. Ms JM Rojas and Dr RL Printz declare no potential conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Rojas, J., Printz, R. & Niswender, K. Insulin detemir attenuates food intake, body weight gain and fat mass gain in diet-induced obese Sprague–Dawley rats. Nutr & Diabetes 1, e10 (2011). https://doi.org/10.1038/nutd.2011.6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nutd.2011.6
