
Abstract
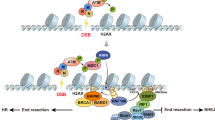
Although both homologous recombination (HR) and nonhomologous end joining can repair DNA double-strand breaks (DSBs), the mechanisms by which one of these pathways is chosen over the other remain unclear. Here we show that transcriptionally active chromatin is preferentially repaired by HR. Using chromatin immunoprecipitation–sequencing (ChIP-seq) to analyze repair of multiple DSBs induced throughout the human genome, we identify an HR-prone subset of DSBs that recruit the HR protein RAD51, undergo resection and rely on RAD51 for efficient repair. These DSBs are located in actively transcribed genes and are targeted to HR repair via the transcription elongation–associated mark trimethylated histone H3 K36. Concordantly, depletion of SETD2, the main H3 K36 trimethyltransferase, severely impedes HR at such DSBs. Our study thereby demonstrates a primary role in DSB repair of the chromatin context in which a break occurs.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others


Accession codes
References
Hartlerode, A.J. & Scully, R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 423, 157–168 (2009).
Beucher, A. et al. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 28, 3413–3427 (2009).
Noon, A.T. et al. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol. 12, 177–184 (2010).
Shibata, A. et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 30, 1079–1092 (2011).
Fattah, F. et al. Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet. 6, e1000855 (2010).
Bothmer, A. et al. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol. Cell 42, 319–329 (2011).
Bouwman, P. et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 17, 688–695 (2010).
Bunting, S.F. et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254 (2010).
Chapman, J.R. et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 49, 858–871 (2013).
Di Virgilio, M. et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339, 711–715 (2013).
Escribano-Díaz, C. et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883 (2013).
Zimmermann, M., Lottersberger, F., Buonomo, S.B., Sfeir, A. & de Lange, T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339, 700–704 (2013).
Sartori, A.A. et al. Human CtIP promotes DNA end resection. Nature 450, 509–514 (2007).
Shibata, A. et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 53, 7–18 (2014).
Botuyan, M.V. et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127, 1361–1373 (2006).
Hartlerode, A.J. et al. Impact of histone H4 lysine 20 methylation on 53BP1 responses to chromosomal double strand breaks. PLoS ONE 7, e49211 (2012).
Oda, H. et al. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol. Cell 40, 364–376 (2010).
Pei, H. et al. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 470, 124–128 (2011).
Sanders, S.L. et al. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119, 603–614 (2004).
Tang, J. et al. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 20, 317–325 (2013).
FitzGerald, J.E., Grenon, M. & Lowndes, N.F. 53BP1: function and mechanisms of focal recruitment. Biochem. Soc. Trans. 37, 897–904 (2009).
Chiolo, I. et al. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144, 732–744 (2011).
Berkovich, E., Monnat, R.J. Jr. & Kastan, M.B. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 9, 683–690 (2007).
Rouet, P., Smih, F. & Jasin, M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl. Acad. Sci. USA 91, 6064–6068 (1994).
Miller, K.M. & Jackson, S.P. Histone marks: repairing DNA breaks within the context of chromatin. Biochem. Soc. Trans. 40, 370–376 (2012).
Soria, G., Polo, S.E. & Almouzni, G. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol. Cell 46, 722–734 (2012).
Wolner, B., van Komen, S., Sung, P. & Peterson, C.L. Recruitment of the recombinational repair machinery to a DNA double-strand break in yeast. Mol. Cell 12, 221–232 (2003).
Shanbhag, N.M., Rafalska-Metcalf, I.U., Balane-Bolivar, C., Janicki, S.M. & Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981 (2010).
Caron, P. et al. Cohesin protects genes against γH2AX Induced by DNA double-strand breaks. PLoS Genet. 8, e1002460 (2012).
Iacovoni, J.S. et al. High-resolution profiling of γH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 29, 1446–1457 (2010).
Massip, L., Caron, P., Iacovoni, J.S., Trouche, D. & Legube, G. Deciphering the chromatin landscape induced around DNA double strand breaks. Cell Cycle 9, 2963–2972 (2010).
Chailleux, C. et al. Quantifying DNA double-strand breaks induced by site-specific endonucleases in living cells by ligation-mediated purification. Nat. Protoc. 9, 517–528 (2014).
Nishimura, K., Fukagawa, T., Takisawa, H., Kakimoto, T. & Kanemaki, M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917–922 (2009).
ENCODE Project Consortium. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 9, e1001046 (2011).
Kim, Y. et al. A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol. 31, 251–258 (2013).
Daugaard, M. et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol. 19, 803–810 (2012).
Butler, J.S. & Dent, S.Y. Chromatin 'resetting' during transcription elongation: a central role for methylated H3K36. Nat. Struct. Mol. Biol. 19, 863–864 (2012).
Edmunds, J.W., Mahadevan, L.C. & Clayton, A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 27, 406–420 (2008).
Dumay, A. et al. Bax and Bid, two proapoptotic Bcl-2 family members, inhibit homologous recombination, independently of apoptosis regulation. Oncogene 25, 3196–3205 (2006).
Huertas, P. & Jackson, S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 284, 9558–9565 (2009).
Yu, X. & Chen, J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell. Biol. 24, 9478–9486 (2004).
Karanam, K., Kafri, R., Loewer, A. & Lahav, G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell 47, 320–329 (2012).
Gandhi, M. et al. Homologous chromosomes make contact at the sites of double-strand breaks in genes in somatic G0/G1-phase human cells. Proc. Natl. Acad. Sci. USA 109, 9454–9459 (2012).
Goodarzi, A.A., Jeggo, P. & Lobrich, M. The influence of heterochromatin on DNA double strand break repair: getting the strong, silent type to relax. DNA Repair (Amst.) 9, 1273–1282 (2010).
Chantalat, S. et al. Histone H3 trimethylation at lysine 36 is associated with constitutive and facultative heterochromatin. Genome Res. 21, 1426–1437 (2011).
Finnegan, D.J. Oogenesis: active heterochromatin. Curr. Biol. 21, R630–R632 (2011).
Lejeune, E. & Allshire, R.C. Common ground: small RNA programming and chromatin modifications. Curr. Opin. Cell Biol. 23, 258–265 (2011).
Probst, A.V. & Almouzni, G. Heterochromatin establishment in the context of genome-wide epigenetic reprogramming. Trends Genet. 27, 177–185 (2011).
Ernst, J. et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 (2011).
Dhayalan, A. et al. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 285, 26114–26120 (2010).
Vezzoli, A. et al. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat. Struct. Mol. Biol. 17, 617–619 (2010).
Miller, K.M. et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 17, 1144–1151 (2010).
Acknowledgements
We thank New England Biolabs for providing AsiSI genomic DNA. We thank B. Lopez (Institut de Cancérologie Gustave-Roussy) for RG37-HR I-SceI GFP cells. We thank V. Benes and the Solexa team at the European Molecular Biology Laboratory Genomic Core Facility, the Beijing Genomic Institute (BGI) and the Genomics core facility at the Cancer Research Institute (CRI) in Cambridge for high-throughput sequencing. We thank the Flow Cytometry platform from the Fédération de Biologie de Toulouse at the Laboratoire de Biologie Cellulaire et Moléculaire du Contrôle de la Prolifération (LBCMCP-FRBT). F.A. is supported by a grant from the Ligue Nationale Contre le Cancer (LNCC); P.C. is supported by a grant from the Association Contre le Cancer (ARC); and S.B. and E.G. are supported by grants from the Fondation pour la Recherche Médicale (FRM). Research in S.P.J.'s laboratory is supported by grants from Cancer Research UK (C6/A11226), the European Research Council and the European Community's Seventh Framework Program (DDResponse) and by core infrastructure funding from Cancer Research UK and the Wellcome Trust. K.M.M. was funded by a Wellcome Trust Project grant and C.K.S. by a Return-to-Europe Federation of European Biochemical Societies fellowship. S.P.J.'s salary is provided by the University of Cambridge and supplemented by Cancer Research UK. Funding to G.L. was provided by grants from the ARC, Agence Nationale pour la Recherche (ANR-09-JCJC-0138), Canceropole Grand Sud Ouest (GSO) and Research Innovation Therapeutic Cancerologie (RITC).
Author information
Authors and Affiliations
Contributions
F.A., P.C., E.G. and V.D. performed experiments. B.B. developed the AID-DIvA cell line. C.K.S. suggested and performed immunofluorescence studies on laser-induced damage. J.S.I. and S.B. performed bioinformatic analyses of the ChIP-seq data. K.M.M. performed XRCC4 ChIP library preparation and sequencing. G.L. conceived and analyzed experiments. F.A., J.S.I., K.M.M., S.P.J. and G.L. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 γH2AX profile around AsiSI-induced DSBs.
ChIP-seq against γH2AX (Epitomics) performed in 4OHT treated DIvA cells. a. A representative γH2AX domain is presented (top panel, chromosome position are indicated in Mbp). A magnification of the γH2AX enrichment at the proximity of the DSB is shown (bottom panel, chromosomal positions are in bp). The DSB is indicated by an arrow. b. Average γH2AX signal on a 20kb window around each annotated AsiSI site on the human genome. c. The γH2AX signal obtained by ChIP-seq, around the DSBs presented Fig. 1b is shown. DSBs are indicated by arrows. RAD51-bound DSBs are indicated in red, and RAD51-unbound DSBs in blue.
Supplementary Figure 2 Binding of RAD51 and XRCC4 at AsiSI-induced DSBs.
a. Duplicate ChIP-seq experiment in 4OHT treated DIvA cells using an anti XRCC4 antibody (Abcam) or an anti-RAD51 antibody (Santa Cruz). The averaged XRCC4 (blue) and Rad51 (red) signal on 10kb around annotated AsiSI sites on the human genome are shown. b. Signals obtained around the four DSBs presented Fig. 1b are shown. Positions are indicated in bp. c. ChIP experiments performed in DIvA cells, treated (filled circles) or not (empty circles) with 4OHT, using an anti-XRCC4 antibody (blue) or an anti-RAD51 antibody (red). ChIP efficiencies (as percent of input immunoprecipitated) were measured by RT-qPCR at 80, 200 and 800bp from an AsiSI induced DSB (DSB-III). Mean and S.e.m (technical replicates n=4) of representative experiment is shown.
Supplementary Figure 3 Measure of cleavage efficiency at selected AsiSI sites, throughout the cell cycle.
Cleavage efficiency (using the protocol described in the online methods) was measured from untreated or 4OHT-treated cells by RT-qPCR for five AsiSI sites either identified as RAD51-bound or RAD51-unbound. The same representative experiment is presented in a, b and c. a. Raw qPCR data, as percent of input pulled down by the streptavidin purification are shown. b. The data presented in (a) were further normalized using an internal control constituted by in vitro AsiSI-digested plasmid added in the ligation reaction. This allows calculating the cutting efficiency at each specific sites as percent of cleavage (the in vitro digested plasmid representing the 100% cleavage). c. Ratios between treated and untreated cells are presented (using the normalized data presented in b). This last representation will be used throughout the manuscript. d. Pull own efficiency was measured by RT-qPCR for the eight AsiSI sites analyzed for the presence of ssDNA in Fig. 3a. Ratios between treated and untreated cells are presented. Mean and s.e.m (technical replicates, n=4) of a representative experiment is shown.
Supplementary Figure 4 Kinetics of DSB induction and disappearance in the AID-DIvA cell line.
a. HA and γH2AX staining in AID-DIvA treated as indicated. b. Measure of cleavage efficiency, at two AsiSI-induced DSBs, in AID-DIvA cells treated as indicated. c. γH2AX staining in AID-DIvA cells treated (or not) with 4OHT for 4h and further incubated with auxin (or not) as indicated. d. γH2AX foci observed in AID-DIvA cells treated as in c, were counted using images aquired with an Array-Scan device. The mean and s.e.m (n=5, biological replicates, about 200 acquired nuclei per experiment) are presented. e. Repair analysis of AsiSI-induced RAD51-unbound (in blue, right panel) or RAD51-bound (in red, left panel) DSBs in AID-DIvA cells transfected with control, RAD51, or XRCC4. A biological replicate from Fig. 3f is shown.
Supplementary Figure 5 HR-prone DSBs are enriched in active chromatin marks and in PolII-S2P.
a. ChIP-seq data of various histones marks, associated with active (top and middle panels) or inactive (bottom panels) chromatin in U20S and K562 cells were retrieved from the ENCODE project, over 4kb surrounding each DSB from the RAD51-bound (red) and RAD51-unbound subsets (blue). The name of the Institute that generated the data are indicated in parentheses. The average and s.e.m are shown. p values (Mann-Whitney) are shown above each graphs. b. Average PolII–S2P signal obtained by ChIP-seq in DIvA cells, around the transcription start sites (TSS) of all genes from the human genome (hg18). c. Similar analysis than for Fig.4b except that all cleaved DSBs (100 best) were divided in two categories of 50 DSBs each based on their ratio RAD51/XRCC4. d. The Pol II–S2P signal obtained around two RAD51-bound DSBs, but far from any annotated gene (RefSeq, indicated in brown) are shown. DSBs are indicated by arrows, and positions are in bp. Feature from in lincRNA (large intergenic non coding RNAs) and TUCP (transcripts of uncertain coding potential) database (UCSC)(left panel), or from annotated pseudogene and non coding RNA (UCSC)(right panel) are indicated in purple.
Supplementary Figure 6 Effect of DRB and actinomycin D on transcription, RAD51 binding and AsiSI-mediated break induction.
ChIP using a PolII-S2P antibody in 4OHT-treated DIvA cells incubated with DRB (a) or actinomycin D (b) PolII-S2P levels were analyzed within a 1kb region around DSBs shown Fig. 4c-d, i.e. three RAD51-bound DSBs (DSB-I; -II; -III), one RAD51-unbound DSB (DSB-1), and two DSBs located far from any gene (DSB-5, -6). c. XRCC4 (blue) and RAD51 (red) ChIP in 4OHT-treated DIvA incubated with Actinomycin D or DRB as indicated. XRCC4 and RAD51 enrichments were respectively analyzed close to (<100bp) and at 800bp from selected DSBs, by RT-qPCR. The mean and s.e.m (n=4 technical replicates) of % of input immunoprecipitated for three DSBs (one RAD51-bound: DSB-III, one RAD51-unbound: DSB-1, and one intergenic: DSB-5) are shown. d. Analysis of cleavage efficiency in DIvA cells incubated with Actinomycin D (or not) (see online Methods). e. Analysis of cleavage efficiency in DIvA cells in G1 or G2 synchronized cells, and in presence or absence of DRB treatment as indicated, at an AsiSI site either identified as RAD51-bound (in red), or RAD51-unbound (in blue). Mean and s.e.m (n=4, technical replicates) of the raw qPCR data of a representative experiment, as percent of input pulled down by the streptavidin purification are shown.
Supplementary Figure 7 Effect of LEDGF and SETD2 siRNAs on H3K36me3, RAD51, XRCC4 and PolII-S2P levels.
DIvA cells were transfected using a control siRNA, a siRNA directed against LEDGF (a-d) or a siRNA directed against SETD2 (e-h). a,e. Relative mRNA level assessed by reverse transcription followed by qPCR. b,f. H3K36me3 ChIP analysed by qPCR at selected sites. c,g XRCC4 (blue) and RAD51 (red) ChIP after 4OHT-treatment respectively analyzed close to (<100bp) and at 800bp from selected DSBs. ChIP efficiencies (as % of input) are shown for 3 DSBs (RAD51 bound (DSB-III), RAD51 unbound (DSB-1), and intergenic (DSB-5)). The mean and s.e.m (n=4, technical replicate) of a representative experiment are presented. d, h PolII-S2P ChIP analyzed within a 1kb window around DSBs. The mean and s.e.m (n=4, technical replicates) of a representative experiment are presented. p values are indicated, ns non-significant (unpaired student test, two sided).
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–8 and Supplementary Tables 1–4 (PDF 943 kb)
Rights and permissions
About this article
Cite this article
Aymard, F., Bugler, B., Schmidt, C. et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol 21, 366–374 (2014). https://doi.org/10.1038/nsmb.2796
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nsmb.2796
This article is cited by
-
HJURP is recruited to double-strand break sites and facilitates DNA repair by promoting chromatin reorganization
Oncogene (2024)
-
PSIP1/LEDGF reduces R-loops at transcription sites to maintain genome integrity
Nature Communications (2024)
-
Multiscale reorganization of the genome following DNA damage facilitates chromosome translocations via nuclear actin polymerization
Nature Structural & Molecular Biology (2023)
-
Chromatin organization drives the search mechanism of nuclear factors
Nature Communications (2023)
-
iMUT-seq: high-resolution DSB-induced mutation profiling reveals prevalent homologous-recombination dependent mutagenesis
Nature Communications (2023)
