
Key Points
-
Most, if not all, cells in humans and mice express the receptor for type I interferons (IFNs). Therefore, these cytokines have a range of direct and indirect effects on various cell types during infection with viruses, bacteria, parasites and fungi.
-
Type I IFNs are important for host defence against viruses, through the induction of antiviral effector molecules that are encoded by IFN-stimulated genes. These IFNs can, however, cause immunopathology in acute viral infections. Conversely, they can lead to immunosuppression and loss of virus control during chronic viral infections.
-
During bacterial infections, low levels of type I IFNs may be required early, to initiate cell-mediated immune responses. By contrast, type I IFNs have been shown to have adverse effects in infections with intracellular bacteria such as Listeria monocytogenes and Mycobacterium tuberculosis.
-
In bacterial infections, high concentrations of type I IFNs may block B cell responses or may lead to the production of immunosuppressive molecules such as interleukin-10.
-
Type I IFNs also antagonize the action of type II IFN (that is, IFNγ) by reducing the responsiveness of macrophages to activation by type II IFN.
-
Another important antagonism is between type I IFNs and interleukin-1. This antagonism was recently shown to be important in M. tuberculosis infection and to be mediated by eicosanoids, in particular prostaglandin E2.
-
Thus, type I IFNs are part of a complex cross-regulatory network, which leads mostly, but not always, to protection of the host against infectious diseases with minimum damage to the host.
Abstract
Type I interferons (IFNs) have diverse effects on innate and adaptive immune cells during infection with viruses, bacteria, parasites and fungi, directly and/or indirectly through the induction of other mediators. Type I IFNs are important for host defence against viruses. However, recently, they have been shown to cause immunopathology in some acute viral infections, such as influenza virus infection. Conversely, they can lead to immunosuppression during chronic viral infections, such as lymphocytic choriomeningitis virus infection. During bacterial infections, low levels of type I IFNs may be required at an early stage, to initiate cell-mediated immune responses. High concentrations of type I IFNs may block B cell responses or lead to the production of immunosuppressive molecules, and such concentrations also reduce the responsiveness of macrophages to activation by IFNγ, as has been shown for infections with Listeria monocytogenes and Mycobacterium tuberculosis. Recent studies in experimental models of tuberculosis have demonstrated that prostaglandin E2 and interleukin-1 inhibit type I IFN expression and its downstream effects, demonstrating that a cross-regulatory network of cytokines operates during infectious diseases to provide protection with minimum damage to the host.
Similar content being viewed by others
Main
There are three distinct interferon (IFN) families. The type I IFN family is a multi-gene cytokine family that encodes 13 partially homologous IFNα subtypes in humans (14 in mice), a single IFNβ and several poorly defined single gene products (IFNɛ, IFNτ, IFNκ, IFNω, IFNδ and IFNζ)1. The type II IFN family consists of a single gene product, IFNγ, that is predominantly produced by T cells and natural killer (NK) cells, and can act on a broad range of cell types that express the IFNγ receptor (IFNγR)2. The type III IFN family comprises IFNλ1, IFNλ2 and IFNλ3 (also known as IL-29, IL-28A and IL-28B, respectively) and the recently identified IFNλ4 (Refs 3,4), which have similar functions to cytokines of the type I IFN family but restricted activity, as the expression of their receptor is largely restricted to epithelial cell surfaces5. Indeed, immune cells are largely unresponsive to IFNλ (reviewed in Refs 5,6). This Review focuses on IFNα and IFNβ (hereafter referred to as IFNα/β), which are the best-defined and most broadly expressed type I IFNs. These cytokines are best known for their ability to induce an antiviral state in both virus-infected cells and uninfected, bystander cells, by inducing a programme of gene transcription that interferes with multiple stages of the viral replication cycle through various mechanisms7. However, IFNα/β have numerous additional functions that influence the innate and adaptive immune responses not only to viruses but also to bacterial pathogens and other pathogens. The outcome of the IFNα/β response during infectious disease is highly context dependent. Different conditions are induced during specific infections and affect when and where IFNα/β signals are delivered, as well as the signalling pathways that are triggered downstream of the type I IFN receptor (IFNAR). This, in turn, influences which IFN-stimulated genes (ISGs) are activated or repressed. Overall, this can lead to beneficial or detrimental outcomes for the host. In this Review, we discuss IFNα/β-mediated effects on the host response during various infectious diseases and the mechanisms involved in conferring these effects.
Type I IFN production and signalling
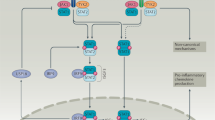
Induction of IFNα/β production. Almost all cells in the body can produce IFNα/β, and this usually occurs in response to the stimulation of receptors known as pattern recognition receptors (PRRs) by microbial products. These receptors are located on the cell surface, in the cytosol or in endosomal compartments. They recognize foreign nucleic acids and self DNA (which are generally not found in the cytosol), as well as a limited number of other non-nucleic-acid pathogen-associated molecular patterns (PAMPs). The RNA helicases retinoic acid-inducible gene I (RIG-I; also known as DDX58) and melanoma differentiation-associated gene 5 (MDA5; also known as IFIH1) are the main cytosolic receptors that are responsible for the recognition of RNA, and they may recognize certain AT-rich DNA motifs, although this is controversial (reviewed in Ref. 8). These receptors are highly associated with the induction of type I IFNs (Fig. 1). Other DNA motifs in the cytosol can be recognized by various receptors, including DNA-dependent activator of IFN-regulatory factors (DAI; also known as ZBP1), the DEAD box and DEAH box (DEXD/H box) helicases, and the recently described receptor cytosolic GAMP synthase (cGAS; also known as MB21D1) (reviewed in Refs 8,9), all of which are highly associated with the induction of type I IFN production. Finally, the cytosolic molecular sensors NOD-containing protein 1 (NOD1) and NOD2 are expressed by various cell types and recognize nucleic acids and other ligands, which can lead to IFNα/β production10,11,12 (reviewed in Ref. 13).

Recognition of microbial products by a range of cell-surface and intracellular pattern recognition receptors, including Toll-like receptors (TLRs) and retinoic acid-inducible gene I (RIG-I), can lead to induction of the genes encoding type I interferons (IFNs), which is mediated by several distinct signalling pathways. On the binding of type I IFNs to their receptor (IFNAR), multiple downstream signalling pathways can be induced, leading to a diverse range of biological effects. The canonical signal transducer and activator of transcription 1 (STAT1)–STAT2–IFN-regulatory factor 9 (IRF9) signalling complex (also known as the IFN-stimulated gene factor 3 (ISGF3) complex) binds to IFN-stimulated response elements (ISREs) in gene promoters, leading to induction of a large number of IFN-stimulated genes (ISGs). Type I IFNs can also signal through STAT1 homodimers, which are more commonly associated with the IFNγ-mediated signalling pathway. Other STAT heterodimers and homodimers may also be activated downstream, including STAT3, STAT4 and STAT5. Other signalling pathways that do not rely on Janus kinase (JAK) and/or STAT activity may also be activated, including mitogen-activated protein kinases (MAPKs) and the phosphoinositide 3-kinase (PI3K) pathway, thereby leading to diverse effects on the cell. Alt-IRF, IRFs other than IRF3 or IRF7; AP-1, activator protein 1; cGAMP, cyclic di-GMP-AMP; cGAS, cytosolic GAMP synthase; DAI, DNA-dependent activator of IRFs; ER, endoplasmic reticulum; GAS, γ-activated sequence; IKKɛ, IκB kinase-ɛ; MAVS, mitochondrial antiviral signalling protein; MDA5, melanoma differentiation-associated gene 5; MYD88, myeloid differentiation primary response protein 88; NF-κB, nuclear factor-κB; NOD2, NOD-containing protein 2; STING, stimulator of IFN genes; TBK1, TANK-binding kinase 1; TRAF, TNF receptor-associated factor; TRAM, TLR adaptor molecule (also known as TICAM2); TRIF, TIR domain-containing adaptor protein inducing IFNβ; TYK2, tyrosine kinase 2.
In addition to these cytosolic receptors, several Toll-like receptors (TLRs) activate pathways that lead to IFNα/β production. Of the cell-surface TLRs, TLR4, which recognizes lipopolysaccharide from bacteria, is the most potent type I IFN inducer and signals through the adaptor protein TIR domain-containing adaptor protein inducing IFNβ (TRIF; also known as TICAM1). In endosomal compartments, TLR3, TLR7 and TLR8, and TLR9 respond to double-stranded RNA, single-stranded RNA and unmethylated CpG DNA, respectively14.
Diverse pathways downstream of these receptors transduce signals that converge on a few key molecules, such as the IFN-regulatory factor (IRF) family of transcription factors, that activate the transcription of genes encoding IFNα/β. In most cases, IRF3 and IRF7 are the fundamental IRFs that are required, although others (such as IRF1, IRF5 and IRF8) can also induce IFNA/B gene transcription. The central tenet of IFNα/β production is that the IFNB and IFNA4 genes are induced in an initial wave of transcription that relies on IRF3. This initial IFN burst triggers the transcription of IRF7, which then mediates a positive feedback loop, leading to the induction of a second wave of gene transcription, including additional IFNα-encoding genes15,16. Nuclear factor-κB (NF-κB) is also required as a cofactor, although there is some disagreement about the importance of this pathway in IFNα/β production15. Immediately upstream of the IRFs, the kinases IκB kinase-ɛ (IKKɛ; encoded by IKBKE) and TANK-binding kinase 1 (TBK1) are responsible for the phosphorylation of IRF3 and IRF7. The cytosolic RNA sensors RIG-I and MDA5 rely on the adaptor mitochondrial antiviral signalling protein (MAVS; also known as IPS1 or VISA) to activate TBK1, whereas stimulator of IFN genes (STING; also known as TMEM173) is an important mediator of much of the response to cytosolic DNA9. TLR3 and TLR4 use the adaptor molecule TRIF, which activates TBK1, leading to the activation of IRF3. TLR7 and TLR9 are preferentially expressed by plasmacytoid dendritic cells (pDCs) and transduce signals for IFNα/β production through myeloid differentiation primary response protein 88 (MYD88) rather than TRIF, and the potent production of IFNα/β by pDCs is due to constitutive expression of IRF7 and to retention of the MYD88–IRF7 complex in endosomes14,16.
Type I IFN signalling and induction of ISGs. IFNβ and all of the IFNα subtypes bind to, and signal through, a heterodimeric transmembrane receptor composed of the subunits IFNAR1 and IFNAR2. Ligation of IFNAR activates the receptor-associated protein tyrosine kinases Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2). In the canonical pathway of IFNα/β-mediated signalling, activated JAK1 and TYK2 phosphorylate signal transducer and activator of transcription 1 (STAT1) and STAT2 molecules that are present in the cytosol, leading to the dimerization, nuclear translocation and binding of these molecules to IRF9 to form the ISG factor 3 (ISGF3) complex. This complex then binds to IFN-stimulated response elements in ISG promoters, leading to the activation of ISG transcription (reviewed in Ref. 17). In this manner, IFNα/β induces the expression of several hundred ISGs, a large number of which function to induce an antiviral state within the cell.
IFNα/β-mediated signalling is not limited to this canonical pathway, however. In addition to signalling through STAT1–STAT2 heterodimers, IFNα/β can signal through STAT1 homodimers, which are more commonly associated with IFNγ-mediated signalling and bind to γ-activated sequences in gene promoters17. IFNα/β can also signal through STATs that are usually associated with other cytokine-mediated signalling pathways, including STAT3, STAT4, STAT5A and STAT5B. The phosphoinositide 3-kinase (PI3K)–mammalian target of rapamycin (mTOR) pathway and multiple mitogen-activated protein kinase (MAPK) pathways can also be activated downstream of IFNAR. This diversity of signalling pathways may in part explain the broad effects of IFNα/β-mediated signalling, as it allows the transcription of a broad range of genes in addition to those dedicated to viral restriction (reviewed in Ref. 17). These include genes that encode cytokines and chemokines, antibacterial effectors, pro-apoptotic and anti-apoptotic molecules, and molecules involved in metabolic processes18 (Fig. 1).
Protective effects in viral infection
Virus restriction in vitro. IFNs were named for their ability to restrict (that is, to 'interfere' with) viral replication in vertebrate cells, which has now been shown for many viruses both in human and mouse cells and cell lines7. The ability of IFNα/β to restrict viral replication is largely attributable to the induction of ISGs. These genes are either expressed constitutively in cells in response to low levels of IFNα/β in the microenvironment or, more commonly, in response to IFNα/β produced in response to infection, during which IFNα/β promote an antiviral state in bystander cells and restrict the viral replication cycle in cells that have already been infected7. The fact that most viruses devote part of their limited genome to mechanisms that perturb IFNα/β production and/or IFNα/β-mediated signalling, thereby preventing ISGs from being induced, illustrates the importance of this cytokine family in host cell protection against viral infection19.
The mechanisms of action of many of these ISGs have been described. Some of the best known are myxovirus resistance 1 (MX1), IFN-inducible double-stranded RNA-dependent protein kinase (PKR; encoded by EIF2AK2), 2′-5′-oligoadenylate synthetase (OAS), IFN-induced transmembrane proteins (IFITMs), apolipoprotein B mRNA-editing enzyme catalytic polypeptide 1 (APOBEC1)7 and the tripartite motif-containing (TRIM) family of molecules7,20. These ISGs have been reviewed in great detail elsewhere and are therefore not discussed further here7. However, it is worth noting interesting recent work aimed at understanding this acute ISG response at a broader level by defining the transcriptional programmes of ISGs that are induced by different viruses21. These studies reveal that specific sets of induced ISGs are effective in different viral infections.
Virus restriction in vivo. Studying IFNAR1-deficient mice has provided definitive proof that IFNα/β mediate potent protection against viruses in vivo22, although previous studies in which exogenous IFN was experimentally used to treat viral infections also strongly suggested this property of IFNα/β23. Ifnar1−/− mice were found to be susceptible to infection with four viruses — vesicular stomatitis virus (VSV), Semliki forest virus, vaccinia virus and lymphocytic choriomeningitis virus (LCMV) — a list that, interestingly, does not include influenza virus, as it was not tested in this study. Subsequently, Stat1−/− mice were shown to be highly susceptible to influenza virus, but the role of IFNAR1 in influenza virus infection — as tested in Ifnar1−/− mice — was less clear24,25,26,27.
This discrepancy was explained when mice that were deficient in both IFNAR1 and IFNλR (Ifnar1−/−Ifnlr−/− mice) were shown to be unable to control influenza virus infection, whereas IFNAR1-deficient mice and IFNλR-deficient mice had a mild phenotype28,29. This finding suggests that there is redundancy between the type I and type III IFN systems, which both require STAT1 downstream of their respective receptors. Only Stat1−/− and Ifnar1−/−Ifnlr−/− mice lack all IFN responsiveness in both haematopoietic and epithelial cells; Ifnar1−/− mice retain type III IFN-mediated signalling in the epithelium and can partially control influenza virus infection29. In addition, when both type I and type III IFN-mediated signalling is deficient only in epithelial cells, mice succumb to influenza virus infection30.
Naturally occurring mutations in the JAK and STAT genes in humans have provided further evidence of the importance of IFNs in host protection against viruses, as well as other types of pathogen, although the relative contribution of type I and type III IFNs is unclear, given that these mutations affect signalling downstream of both IFN receptors31,32. That the IFNα/β and IFNλ pathways often intersect in antiviral responses is supported by studies of patients who are infected with hepatitis C virus (HCV). In these patients, single nucleotide polymorphisms in the interleukin-28 (IL28) locus (which encodes IFNλ subtypes) are predictive of a successful response to treatment with IFNα (or the drug ribavirin), which is associated with a sustained virological response and clearance of the virus33,34,35,36. Recently, a new type III IFN (IFNλ4) has been identified and associated with impairment of spontaneous clearance of HCV3,4.
Recently, during simian immunodeficiency virus (SIV) transmission and acute infection of rhesus macaques, blockade of IFNAR signalling was found to reduce antiviral gene expression, increase the SIV reservoir size and accelerate CD4+ T cell depletion, with progression to AIDS despite a decrease in T cell activation37. Conversely, administration of recombinant IFNα2a initially upregulated the expression of antiviral genes and prevented systemic infection in these animals. However, with continued IFNα2a treatment, animals became desensitized to IFNα/β, and antiviral gene expression decreased, resulting in an increased SIV reservoir size and accelerated CD4+ T cell loss. This study indicates that the timing of IFN-induced innate responses in acute SIV infection markedly affects the overall disease course and outweighs the detrimental consequences of increased immune activation37, and this is likely to be the case for most infections.
So far, relatively few downstream effector ISGs (that is, molecules that are downstream of, but not involved in, the IFN-mediated signalling cascade) have been shown to control viral infection in humans. However, recent studies38,39 found that the ISG IFITM3 controls influenza virus infection in mice in vivo. They also found that an allele of IFITM3 that renders the protein ineffective at restricting the virus in cells in vitro is over-represented in patients requiring hospitalization due to influenza virus infection38 and among patients suffering from severe infection with pandemic influenza virus39. The ISG MX1 also has important antiviral functions in influenza virus infection. Most inbred mouse strains have deletions or point mutations in Mx1 (Ref. 40), and reintroduction of a functional gene into deficient mouse strains markedly increases their resistance to influenza virus infection41. In keeping with this finding, type I IFNs have been shown to provide protection against influenza A virus infection in the presence of MX1 (Ref. 26). However, it should be noted that the strongest phenotype of susceptibility to influenza virus infection has been observed in mice carrying deletions in both the type I and type III IFN receptors29. Human MX1 has antiviral effects in vitro, but whether polymorphisms in the MX1 gene affect susceptibility to influenza virus infection in the human population has not been investigated42.
Enhanced action of dendritic cells and monocytes. The effects of IFNα/β on the host response to infection are not limited to the acute, cell-intrinsic antiviral response described above. IFNα/β have effects on both the innate and adaptive cellular immune response. By contrast, the effects of type III IFNs are largely limited to non-haematopoietic cells, owing to the restricted expression of IFNλR. IFNα/β affect myeloid cells, B cells, T cells and NK cells, thereby enhancing the immune response, more effectively resolving viral infection and improving the generation of memory responses that will allow responses to future viral challenges.
Myriad studies in both human and mouse systems indicate that IFNα/β are involved at various stages in the activation of adaptive immune cell responses by dendritic cells (DCs), either activating or inhibiting these cells depending on the context. IFNα/β variously inhibit or promote the differentiation of precursors into DCs43,44,45,46, and some viruses, such as measles virus and LCMV, can exploit this property to reduce the DC pool47. However, IFNα/β seem to have an activating effect on immature committed DCs, enhancing the cell-surface expression of MHC molecules and co-stimulatory molecules, such as CD80 and CD86, which is associated with an increased ability to stimulate T cells47,48,49. It has also been observed that IFNα/β promote the ability of DCs to cross-present antigens during viral infections, such as vaccinia virus and LCMV infections50,51,52. IFNα/β may also promote the migration of DCs to lymph nodes, through upregulating chemokine receptor expression, thus promoting T cell activation53,54.
DCs are potent producers of IL-12, which is crucial for driving T helper 1 (TH1)-type responses during some bacterial and viral infections, and important for IFNγ production by T cells and NK cells. In some settings, IFNα/β-mediated signalling has been shown to be necessary for IL-12 production by DCs following PRR stimulation55. However, high but physiological levels of IFNα/β strongly inhibit IL-12 production during murine cytomegalovirus (MCMV) and LCMV infections56,57. This suppression of IL-12 production may have developed to favour optimal cytotoxic responses by T cells and NK cells in response to virus, while limiting the pathological effects of excessive IL-12 production56,57,58,59. However, in other situations in which IL-12 production is crucial to the host response, such as during infection with intracellular bacteria, certain pathogens may be able to exploit the suppression of IL-12 by IFNα/β for their own benefit (discussed below).
Promotion of CD4+ and CD8+ T cell responses. In addition to affecting DCs in a manner that drives or inhibits T cell activation as a downstream consequence, IFNα/β can act directly on both CD4+ and CD8+ T cells, influencing their function. IFNα/β have been described to have inhibitory and stimulatory effects on T cell survival and proliferation, cytokine (IFNγ) production, cytotoxic function and memory formation. Detailed dissection of these effects has revealed that these diverse outcomes are controlled by differential levels and differential activation of STAT molecules downstream of IFNAR.
In CD4+ T cells, IFNα/β enhance the ability to help B cells60, as well as survival, and thus clonal expansion in response to viral (LCMV) but not bacterial infection61. In human T cells, IFNα/β promote differentiation into IFNγ-producing TH1 cells62. In LCMV infection, depletion of CD4+ T cells has been shown to prevent lethality in LCMV-infected STAT1-deficient mice and to be associated with a reduction in tissue immunopathology63. In West Nile virus infection, IFNAR signalling controls CD4+ regulatory T cell differentiation, which suggests further effects on CD4+ T cell differentiation and function64. In addition, lymphocyte responses to type I IFNs may be reduced during viral infection, as type I IFNs have been shown to inhibit lymphocyte egress from lymphoid organs during LCMV infection65.
IFNα/β can promote growth-inhibitory signals in CD8+ T cells66,67,68, in line with the known, STAT1-dependent, antiproliferative effects of IFNα/β69,70,71; however, in activated CD8+ T cells and during viral (LCMV and VSV) infection, IFNα/β can also promote the survival and clonal expansion of the CD8+ T cell pool72,73,74,75,76. One possible explanation for these opposing findings may relate to differential STAT signalling downstream of IFNAR because in STAT1-deficient T cells, IFNα/β provide pro-survival and mitogenic signals, possibly through STAT3 and STAT5, rather than antiproliferative signals through STAT1 (Refs 71,77). Furthermore, activated CD8+ T cells 'escape' the antiproliferative effects of IFNα/β during viral (LCMV) infection by expressing lower total levels of STAT1 (Ref. 78). With regard to CD8+ T cell function, cytotoxicity is positively regulated by IFNα/β75,79,80, and IFNγ production is both positively81,82 and negatively83 affected by IFNα/β. This dichotomous outcome depends on the relative levels of STATs, with dominant STAT1 driving inhibition of IFNγ production but STAT4 activation promoting IFNγ production82,83. Therefore, the levels of IFNα/β expressed during a specific infection, the relative strength of the signalling pathways induced and the kinetics of this signalling seem to determine the nature of the CD8+ T cell response that develops76,84. Indeed, it is likely that both the quantity and the timing of type I IFN delivery may be crucial for the consequent adaptive immune responses to infection, as previously reported85.
IFNα/β also influence the differentiation and function of memory CD8+ T cells. By affecting the initial expansion of the T cell pool after infection with viruses such as vaccinia virus, VSV and LCMV, IFNα/β also determine the size of the downstream memory T cell pool74,84,86. Furthermore, IFNα/β support memory T cell effector function and trafficking during secondary infection in several ways, including: driving the cytotoxicity of circulating memory T cells that are recruited to the lungs during respiratory infection with Sendai virus87; promoting chemokine production for the correct trafficking of central memory T cells during recall responses to LCMV88; and driving inflammatory monocytes to produce factors such as IL-15 and IL-18, which support memory CD8+ T cell survival and function in infections, including MCMV infection89. Finally, two recent studies indicate that type I IFNs can protect T cells against NK cell-mediated killing, through inducing the expression of inhibitory NK cell receptor ligands on the target T cells90,91.
Enhancement of NK cell responses. Similarly to their effects on T cells, IFNα/β promote the function and survival of NK cells, through both direct and indirect means. The inflammatory conditions induced by specific viral infections seem to dictate the degree to which direct or indirect effects of IFNα/β modulate NK cell function and which NK cell function is affected. During both influenza virus92 and vaccinia virus93 infections, the direct action of IFNα/β on NK cells is required for the activation and expression of cytolytic effector functions and the production of IFNγ by NK cells. By contrast, in MCMV infection, IFNα/β-mediated signalling through STAT1 is required for NK cell accumulation and cytolytic function but not for IFNγ production94. These effects have also been described to be mediated indirectly through IL-15, with similar findings in TLR-stimulated mice95, although others have reported no requirement for IL-15 (Ref. 96). A recent study investigating the transcriptional response of NK cells and DCs during MCMV infection supports a largely IL-15-dependent role for IFNα/β in this infection97. In this study, the NK cell transcriptional response revealed a relatively weak IFNα/β-responsive profile but a distinct and prolific IL-15-dependent response, whereas DCs had high levels of IFNα/β-inducible gene expression97.
As with T cells, the ability of IFNα/β to induce or restrict IFNγ production by NK cells is related to differential STAT1 and STAT4 signalling. High levels of STAT1-dependent signalling inhibit IFNγ production by NK cells, whereas high basal levels of STAT4 prime NK cells for IFNγ production83,98. Accordingly, the kinetics and levels of IFNα/β production and signalling during infection with viruses such as LCMV and MCMV modulate the IFNγ response by NK cells99.
Enhancement of B cell responses. B cells have an important role in the resolution of many viral infections, largely through the production of neutralizing antibodies. Whereas some studies100,101,102 indicate that IFNα/β may impair the survival and development of precursor and immature B cells, committed B cells seem to benefit from the presence of IFNα/β for various functions.
Similarly to findings with viral protein antigens60,103,104, IFNα/β can promote B cell activation and antibody responses, including class switching, during viral infection. Within the first 48 hours of influenza virus infection, early activation of B cells has been shown to be mediated by IFNAR signalling, resulting in upregulation of activation markers and alteration of the transcriptional response105,106,107. This response involved only the respiratory tract B cells and not systemic B cells105,106, and affected both the magnitude and quality of the antibody response105. IFNα/β have also been reported to 'fine-tune' B cell antibody class switching between IgG subtypes during influenza virus infection108. Interestingly, although IFNα/β seem to be beneficial for the antibody response early in infection, at least one study has found that at late time points after influenza virus infection, the antibody titres are higher in IFNAR-deficient mice than in wild-type mice, although the underlying biology has not been explored27.
Similarly to influenza virus infection, IFNα/β are important for early B cell responses during VSV infection109 and for class switching110. Likewise, during West Nile virus infection, IFNα/β are required for B cell activation in the lymph nodes but not in the spleen of infected animals111. Moreover, recent work on VSV infection shows that rather than acting as targets of IFNα/β, B cells in the lymph nodes produced lymphotoxin, driving a protective macrophage phenotype. In the absence of this lymphotoxin, the host-protective IFNα/β were not produced and the mice succumbed to VSV infection112.
Detrimental effects in viral infection
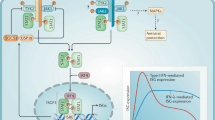
Chronic viral infection. As described above, IFNs contribute to antiviral protection through the induction of an ISG-based cellular antiviral programme and through enhancing immune responses for the efficient termination of infection. However, there is an increasing appreciation that IFNα/β can also be harmful in virus infection, either by inducing immunosuppressive effects that impede viral control113 or by triggering inflammation and tissue damage that exacerbate disease114 (Fig. 2).

a | Infected cells of the vertebrate body produce type I interferons (IFNs) in response to viral infection and/or contact with viral products. Feedback of type I IFNs onto infected and bystander cells leads to the induction of IFN-stimulated genes (ISGs), which function to block the viral replication cycle. Type I IFNs are also produced by, and act on, innate immune cells, including professional antigen-presenting cells (APCs), in response to viral infection and viral products. Type I IFNs acting on APCs can enhance the antigen-presenting function of these cells. They can also enhance the antiviral function of adaptive immune cells, including B cells, T cells and natural killer (NK) cells, which act to restrict viral infection through the production of antibody (B cells) and cytotoxic responses (T cells and NK cells). b | During chronic viral infection, type I IFNs can induce the production of immunosuppressive cytokines such as interleukin-10 (IL-10). They can also induce APCs to express ligands (such as programmed cell death 1 ligand 1 (PDL1)) for T cell-inhibitory receptors (such as PD1, the PDL1 receptor). These factors lead to the suppression of T cell function and failure to clear infection. c | During acute viral infections such as with influenza virus, type I IFN production by myeloid cells, such as plasmacytoid dendritic cells (pDCs) and inflammatory monocytes, leads to the upregulation of expression of both the death ligand TNF-related apoptosis-inducing ligand (TRAIL) on inflammatory monocytes and the TRAIL receptor death receptor 5 (DR5) on epithelial cells. TRAIL-expressing inflammatory monocytes then induce immunopathology and host morbidity and/or mortality through killing epithelial cells. TCR, T cell receptor.
Comparisons of SIV infection in primate species that develop AIDS-like disease and species without disease symptoms indicate that strong IFNα/β responses occur only during pathogenic infection in macaques, whereas natural SIV hosts, without disease progression, have weaker IFNα/β responses115,116. Similar findings have been made in individuals infected with HIV; rapid progressors show stronger IFNα/β signatures than viraemic non-progressors117. These studies suggest a link between sustained IFNα/β levels and disease progression, but the mechanisms involved are as yet unclear. One possibility is that IFN-induced chronic inflammation and immune system activation facilitate the recruitment of target CD4+ T cells and thereby the spread of HIV. Another possibility is that the immunosuppressive effect of IFNα/β113 reduces T cell clonal expansion (through STAT1 signalling) and the ability of T cells to restrict HIV. The negative effects of IFNα/β on CD8+ T cell proliferation may depend on the timing of IFN exposure. Exposure before an antigenic stimulus is suppressive, whereas simultaneous exposure is stimulatory68. It has also been demonstrated in mice that transfer of antigen-specific CD8+ T cells or treatment with polyinosinic–polycytidylic acid (poly(I:C)) causes IFNα/β-dependent apoptosis and thus attrition of bystander CD8+ T cells118. Similar type I and type III IFN-dependent suppression has been shown in vitro for human CD4+ T cells co-cultured with monocyte-derived DCs infected with respiratory syncytial virus119. The signalling mechanisms that control whether T cell clonal expansion is limited after exposure to IFNα/β are relatively well described (for examples, see Refs 78,120), whereas the outcomes of viral infection in the presence of this IFNα/β-mediated suppression require more investigation.
TNF-related apoptosis-inducing ligand (TRAIL; also known as TNFSF10) and its receptor death receptor 5 (DR5; also known as TNFRSF10B) have been suggested as candidates that link high IFNα/β levels to lymphocyte death. For example, in a study of individuals infected with HIV, the IFNα/β expression by pDCs and the TRAIL and DR5 expression levels in tonsil tissue were higher in progressors than in non-progressors121. Similarly, an in vitro study showed that HIV caused IFNα/β-mediated upregulation of TRAIL expression by pDCs, enabling these cells to induce TRAIL-dependent CD4+ T cell apoptosis122. DR5 expression has also been found to be increased on CD4+ T cells in the blood of HIV-infected individuals123, and B cells undergo apoptosis in a TRAIL-dependent manner in HIV infection124. In another chronic viral infection (HCV), it has been shown in the human hepatoma cell line Huh-7 that caspase 8, DR5 and TRAIL function alone or together to increase apoptosis in response to exogenously added type I IFNs125,126,127. However, the extent to which these mechanisms are mediating immunosuppression and/or immunopathology in patients with hepatitis will require further investigation.
Two recent in vivo studies have identified suppressive mechanisms involved in the harmful effects of IFNα/β in chronic viral infection128,129. Blocking IFN-mediated signalling, through antibody administration or receptor deficiency, improved CD4+ T cell-mediated virus control in chronic infection with LCMV clone 13. Furthermore, IFNα/β reduced T cell responses, through the induction of immunosuppressive genes such as those encoding IL-10 and programmed cell death 1 ligand 1 (PDL1; also known as CD274).
Acute viral infection. As discussed above, both type I and type III IFNs contribute to protection against influenza virus infection. The disease-promoting effects of IFNα/β in an acute viral infection, such as influenza virus infection, were discovered more recently and were perhaps more surprising given the well-established antiviral activities of these IFNs. It was shown that severe influenza virus infection is associated with TRAIL-mediated epithelial cell damage130 and that IFNα/β can induce TRAIL expression by inflammatory monocytes131. Similarly, exposure to influenza virus was shown to induce TRAIL expression by human pDCs in vitro132, but the involvement of IFNα/β was not assessed in this study.
When inbred mouse strains (MX1 deficient) were ranked according to susceptibility to influenza virus and their IFNα/β levels were assessed, susceptible strains were found to have a stronger and more sustained IFNα/β signal than resistant strains, even at early time points when no differences in virus titres were detected114. Higher pDC numbers and higher levels of pro-inflammatory cytokines were found in susceptible strains compared with resistant strains, and blocking the IFNα/β signal in susceptible strains, through receptor deficiency or pDC removal, reduced the inflammation and lung damage, resulting in improved survival114. The pathogenic mechanism downstream of type I IFNs was found to be upregulation of TRAIL expression by monocytes and DR5 expression by epithelial cells114. Thus, excessive levels of IFNα/β can contribute to immunopathology in severe influenza virus infection, mainly by inducing immune cell-mediated tissue damage, although the response in MX1-sufficient mice remains to be studied.
In addition to TRAIL, expression of the apoptosis-inducing ligand CD95 ligand (CD95L; also known as FASL) has been shown to be upregulated in an IFN-dependent manner in severe influenza virus infection, and the presence of a functional mutation in the CD95L gene or blockade of the CD95–CD95L interaction has been found to reduce the mortality after high-dose influenza virus infection133. In contrast to the effects in chronic viral infection, it seems that most of the disease-promoting effects of IFNα/β in acute influenza virus infection involve the induction of immunopathology rather than the suppression of the antiviral adaptive immune response, as the virus titres are mostly unaffected. However, IFNα/β-dependent PDL1 expression by influenza virus-infected airway epithelial cells has been shown to suppress the function of T cells expressing programmed cell death 1 protein 1 (PD1; also known as PDCD1)134. Similarly, influenza virus-induced TRAIL expression by mouse CD8+ T cells has been found to control the magnitude of the CD8+ T cell response135 (although the role of IFNs in this mechanism was not assessed in this study), indicating that immunosuppressive pathways similar to those in chronic viral infection are also active in acute infections such as influenza virus infection.
In conclusion, a theme emerges: IFNα/β mediate the upregulation of expression of apoptosis-inducing proteins, which, if expressed by non-haematopoietic somatic cells, mediate tissue damage. The same molecules, when induced on immune cells by IFNα/β, can contribute to immunosuppression in a similar manner to PDL1 and IL-10. Therefore, depending on the pathogen, the host and the context, type I IFNs can have protective effects in viral infection or can contribute to immunosuppression or immunopathology (Fig. 2).
Protective effects in bacterial infection
As seen in viral infection, IFNα/β can be protective or detrimental to the host during bacterial infection in a bacterium-specific manner, although less is known about the role of these IFNs in bacterial infections than in viral infections136. Immunity to intracellular bacteria relies on TH1 cell responses, which activate macrophages and other phagocytic cells to kill intracellular bacteria. By contrast, immunity to extracellular bacteria typically requires a combination of antibody responses, activation of phagocytic cells (such as neutrophils) and TH17 cell responses.
Many of the cytokines and chemokines responsible for coordinating these responses are IFN inducible (mainly through IFNγ), as are many of the antibacterial effector molecules, such as indoleamine 2,3-dioxygenase (IDO), inducible nitric oxide synthase (iNOS; also known as NOS2), immunoresponsive genes and guanylate-binding proteins136. Conversely, under different conditions, IFNα/β can inhibit the induction of many of these host antibacterial effector mechanisms, chemokines and pro-inflammatory cytokines. The mechanisms by which IFNα/β promote host protection or susceptibility to bacterial pathogens are as yet poorly defined, and the factors that determine whether a response will be protective or pathogenic are not yet fully understood.
Some of the earliest reports of a protective role of IFNs were in infection with chlamydial species. Treatment with exogenous IFNs or IFN-inducing agents such as poly(I:C) was shown to protect mice against Chlamydia trachomatis infection137 and to inhibit intracellular replication of C. trachomatis in various human and mouse cell types138. This protection resulted from IDO-mediated depletion of intracellular l-tryptophan, thereby reducing the availability of this amino acid to intracellular pathogens and thus impeding their survival139. IFNα/β may also be involved in protection against Chlamydia pneumoniae infection, through a cooperative interaction with IFNγ that induces antimicrobial effectors and thereby suppresses bacterial survival140,141. However, IFNα/β are not universally protective against chlamydial species, as Ifnar1−/− mice are protected against Chlamydia muridarum infection, showing longer survival and lower bacterial loads than wild-type controls142.
IFNα/β also protect macrophages and lung epithelial cells in vitro against infection with Legionella pneumophila, the causative agent of Legionnaires' disease143,144,145. Ifnar1−/− macrophages have been found to have higher bacterial loads than wild-type cells144, and the treatment of both cell types with IFNα/β has been shown to restrict intracellular bacterial growth143,144,145. The mechanisms underlying this protective effect have not been fully elucidated but were found to be STAT1, STAT2 and STAT3 independent in macrophages, and were associated with polarization towards a classically activated M1 macrophage phenotype and the induction of iNOS expression144. Similar inhibition of bacterial growth has been observed in IFNα/β-treated human macrophages infected with Bacillus anthracis, suggesting that IFNα/β have a protective role against anthrax146.
In addition to promoting the restriction of bacterial growth and bacterial killing within cells, IFNα/β may prevent or reduce cellular invasion by invasive gut bacteria, such as Shigella flexneri and Salmonella enterica subsp. enterica serovar Typhimurium. Treatment with IFNα/β increased the survival of mice infected with S. flexneri or S. Typhimurium and reduced the invasion of their intestinal epithelial cells in vivo, as well as the invasion of fibroblasts in vitro147,148.
A protective role for IFNα/β has also been reported in mouse models of group B streptococcus, Streptococcus pneumoniae, Escherichia coli, Helicobacter pylori and Streptococcus pyogenes infections12,149,150,151, as well as in a model of caecal ligation and puncture152. In all of these infections, Ifnar1−/− mice had a shorter survival and/or more bacterial growth than wild-type controls. By contrast, type I IFNs have been shown to have adverse effects in colon ascendens stent peritonitis, which is a model of peritoneal sepsis153.
In the case of the immune response to group B streptococcus, E. coli and S. pneumoniae, IFNα/β-mediated signalling contributed to the optimal activation of macrophages, in terms of their ability to produce tumour necrosis factor (TNF) and nitric oxide, although the plasma TNF and IL-6 levels during in vivo infection were much higher in Ifnar1−/− mice than in wild-type controls, which may reflect greater inflammation as a result of the higher bacterial burden in the knockout mice or may reflect multiple effects of IFNα/β at the systemic level versus the local level149. IFNα/β may also contribute to the production of host-protective cytokines during S. Typhimurium infection, inducing strong IFNγ production in an IL-12-independent manner, although the direct contribution of this response to host protection has not been established154.
IFNα/β-mediated signalling downstream of NOD1 signalling has been shown to have a role in protecting intestinal epithelial cells against H. pylori infection12. Although the mechanism of protection was not fully elucidated, impairment of chemokine and IFNγ production in the absence of IFNα/β-mediated signalling was implicated. In addition, the importance of the correct recruitment of host-protective phagocytic cells by IFNα/β- dependent chemokine production has been highlighted by results from a caecal ligation and puncture model of infection152. In this model, Ifnar1−/− mice have a shorter survival and elevated bacteraemia compared with wild-type control mice. These differences were associated with decreased levels of CXC-chemokine ligand 10 (CXCL10) and with reduced neutrophil numbers and function. Treatment of Ifnar1−/− mice with recombinant CXCL10 rescued them from fatal infection and restored neutrophil function. Conversely, during subcutaneous S. pyogenes infection, Ifnar1−/− mice had increased tissue damage and a shorter survival after infection than did wild-type mice, and these were associated with uncontrolled neutrophilia at the disease site, although whether neutrophils had a detrimental role in this case was not confirmed152.
Therefore, the induction of cell-intrinsic immunity to kill bacteria or prevent their invasion and the regulation of chemokines, pro-inflammatory cytokines and phagocytic cells, are all implicated as mechanisms by which IFNα/β suppress bacterial infection, with the exact mechanisms involved being dependent on the pathogen.
Detrimental effects in bacterial infection
Perhaps the two best-described examples of a harmful role for IFNα/β are in infections with Listeria monocytogenes and Mycobacterium tuberculosis. These pathogens are intracellular, preferentially infect macrophages and require broadly similar immune responses for their control.
Infection with L. monocytogenes. Three research groups initially described the first important mechanism of host immunosuppression by IFNα/β in bacterial infections: Ifnar1−/− mice are resistant to L. monocytogenes infection, with a longer survival, and lower spleen and liver bacterial loads after infection than wild-type mice155,156,157. The main mechanism attributed to this resistance was reduced apoptotic cell death, particularly of lymphocytes, with IFNα/β sensitizing these cells to the L. monocytogenes virulence factor listeriolysin O and resultant cell death in wild-type mice156,157,158. This reduced cell death was also associated with lower levels of expression of IFN-inducible apoptosis-associated genes, such as TRAIL, p53 and death domain-associated protein 6 (DAP6; also known as DAXX), in infected Ifnar1−/− mice157. Subsequent induction of immunosuppressive cytokines, particularly IL-10, after this large-scale apoptosis of lymphoid cells was suggested as the mechanism by which lymphocyte apoptosis led to the IFNα/β-dependent increase in susceptibility to infection158.
Decreased expression of pro-apoptotic genes has also been reported in infected Ifnar1−/− bone marrow-derived macrophages compared with wild-type cells157. Several other reports have also suggested that macrophages are targets of IFNα/β-induced cell death following L. monocytogenes infection159,160,161. This cell death can take the form of apoptosis that is STAT1 dependent but iNOS and PKR independent159 or of necrotic cell death that is iNOS dependent but TRAIL and PKR independent160,161 and is related to STAT1-dependent breakdown of the plasma membrane160. The death of myeloid cells may be involved in pathology in vivo, as increased levels of host-protective TNF- and iNOS-producing DCs (TIP-DCs) have been reported in Ifnar1−/− mice following L. monocytogenes infection155. However, the overall role of TIP-DCs in this infection may be ambiguous, as it was shown in IFNβ reporter mice that they are also an important source of IFNβ during infection162. Interestingly, CD11b+ DCs seem to be one of the main IFNβ-producing cells during L. monocytogenes infection163. This finding might suggest that IFNα/β production is a method of self-regulation by immune cells, which in this case is subverted by L. monocytogenes for its own advantage. However, whether TIP-DCs, as well as CD11b+ DCs, are themselves targets of IFNα/β-induced cell death remains unclear.
A second important mechanism of host immunosuppression by IFNα/β was elucidated in later studies. During infection with pathogens such as L. monocytogenes, the activation of macrophages by T cell-derived and/or NK cell-derived IFNγ is crucial for inducing antimicrobial pathways and for the subsequent eradication of the intracellular bacteria136. Although IFNα/β can induce some of these antimicrobial pathways in particular circumstances, it has now been shown that during infection with L. monocytogenes, IFNα/β potently inhibit these pathways by blocking the responsiveness of macrophages to IFNγ164. This block in responsiveness results from downregulation of IFNγR expression by macrophages164, owing to silencing of new transcription from the gene encoding IFNγR (Ifngr1) by repressive transcriptional regulators165.
Infection with M. tuberculosis. Studies performed in patients and mouse models of infection collectively point to a detrimental role of IFNα/β during tuberculosis. Several studies have reported a decreased bacterial load and/or improved host survival in the absence of IFNα/β-mediated signalling166,167,168,169. However, these changes have not been universally observed170, and there has not always been concordance between studies regarding bacterial load and survival data. It is likely that the differences between studies result from differences in experimental protocols, and in the genetics of the host and the M. tuberculosis strain used.
The importance of type I IFNs as a potentially detrimental factor during tuberculosis was suggested by studies of patient cohorts from the United Kingdom and South Africa171. Patients with active tuberculosis had a prominent whole blood IFNα/β-inducible transcriptional profile that correlated with the extent of radiographic disease and diminished with successful treatment171. Several other studies have since verified these findings in additional patient cohorts from Africa172,173 and Indonesia174, suggesting that this IFNα/β-inducible signature is broadly applicable to humans and may be detrimental.
IFNα/β overexpression during M. tuberculosis infection in experimental mouse models has provided additional robust evidence for the detrimental effects of the IFNα/β system during tuberculosis. Studies of infection with hyper-virulent M. tuberculosis strains showed a correlation between increased levels of IFNα/β and increased virulence166,167,169. Direct instillation of IFNα/β into the lungs during infection was also injurious to the host169. Similarly, enhanced induction of IFNα/β expression during M. tuberculosis infection via administration of a TLR3 ligand derivative led to an increased severity of infection175,176. Likewise, deletion of the gene encoding a negative regulator of IFNα/β, MAPK kinase kinase 8 (MAP3K8; also known as TPL2), that functions downstream of TLRs led to increased levels of IFNα/β production and increased bacterial burdens177, and these increases were abrogated in Map3k8−/−Ifnar1−/− (double knockout) mice during M. tuberculosis or L. monocytogenes infection. Control of the bacterial load in Map3k8−/−Ifnar1−/− mice was correlated with reduced IL-10 levels and increased IL-12 levels in the serum. Finally, concurrent co-infection of mice with influenza A virus and M. tuberculosis results in increased bacterial loads in an IFNα/β-dependent manner178, as seen for other pathogen co-infections as outlined in Box 1.
The mechanisms that mediate the IFNα/β-driven exacerbation of disease are not fully understood but seem to be multifactorial. Data from investigations of hyper-virulent M. tuberculosis strains initially suggested that the suppression of pro-inflammatory cytokines and of TH1-type immunity are important166,167,169, and there is good evidence both in human cells and in mouse models that IFNα/β suppress the production of host-protective cytokines following M. tuberculosis infection. The production of IL-1α and IL-1β, which are crucial for host defence against M. tuberculosis179, is inhibited by IFNα/β, both in vitro in infected human and mouse cells and in vivo in mouse models176,180,181,182. This finding is in line with a previous study using lipopolysaccharide that showed that IFNα/β can potently inhibit the NOD-, LRR- and pyrin domain-containing 1 (NLRP1) and NLRP3 inflammasomes, which are responsible for the post-translational maturation of IL-1β183.
In addition, cell-intrinsic type I IFN signals have been shown to negatively regulate iNOS production by pulmonary myeloid cells, particularly TIP-DCs176. The production of other pro-inflammatory cytokines such as TNF and IL-12 is also negatively affected177,180,182 (K.M.-B. and A.S., unpublished observations). The induction of the immunosuppressive cytokines IL-10 and IL-1 receptor antagonist by IFNα/β seems to have an important role in this suppression of pro-inflammatory cytokines176,177,180,182.
In turn, IL-1α and IL-1β have recently been shown to inhibit IFNα/β induction in mouse and human macrophages, and when IL-1 was present in IFNα/β-treated cultures, it also suppressed the pro-bacterial effects downstream of IFNβ184. Interestingly, IL-1-induced prostaglandin E2 was also able to potently inhibit IFNα/β in this context184, as observed previously in lipopolysaccharide-induced IFNα/β responses185 and more recently during influenza virus infection186. Moreover, investigating the effects of prostaglandin E2 during M. tuberculosis infection, either by directly administering this prostanoid or by increasing its level through 5-lipoxygenase blockade with zileuton, reversed poly(I:C)-mediated IFNα/β-driven mortality184.
Similarly to the findings in L. monocytogenes infection, the repression of innate cell responsiveness to IFNγ is emerging as an important mechanism of IFNα/β-mediated immunosuppression during mycobacterial infection180,182,187. However, direct downregulation of IFNγR expression may not be the central mechanism by which IFNα/β exert their effects on IFNγ activity176. Instead, in both mouse and human cells, it has been shown that IFNα/β potently suppress the ability of macrophages to upregulate antimycobacterial effector molecules and to restrict bacterial growth, in response to both Mycobacterium leprae and M. tuberculosis180,187 (F.M., J. Ewbank and A.O., unpublished observations; K.M.-B., unpublished observations). The importance of this mechanism of action of IFNα/β is further suggested by experiments using Ifngr1−/−Ifnar1−/− mice, which suggest that IFNα/β contribute to host protection in the absence of the IFNγ pathway188. Furthermore, the observation of naturally occurring mutations in the host-protective gene ISG15 in humans suggests that IFNα/β can induce host-protective responses to mycobacterial infection, although the circumstances under which IFNα/β induce this gene during M. tuberculosis infection are unclear189.
Additionally, the production of innate cytokines such as IL-12p70 has also been shown to be suppressed by IFNα/β during M. tuberculosis infection180,182,187 (K.M.-B., unpublished observations). This suppression could result from the presence of IL-10, the downregulation of IFNγR and/or the induction of negative regulators of IFN-mediated signalling such as protein arginine methyltransferase 1 (PRMT1)180,182,187. Finally, IFNα/β, possibly by influencing chemokine expression, have been shown to be involved in the generation and trafficking of M. tuberculosis-permissive innate cells to the lungs in a mouse model, thus contributing to the exacerbation of infection175,190.
Infection with Francisella tularensis and Francisella tularensis subsp . novicida. The facultative intracellular bacterium F. tularensis and the subspecies F. tularensis subsp. novicida, which is highly pathogenic in mice, have been investigated for a possible role of IFNα/β in the immune response to infection191,192,193. Two studies found that type I IFNs were necessary for activation of the inflammasome during F. t. novicida192 or F. tularensis191 infection and that the AIM2 inflammasome, in turn, was necessary for host protection against F. tularensis191. This finding is in contrast to those of another study showing that IFNα/β inhibit inflammasomes (see above) and that type I IFN-dependent AIM2 inflammasomes were triggered in vitro during mycobacterial infection but that their role is unclear in vivo194, suggesting that IFNα/β may have differential effects on inflammasome activity, depending on the type of inflammasome involved.
Similarly to infection with L. monocytogenes, IFNα/β have been shown to be involved in the apoptosis of macrophages during F. t. novicida infection192, although this cell death did not correlate with the outcome for the host. Despite these data indicating that IFNα/β may mediate some host-protective mechanisms during these infections, a comparison of wild-type and Ifnar1−/− mice infected with F. t. novicida revealed that IFNα/β are detrimental to the host, restricting the development of a protective IL-17-producing γδ T cell response193.
Infection with other bacteria. A limited range of studies further implicate IFNα/β in enhancing susceptibility to various other bacterial agents. IFNα/β have been suggested to be detrimental factors during Whipple's disease (caused by Tropheryma whipplei), diverting macrophages to an alternatively polarized, permissive state and promoting macrophage apoptosis195.
IFNα/β are also detrimental during Brucella abortus infection, with Ifnar1−/− mice having lower bacterial loads than wild-type controls196. Bacterial control in these mice is correlated with increased IFNγ and nitric oxide production, and reduced TRAIL expression and apoptosis196. Ifnar1−/− mice are also reportedly more resistant to infection with the plague agent Yersinia pestis197. This resistance was associated with an increased number of neutrophils and enhanced function of phagocytic cells197. In contrast to earlier reports147,154, it has been found that IFNα/β were harmful to the host during S. Typhimurium infection198. Protection in these mice was associated with macrophage resistance to necroptosis rather than to alterations in cytokine production or inflammasome activation.
IFNα/β have also been implicated in mediating deleterious inflammation during infection with a large range of Gram-negative bacteria through the activation of caspase 11, leading to the production of IL-1β and IL-18, and caspase 1-independent cell death199. Another study also found a role for IFNα/β in inducing the activation of caspase 11 during S. Typhimurium infection. This activation resulted in macrophage cell death that was injurious to the host, but only in the absence of caspase 1, which was required for the antibacterial function of neutrophils200.
IFNα/β are detrimental for the host during Staphylococcus aureus infection, with more Ifnar1−/− mice than wild-type mice surviving after intranasal infection201. Protection correlated with an increased proportion of CD11c+ cells within the total population of airway and lung immune cells, and reduced pro-inflammatory cytokine production in the lungs.
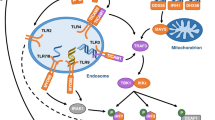
In conclusion, IFNα/β may contribute to host protection against bacterial infection by upregulating antimicrobial effectors, such as IDO, iNOS and pro-inflammatory cytokines. Conversely, IFNα/β may impair the host response to bacteria by eliciting the production of IL-10 and IL-1 receptor antagonist, suppressing pro-inflammatory cytokine production, inducing immune cell death (including apoptosis) and restricting host responses to IFNγ (Figs 3,4).

Low level autocrine interferon-α/β (IFNα/β)-mediated signalling primes the production of interleukin-10 (IL-10), pro-inflammatory cytokines and antimicrobial effector mechanisms. Type I IFNs induce IL-1 receptor antagonist (IL-1RA), which in turn inhibits IL-1-mediated signalling. IL-10 mediates a negative feedback loop, suppressing the production of pro-inflammatory cytokines, including IL-12, tumour necrosis factor (TNF) and IL-1α/β. On infection, high levels of IFNα/β, which affect myeloid cells, can be contributed by autocrine production, as well as from exocrine cellular sources. IFNα/β can also suppress pro-inflammatory cytokine production in an IL-10-independent manner. A major type I IFN-suppressive mechanism is downregulation of the IFNγ receptor (IFNγR), thus abrogating IFNγ-dependent host-protective immune responses. IFNα/β-mediated signalling can promote the production of high levels of IL-10, as well as the induction of pro-apoptotic factors. IL-1α and IL-1β induce cyclooxygenase 2 (COX2)-dependent prostaglandin E2 (PGE2). PGE2 and IL-1 inhibit type I IFN expression and the downstream effects. IFNAR, type I IFN receptor; IL-1R, IL-1 receptor.

The diagram indicates the mechanistic processes that are influenced by interferon-α/β (IFNα/β) during bacterial infections. The small vertical arrows indicate whether IFNα/β promote (arrow pointing upwards), suppress (arrow pointing downwards) or have variable, context-dependent effects (two arrows) on the associated process. For each process, the organisms that cause infections in which IFN-mediated effects may occur are shown. In green are those infections in which IFNα/β are thought to be protective, in red are those in which IFNα/β have host-detrimental effects, and in purple are those in which IFNα/β have both host protective and detrimental effects. For example, IFNα/β have variable effects on chemokine production and cell migration. In Streptococcus pyogenes infection (in which IFNα/β are protective), IFNα/β have promoting effects on chemokine production and cell migration. B. abortus, Brucella abortus; C. albicans, Candida albicans; CLP, caecal ligation and puncture; C. neoformans, Cryptococcus neoformans; C. pneumoniae, Chlamydia pneumoniae; C. trachomatis, Chlamydia trachomatis; E. coli, Escherichia coli; F. t. novicida, Francisella tularensis subsp. novicida; H. pylori, Helicobacter pylori; IDO, indoleamine 2,3-dioxygenase; IL, interleukin; IL-1RA, IL-1 receptor antagonist; iNOS, inducible nitric oxide synthase; L. major, Leishmania major; L. monocytogenes, Listeria monocytogenes; L. pneumophila, Legionella pneumophila; M. leprae, Mycobacterium leprae; M. tuberculosis, Mycobacterium tuberculosis; P. berghei, Plasmodium berghei; PDL1, programmed cell death 1 ligand 1; S. aureus, Staphylococcus aureus; S. flexneri, Shigella flexneri; S. pneumoniae, Streptococcus pneumoniae; S. Typhimurium, Salmonella enterica subsp. enterica serovar Typhimurium; TNF, tumour necrosis factor; T. whipplei, Tropheryma whipplei; Y. pestis, Yersinia pestis.
Effects in parasitic and fungal infection
Analyses of the effects of IFNα/β on the course of disease during parasitic and fungal infections have been relatively limited, with most work carried out in Leishmania major, Plasmodium spp. and Trypanosoma cruzi models of parasite infection and Candida spp. (yeast) models of fungal infection (Fig. 4).
Parasitic infection. Work conducted during the late 1990s and early 2000s elucidated an important role for IFNα/β in inducing iNOS expression during L. major infection202,203,204. Interestingly, it was noted that high levels of IFNα/β actually impaired iNOS induction, implicating IFN levels as important in determining whether IFNα/β had a host-protective or pathogenic role203,204. More recent work with different strains of Leishmania spp. suggests a detrimental role for IFNα/β, through inhibiting macrophage function and regulating neutrophil number and function205,206.
During malaria, IFNα/β can have either a host- protective or detrimental effect, depending on both the stage of infection and the species of infecting Plasmodium. In the blood stages of infection with the mouse malaria parasites Plasmodium berghei and Plasmodium chabaudi, IFNα/β enhance infection through inhibiting CD4+ T cell function207. By contrast, studies of Plasmodium yoelii infection indicate a protective role for IFNα/β, possibly through inhibiting reticulocytosis, a condition in which immature red blood cells accumulate208. Treatment with recombinant IFNα also has been reported to protect mice from developing the cerebral malaria induced by the P. berghei strain ANKA, in part through enhancing the TH1 cell response209. However, using Ifnar1−/− mice, another study has reported only a minor influence of IFNα/β during acute P. chabaudi infection210. An interesting recent report has shown that during the liver stage of infection, P. berghei induces an IFNα/β response that is essential for host protection211. This protection, mediated through cytosolic recognition of parasite RNA by the PRR MDA5, was associated with IFNα/β-dependent recruitment of leukocytes to infectious foci. It remains to be seen whether this host resistance-promoting function of IFNα/β in the liver stages of malaria is specific to the parasite species and whether it occurs in human malarial infection.
Studies of infection with the protozoan parasite T. cruzi show various effects of IFNα/β on host immunity, including positive effects212,213,214, negative effects215 and no difference216. The reasons for these differences are not fully understood but may relate to the route of infection, as studies showing a positive role for IFNα/β used the intraperitoneal route212,213,214, whereas those showing a negative role used intradermal infection215. The levels of IFNα/β-mediated signalling that are induced may also be crucial, as Ifnar1−/− mice reportedly succumbed earlier than wild-type mice, yet mice lacking the ubiquitin-specific protease UBP43, which are hyper-responsive to IFNα/β, were also more susceptible than wild-type mice214. Finally, the relative balance between the effects on the innate immune response and the adaptive immune response seems to be important. In the absence of the innate immune signalling molecules MYD88 and/or TRIF, IFNα/β are important for host protection213, as well as for nitric oxide generation212. However, IFNα/β also inhibit the production of the host protective cytokine IFNγ during T. cruzi infection215, and this cytokine is most probably produced by T cells, because NK cells reportedly do not require IFNα/β for IFNγ production in this infection216.
Fungal infection. Studies of IFNα/β during fungal infection have generated conflicting results. Several findings suggest that IFNα/β have a host-protective contribution to immunity to Candida albicans, Saccharomyces cerevisiae and Cryptococcus neoformans217,218,219. IFNα/β-mediated signalling has been found to be required for various processes, including inducing the reactive oxygen intermediates that are necessary for the killing of C. albicans by phagocytic cells218, for maintaining a TH1-like immune response (high IFNγ, TNF, iNOS and CXCL10 levels) to C. neoformans217 and for attracting leukocytes (particularly neutrophils) to the disease site during C. albicans infection219. Interestingly, another study of C. albicans infection, in wild-type and Ifnar1−/− mice, found a similar requirement for IFN-mediated signalling for attracting neutrophils and inflammatory monocytes to the disease site; however, in this study, these cells had no effect on fungal burden but rather caused lethal immunopathology220. The reason for these opposing findings is unclear; however, given the very similar infection protocols used, it is possible that the differences are due to variations in the microbiota at different animal facilities. IFNα/β have also been found to mediate the poly(I:C) sensitization of mice to C. albicans, through suppressing IL-1β183. IFNα/β have also been implicated in sensitizing the host in infections with Candida glabrata and Histoplasma capsulatum, although the mechanism was not investigated in these cases221,222.
Studies of humans with inherited errors in immune signalling components may provide the strongest clues to the role of IFNα/β in fungal infections. Whole exome sequencing and genome-wide association studies looking for the genetic aetiologies of chronic mucocutaneous candidiasis have identified mutations in STAT1 in some patients223,224 (reviewed in Ref. 31). The same STAT1 mutations were also found in patients with disseminated disease caused by other fungal pathogens such as H. capsulatum31. Interestingly, these mutations are gain-of-function and dominant, suggesting that IFNα/β potentially has a detrimental role in the response to fungal infection, possibly through suppressing TH17 cell responses223. However, other cytokines that depend on STAT1 for signalling, such as IFNγ and IL-27, may also be responsible (Fig. 4).
Closing remarks
Type I IFNs are among the first cytokines whose production is induced by a plethora of cells during infection. Owing to the broad distribution of expression of IFNAR, IFNα/β have wide-ranging effects, on epithelial cells and innate and adaptive immune cells. The net effect of IFNα/β on protection or pathogenesis during infection is determined by the type and dose of pathogen, as well as by the genetic background of the host and possibly the microbiota (Box 2). Progress is needed to better understand, first, the precise regulation of the induction of IFNα/β at the transcriptional and post-transcriptional levels and, second, the factors that determine responsiveness to IFNα/β. Such knowledge will allow researchers to uncover mechanisms to harness the immune response for maximum host protection with minimum damage.
References
Pestka, S., Krause, C. D. & Walter, M. R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202, 8–32 (2004).
Schoenborn, J. R. & Wilson, C. B. Regulation of interferon-γ during innate and adaptive immune responses. Adv. Immunol. 96, 41–101 (2007).
O'Brien, T. R., Prokunina-Olsson, L. & Donnelly, R. P. IFN-λ4: the paradoxical new member of the interferon λ family. J. Interferon Cytokine Res. 34, 829–838 (2014).
Prokunina-Olsson, L. et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nature Genet. 45, 164–171 (2013).
Witte, K., Witte, E., Sabat, R. & Wolk, K. IL-28A, IL-28B, and IL-29: promising cytokines with type I interferon-like properties. Cytokine Growth Factor Rev. 21, 237–251 (2010).
Durbin, R. K., Kotenko, S. V. & Durbin, J. E. Interferon induction and function at the mucosal surface. Immunol. Rev. 255, 25–39 (2013).
Yan, N. & Chen, Z. J. Intrinsic antiviral immunity. Nature Immunol. 13, 214–222 (2012).
Goubau, D., Deddouche, S. & Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 38, 855–869 (2013).
Paludan, S. R. & Bowie, A. G. Immune sensing of DNA. Immunity 38, 870–880 (2013).
Leber, J. H. et al. Distinct TLR- and NLR-mediated transcriptional responses to an intracellular pathogen. PLoS Pathog. 4, e6 (2008).
Pandey, A. K. et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog. 5, e1000500 (2009).
Watanabe, T. et al. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J. Clin. Invest. 120, 1645–1662 (2010).
Moreira, L. O. & Zamboni, D. S. NOD1 and NOD2 signaling in infection and inflammation. Front. Immunol. 3, 328 (2012).
Moynagh, P. N. TLR signalling and activation of IRFs: revisiting old friends from the NF-κB pathway. Trends Immunol. 26, 469–476 (2005).
Honda, K., Takaoka, A. & Taniguchi, T. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360 (2006).
Tamura, T., Yanai, H., Savitsky, D. & Taniguchi, T. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 26, 535–584 (2008).
Ivashkiv, L. B. & Donlin, L. T. Regulation of type I interferon responses. Nature Rev. Immunol. 14, 36–49 (2014). This review is a perfect prelude to the present review and describes the molecular mechanisms of regulation of type I IFNs in more detail.
Rauch, I., Muller, M. & Decker, T. The regulation of inflammation by interferons and their STATs. JAKSTAT 2, e23820 (2013).
Versteeg, G. A. & Garcia-Sastre, A. Viral tricks to grid-lock the type I interferon system. Curr. Opin. Microbiol. 13, 508–516 (2010).
McNab, F. W., Rajsbaum, R., Stoye, J. P. & O'Garra, A. Tripartite-motif proteins and innate immune regulation. Curr. Opin. Immunol. 23, 46–56 (2011).
Diamond, M. S. & Schoggins, J. W. Host restriction factor screening: let the virus do the work. Cell Host Microbe 14, 229–231 (2013).
Muller, U. et al. Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921 (1994).
Haller, O., Arnheiter, H., Gresser, I. & Lindenmann, J. Virus-specific interferon action. Protection of newborn Mx carriers against lethal infection with influenza virus. J. Exp. Med. 154, 199–203 (1981).
Durbin, J. E. et al. Type I IFN modulates innate and specific antiviral immunity. J. Immunol. 164, 4220–4228 (2000).
Garcia-Sastre, A. et al. The role of interferon in influenza virus tissue tropism. J. Virol. 72, 8550–8558 (1998).
Koerner, I., Kochs, G., Kalinke, U., Weiss, S. & Staeheli, P. Protective role of β interferon in host defense against influenza A virus. J. Virol. 81, 2025–2030 (2007).
Price, G. E., Gaszewska-Mastarlarz, A. & Moskophidis, D. The role of α/β and γ interferons in development of immunity to influenza A virus in mice. J. Virol. 74, 3996–4003 (2000).
Mordstein, M. et al. λ Interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol. 84, 5670–5677 (2010).
Mordstein, M. et al. Interferon-λ contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 4, e1000151 (2008). This study demonstrates the redundant roles of type I and type III IFNs in the anti-influenza virus response, clarifying the confusion arising from earlier literature that reported that type I IFNs cannot account for the requirement for STAT1 signalling in protection against influenza virus infection.
Crotta, S. et al. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog. 9, e1003773 (2013). This study demonstrates the redundant roles of type I and type III IFN signalling in epithelial cells in the anti-influenza virus response, clarifying the confusion arising from earlier literature over protection against influenza virus infection.
Casanova, J. L., Holland, S. M. & Notarangelo, L. D. Inborn errors of human JAKs and STATs. Immunity 36, 515–528 (2012).
Zhang, S. Y. et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-α/β, IFN-γ, and IFN-λ in host defense. Immunol. Rev. 226, 29–40 (2008).
Suppiah, V. et al. IL28B is associated with response to chronic hepatitis C interferon-α and ribavirin therapy. Nature Genet. 41, 1100–1104 (2009).
Tanaka, Y. et al. Genome-wide association of IL28B with response to pegylated interferon-α and ribavirin therapy for chronic hepatitis C. Nature Genet. 41, 1105–1109 (2009).
Ge, D. et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461, 399–401 (2009).
Thomas, D. L. et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 461, 798–801 (2009).
Sandler, N. G. et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 511, 601–605 (2014).
Everitt, A. R. et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484, 519–523 (2012). This study provided the first evidence of host genetics ( IFITM3 ) contributing to susceptibility to influenza virus infection in humans.
Zhang, Y. H. et al. Interferon-induced transmembrane protein-3 genetic variant rs12252-C is associated with severe influenza in Chinese individuals. Nature Commun. 4, 1418 (2013). This is a follow-up study to reference 38, showing that IFITM3 variants that contribute to the severity of influenza virus infection are predominant in the Chinese population.
Staeheli, P., Grob, R., Meier, E., Sutcliffe, J. G. & Haller, O. Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol. Cell. Biol. 8, 4518–4523 (1988).
Horisberger, M. A., Staeheli, P. & Haller, O. Interferon induces a unique protein in mouse cells bearing a gene for resistance to influenza virus. Proc. Natl Acad. Sci. USA 80, 1910–1914 (1983).
Horby, P., Nguyen, N. Y., Dunstan, S. J. & Baillie, J. K. The role of host genetics in susceptibility to influenza: a systematic review. PLoS ONE 7, e33180 (2012).
Dauer, M. et al. Interferon-α disables dendritic cell precursors: dendritic cells derived from interferon-α-treated monocytes are defective in maturation and T-cell stimulation. Immunology 110, 38–47 (2003).
Lapenta, C. et al. Potent immune response against HIV-1 and protection from virus challenge in hu-PBL-SCID mice immunized with inactivated virus-pulsed dendritic cells generated in the presence of IFN-α. J. Exp. Med. 198, 361–367 (2003).
Santini, S. M. et al. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J. Exp. Med. 191, 1777–1788 (2000).
Santodonato, L. et al. Monocyte-derived dendritic cells generated after a short-term culture with IFN-α and granulocyte-macrophage colony-stimulating factor stimulate a potent Epstein-Barr virus-specific CD8+ T cell response. J. Immunol. 170, 5195–5202 (2003).
Hahm, B., Trifilo, M. J., Zuniga, E. I. & Oldstone, M. B. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity 22, 247–257 (2005).
Ito, T. et al. Differential regulation of human blood dendritic cell subsets by IFNs. J. Immunol. 166, 2961–2969 (2001).
Montoya, M. et al. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 99, 3263–3271 (2002).
Le Bon, A. et al. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nature Immunol. 4, 1009–1015 (2003).
Le Bon, A. et al. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J. Immunol. 176, 4682–4689 (2006).
Spadaro, F. et al. IFN-α enhances cross-presentation in human dendritic cells by modulating antigen survival, endocytic routing, and processing. Blood 119, 1407–1417 (2012).
Parlato, S. et al. Expression of CCR-7, MIP-3β, and Th-1 chemokines in type I IFN-induced monocyte-derived dendritic cells: importance for the rapid acquisition of potent migratory and functional activities. Blood 98, 3022–3029 (2001).
Rouzaut, A. et al. Dendritic cells adhere to and transmigrate across lymphatic endothelium in response to IFN-α. Eur. J. Immunol. 40, 3054–3063 (2010).
Gautier, G. et al. A type I interferon autocrine–paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J. Exp. Med. 201, 1435–1446 (2005).
Cousens, L. P., Orange, J. S., Su, H. C. & Biron, C. A. Interferon-α/β inhibition of interleukin 12 and interferon-γ production in vitro and endogenously during viral infection. Proc. Natl Acad. Sci. USA 94, 634–639 (1997).
Dalod, M. et al. Interferon α/β and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J. Exp. Med. 195, 517–528 (2002).
Orange, J. S., Wolf, S. F. & Biron, C. A. Effects of IL-12 on the response and susceptibility to experimental viral infections. J. Immunol. 152, 1253–1264 (1994).
Orange, J. S. et al. Mechanism of interleukin 12-mediated toxicities during experimental viral infections: role of tumor necrosis factor and glucocorticoids. J. Exp. Med. 181, 901–914 (1995).
Le Bon, A. et al. Enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J. Immunol. 176, 2074–2078 (2006).
Havenar-Daughton, C., Kolumam, G. A. & Murali-Krishna, K. The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J. Immunol. 176, 3315–3319 (2006).
Brinkmann, V., Geiger, T., Alkan, S. & Heusser, C. H. Interferon α increases the frequency of interferon γ-producing human CD4+ T cells. J. Exp. Med. 178, 1655–1663 (1993).
Hofer, M. J. et al. Mice deficient in STAT1 but not STAT2 or IRF9 develop a lethal CD4+ T-cell-mediated disease following infection with lymphocytic choriomeningitis virus. J. Virol. 86, 6932–6946 (2012).
Lazear, H. M., Pinto, A. K., Vogt, M. R., Gale, M. Jr & Diamond, M. S. β-Interferon controls West Nile virus infection and pathogenesis in mice. J. Virol. 85, 7186–7194 (2011).
Shiow, L. R. et al. CD69 acts downstream of interferon-α/β to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440, 540–544 (2006).
Petricoin, E. F. et al. Antiproliferative action of interferon-α requires components of T-cell-receptor signalling. Nature 390, 629–632 (1997).
Kaser, A., Nagata, S. & Tilg, H. Interferon α augments activation-induced T cell death by upregulation of Fas (CD95/APO-1) and Fas ligand expression. Cytokine 11, 736–743 (1999).
Marshall, H. D., Urban, S. L. & Welsh, R. M. Virus-induced transient immune suppression and the inhibition of T cell proliferation by type I interferon. J. Virol. 85, 5929–5939 (2011).
Bromberg, J. F., Horvath, C. M., Wen, Z., Schreiber, R. D. & Darnell, J. E. Jr. Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon α and interferon γ. Proc. Natl. Acad. Sci. USA 93, 7673–7678 (1996).
Lee, C. K., Smith, E., Gimeno, R., Gertner, R. & Levy, D. E. STAT1 affects lymphocyte survival and proliferation partially independent of its role downstream of IFN-γ. J. Immunol. 164, 1286–1292 (2000).
Tanabe, Y. et al. Role of STAT1, STAT3, and STAT5 in IFN-α/β responses in T lymphocytes. J. Immunol. 174, 609–613 (2005).
Marrack, P., Kappler, J. & Mitchell, T. Type I interferons keep activated T cells alive. J. Exp. Med. 189, 521–530 (1999).
Aichele, P. et al. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J. Immunol. 176, 4525–4529 (2006).
Kolumam, G. A., Thomas, S., Thompson, L. J., Sprent, J. & Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 202, 637–650 (2005).
Curtsinger, J. M., Valenzuela, J. O., Agarwal, P., Lins, D. & Mescher, M. F. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 174, 4465–4469 (2005).
Keppler, S. J., Rosenits, K., Koegl, T., Vucikuja, S. & Aichele, P. Signal 3 cytokines as modulators of primary immune responses during infections: the interplay of type I IFN and IL-12 in CD8 T cell responses. PLoS ONE 7, e40865 (2012).
Gimeno, R., Lee, C. K., Schindler, C. & Levy, D. E. Stat1 and Stat2 but not Stat3 arbitrate contradictory growth signals elicited by α/β interferon in T lymphocytes. Mol. Cell. Biol. 25, 5456–5465 (2005).
Gil, M. P., Salomon, R., Louten, J. & Biron, C. A. Modulation of STAT1 protein levels: a mechanism shaping CD8 T-cell responses in vivo. Blood 107, 987–993 (2006).
Agarwal, P. et al. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J. Immunol. 183, 1695–1704 (2009).
Marshall, H. D., Prince, A. L., Berg, L. J. & Welsh, R. M. IFN-α/β and self-MHC divert CD8 T cells into a distinct differentiation pathway characterized by rapid acquisition of effector functions. J. Immunol. 185, 1419–1428 (2010).
Cousens, L. P. et al. Two roads diverged: interferon α/β- and interleukin 12-mediated pathways in promoting T cell interferon γ responses during viral infection. J. Exp. Med. 189, 1315–1328 (1999).
Nguyen, K. B. et al. Critical role for STAT4 activation by type 1 interferons in the interferon-γ response to viral infection. Science 297, 2063–2066 (2002).
Nguyen, K. B. et al. Interferon α/β-mediated inhibition and promotion of interferon γ: STAT1 resolves a paradox. Nature Immunol. 1, 70–76 (2000).
Thompson, L. J., Kolumam, G. A., Thomas, S. & Murali-Krishna, K. Innate inflammatory signals induced by various pathogens differentially dictate the IFN-I dependence of CD8 T cells for clonal expansion and memory formation. J. Immunol. 177, 1746–1754 (2006).
Pinto, A. K. et al. A temporal role of type I interferon signaling in CD8+ T cell maturation during acute West Nile virus infection. PLoS Pathog. 7, e1002407 (2011).
Ramos, H. J. et al. Reciprocal responsiveness to interleukin-12 and interferon-α specifies human CD8+ effector versus central memory T-cell fates. Blood 113, 5516–5525 (2009).
Kohlmeier, J. E., Cookenham, T., Roberts, A. D., Miller, S. C. & Woodland, D. L. Type I interferons regulate cytolytic activity of memory CD8+ T cells in the lung airways during respiratory virus challenge. Immunity 33, 96–105 (2010).
Sung, J. H. et al. Chemokine guidance of central memory T cells is critical for antiviral recall responses in lymph nodes. Cell 150, 1249–1263 (2012).
Soudja, S. M., Ruiz, A. L., Marie, J. C. & Lauvau, G. Inflammatory monocytes activate memory CD8+ T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity 37, 549–562 (2012).
Crouse, J. et al. Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity 40, 961–973 (2014).
Xu, H. C. et al. Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity 40, 949–960 (2014).
Hwang, I. et al. Activation mechanisms of natural killer cells during influenza virus infection. PLoS ONE 7, e51858 (2012).
Martinez, J., Huang, X. & Yang, Y. Direct action of type I IFN on NK cells is required for their activation in response to vaccinia viral infection in vivo. J. Immunol. 180, 1592–1597 (2008).
Nguyen, K. B. et al. Coordinated and distinct roles for IFN-α/β, IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 169, 4279–4287 (2002).
Lucas, M., Schachterle, W., Oberle, K., Aichele, P. & Diefenbach, A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 26, 503–517 (2007).
Sun, J. C., Ma, A. & Lanier, L. L. IL-15-independent NK cell response to mouse cytomegalovirus infection. J. Immunol. 183, 2911–2914 (2009).
Baranek, T. et al. Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe 12, 571–584 (2012).
Miyagi, T. et al. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J. Exp. Med. 204, 2383–2396 (2007).
Mack, E. A., Kallal, L. E., Demers, D. A. & Biron, C. A. Type 1 interferon induction of natural killer cell γ interferon production for defense during lymphocytic choriomeningitis virus infection. MBio 2, e00169-11 (2011).
Wang, J., Lin, Q., Langston, H. & Cooper, M. D. Resident bone marrow macrophages produce type 1 interferons that can selectively inhibit interleukin-7-driven growth of B lineage cells. Immunity 3, 475–484 (1995).
Lin, Q., Dong, C. & Cooper, M. D. Impairment of T and B cell development by treatment with a type I interferon. J. Exp. Med. 187, 79–87 (1998).
Bosio, E., Cluning, C. L. & Beilharz, M. W. Low-dose orally administered type I interferon reduces splenic B cell numbers in mice. J. Interferon Cytokine Res. 21, 721–728 (2001).
Le Bon, A. et al. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 14, 461–470 (2001).
Swanson, C. L. et al. Type I IFN enhances follicular B cell contribution to the T cell-independent antibody response. J. Exp. Med. 207, 1485–1500 (2010).
Coro, E. S., Chang, W. L. & Baumgarth, N. Type I IFN receptor signals directly stimulate local B cells early following influenza virus infection. J. Immunol. 176, 4343–4351 (2006).
Chang, W. L. et al. Influenza virus infection causes global respiratory tract B cell response modulation via innate immune signals. J. Immunol. 178, 1457–1467 (2007).
Rau, F. C., Dieter, J., Luo, Z., Priest, S. O. & Baumgarth, N. B7-1/2 (CD80/CD86) direct signaling to B cells enhances IgG secretion. J. Immunol. 183, 7661–7671 (2009).
Heer, A. K. et al. TLR signaling fine-tunes anti-influenza B cell responses without regulating effector T cell responses. J. Immunol. 178, 2182–2191 (2007).
Fink, K. et al. Early type I interferon-mediated signals on B cells specifically enhance antiviral humoral responses. Eur. J. Immunol. 36, 2094–2105 (2006).
Bach, P. et al. Vesicular stomatitis virus glycoprotein displaying retrovirus-like particles induce a type I IFN receptor-dependent switch to neutralizing IgG antibodies. J. Immunol. 178, 5839–5847 (2007).
Purtha, W. E., Chachu, K. A., Virgin, H. W. & Diamond, M. S. Early B-cell activation after West Nile virus infection requires α/β interferon but not antigen receptor signaling. J. Virol. 82, 10964–10974 (2008).
Moseman, E. A. et al. B cell maintenance of subcapsular sinus macrophages protects against a fatal viral infection independent of adaptive immunity. Immunity 36, 415–426 (2012).
Biron, C. A. Interferons α and β as immune regulators — a new look. Immunity 14, 661–664 (2001).
Davidson, S., Crotta, S., McCabe, T. M. & Wack, A. Pathogenic potential of interferon αβ in acute influenza infection. Nature Commun. 5, 3864 (2014). This seminal publication shows that, in contrast to the dogma, type I IFNs can cause morbidity and mortality, as opposed to protection, during influenza virus infection.
Mandl, J. N. et al. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nature Med. 14, 1077–1087 (2008).
Jacquelin, B. et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J. Clin. Invest. 119, 3544–3555 (2009).
Rotger, M. et al. Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. J. Clin. Invest. 121, 2391–2400 (2011).
McNally, J. M. et al. Attrition of bystander CD8 T cells during virus-induced T-cell and interferon responses. J. Virol. 75, 5965–5976 (2001).
Chi, B. et al. α and λ interferon together mediate suppression of CD4 T cells induced by respiratory syncytial virus. J. Virol. 80, 5032–5040 (2006).
Gil, M. P. et al. Regulating type 1 IFN effects in CD8 T cells during viral infections: changing STAT4 and STAT1 expression for function. Blood 120, 3718–3728 (2012).
Herbeuval, J. P. et al. Differential expression of IFN-α and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc. Natl Acad. Sci. USA 103, 7000–7005 (2006).
Hardy, A. W., Graham, D. R., Shearer, G. M. & Herbeuval, J. P. HIV turns plasmacytoid dendritic cells (pDC) into TRAIL-expressing killer pDC and down-regulates HIV coreceptors by Toll-like receptor 7-induced IFN-α. Proc. Natl Acad. Sci. USA 104, 17453–17458 (2007).
Herbeuval, J. P. et al. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood 106, 3524–3531 (2005).
van Grevenynghe, J. et al. Loss of memory B cells during chronic HIV infection is driven by Foxo3a- and TRAIL-mediated apoptosis. J. Clin. Invest. 121, 3877–3888 (2011).
Liedtke, C., Groger, N., Manns, M. P. & Trautwein, C. Interferon-α enhances TRAIL-mediated apoptosis by up-regulating caspase-8 transcription in human hepatoma cells. J. Hepatol. 44, 342–349 (2006).
Shigeno, M. et al. Interferon-α sensitizes human hepatoma cells to TRAIL-induced apoptosis through DR5 upregulation and NF-κB inactivation. Oncogene 22, 1653–1662 (2003).
Toomey, N. L. et al. Induction of a TRAIL-mediated suicide program by interferon α in primary effusion lymphoma. Oncogene 20, 7029–7040 (2001).
Teijaro, J. R. et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340, 207–211 (2013).
Wilson, E. B. et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340, 202–207 (2013). References 128 and 129 were the first to show that type I IFNs contribute to pathogenesis by inducing suppressive mechanisms in chronic LCMV infection.
Herold, S. et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 205, 3065–3077 (2008).
Hogner, K. et al. Macrophage-expressed IFN-β contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog. 9, e1003188 (2013).
Chaperot, L. et al. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J. Immunol. 176, 248–255 (2006).
Fujikura, D. et al. Type-I interferon is critical for FasL expression on lung cells to determine the severity of influenza. PLoS ONE 8, e55321 (2013).
McNally, B., Ye, F., Willette, M. & Flano, E. Local blockade of epithelial PDL-1 in the airways enhances T cell function and viral clearance during influenza virus infection. J. Virol. 87, 12916–12924 (2013).
Brincks, E. L. et al. The magnitude of the T cell response to a clinically significant dose of influenza virus is regulated by TRAIL. J. Immunol. 187, 4581–4588 (2011).
MacMicking, J. D. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nature Rev. Immunol. 12, 367–382 (2012).
Kazar, J., Gillmore, J. D. & Gordon, F. B. Effect of interferon and interferon inducers on infections with a nonviral intracellular microorganism, Chlamydia trachomatis. Infect. Immun. 3, 825–832 (1971).
de la Maza, L. M., Peterson, E. M., Goebel, J. M., Fennie, C. W. & Czarniecki, C. W. Interferon-induced inhibition of Chlamydia trachomatis: dissociation from antiviral and antiproliferative effects. Infect. Immun. 47, 719–722 (1985).
Ishihara, T. et al. Inhibition of Chlamydia trachomatis growth by human interferon-α: mechanisms and synergistic effect with interferon-γ and tumor necrosis factor-α. Biomed. Res. 26, 179–185 (2005).
Rothfuchs, A. G. et al. IFN-α/β-dependent, IFN-γ secretion by bone marrow-derived macrophages controls an intracellular bacterial infection. J. Immunol. 167, 6453–6461 (2001).
Rothfuchs, A. G. et al. STAT1 regulates IFN-αβ- and IFN-γ-dependent control of infection with Chlamydia pneumoniae by nonhemopoietic cells. J. Immunol. 176, 6982–6990 (2006).
Qiu, H. et al. Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J. Immunol. 181, 2092–2102 (2008).
Opitz, B. et al. Legionella pneumophila induces IFNβ in lung epithelial cells via IPS-1 and IRF3, which also control bacterial replication. J. Biol. Chem. 281, 36173–36179 (2006).
Plumlee, C. R. et al. Interferons direct an effective innate response to Legionella pneumophila infection. J. Biol. Chem. 284, 30058–30066 (2009).
Schiavoni, G. et al. Type I IFN protects permissive macrophages from Legionella pneumophila infection through an IFN-γ-independent pathway. J. Immunol. 173, 1266–1275 (2004).
Gold, J. A. et al. Exogenous γ and α/β interferon rescues human macrophages from cell death induced by Bacillus anthracis. Infect. Immun. 72, 1291–1297 (2004).
Bukholm, G., Berdal, B. P., Haug, C. & Degre, M. Mouse fibroblast interferon modifies Salmonella typhimurium infection in infant mice. Infect. Immun. 45, 62–66 (1984).
Niesel, D. W., Hess, C. B., Cho, Y. J., Klimpel, K. D. & Klimpel, G. R. Natural and recombinant interferons inhibit epithelial cell invasion by Shigella spp. Infect. Immun. 52, 828–833 (1986).
Mancuso, G. et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J. Immunol. 178, 3126–3133 (2007).
Parker, D. et al. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. MBio 2, e00016-11 (2011).
Weigent, D. A., Huff, T. L., Peterson, J. W., Stanton, G. J. & Baron, S. Role of interferon in streptococcal infection in the mouse. Microb. Pathog. 1, 399–407 (1986).
Kelly-Scumpia, K. M. et al. Type I interferon signaling in hematopoietic cells is required for survival in mouse polymicrobial sepsis by regulating CXCL10. J. Exp. Med. 207, 319–326 (2010).
Weighardt, H. et al. Type I IFN modulates host defense and late hyperinflammation in septic peritonitis. J. Immunol. 177, 5623–5630 (2006).
Freudenberg, M. A. et al. A murine, IL-12-independent pathway of IFN-γ induction by Gram-negative bacteria based on STAT4 activation by type I IFN and IL-18 signaling. J. Immunol. 169, 1665–1668 (2002).
Auerbuch, V., Brockstedt, D. G., Meyer-Morse, N., O'Riordan, M. & Portnoy, D. A. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J. Exp. Med. 200, 527–533 (2004).
Carrero, J. A., Calderon, B. & Unanue, E. R. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J. Exp. Med. 200, 535–540 (2004).
O'Connell, R. M. et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med. 200, 437–445 (2004). References 155–157 were the first publications demonstrating an adverse effect of type I IFNs in intracellular infection with the bacterium L. monocytogenes.
Carrero, J. A., Calderon, B. & Unanue, E. R. Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J. Exp. Med. 203, 933–940 (2006).
Stockinger, S. et al. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J. Immunol. 169, 6522–6529 (2002).
Zwaferink, H., Stockinger, S., Hazemi, P., Lemmens-Gruber, R. & Decker, T. IFN-β increases listeriolysin O-induced membrane permeabilization and death of macrophages. J. Immunol. 180, 4116–4123 (2008).
Zwaferink, H., Stockinger, S., Reipert, S. & Decker, T. Stimulation of inducible nitric oxide synthase expression by β interferon increases necrotic death of macrophages upon Listeria monocytogenes infection. Infect. Immun. 76, 1649–1656 (2008).
Dresing, P., Borkens, S., Kocur, M., Kropp, S. & Scheu, S. A fluorescence reporter model defines “Tip-DCs” as the cellular source of interferon β in murine listeriosis. PLoS ONE 5, e15567 (2010).
Stockinger, S. et al. Characterization of the interferon-producing cell in mice infected with Listeria monocytogenes. PLoS Pathog. 5, e1000355 (2009).
Rayamajhi, M., Humann, J., Penheiter, K., Andreasen, K. & Lenz, L. L. Induction of IFN-α/β enables Listeria monocytogenes to suppress macrophage activation by IFN-γ. J. Exp. Med. 207, 327–337 (2010).
Kearney, S. J. et al. Type I IFNs downregulate myeloid cell IFN-γ receptor by inducing recruitment of an early growth response 3/NGFI-A binding protein 1 complex that silences ifngr1 transcription. J. Immunol. 191, 3384–3392 (2013).
Manca, C. et al. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J. Interferon Cytokine Res. 25, 694–701 (2005).
Ordway, D. et al. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J. Immunol. 179, 522–531 (2007).
Stanley, S. A., Johndrow, J. E., Manzanillo, P. & Cox, J. S. The type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J. Immunol. 178, 3143–3152 (2007).
Manca, C. et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-α/β. Proc. Natl Acad. Sci. USA 98, 5752–5757 (2001). This study was the first demonstration of type I IFNs contributing to the exacerbation of tuberculosis in experimental mouse models.
Cooper, A. M., Pearl, J. E., Brooks, J. V., Ehlers, S. & Orme, I. M. Expression of the nitric oxide synthase 2 gene is not essential for early control of Mycobacterium tuberculosis in the murine lung. Infect. Immun. 68, 6879–6882 (2000).
Berry, M. P. et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466, 973–977 (2010). This study provided the first evidence that type I IFN-mediated signalling is associated with active tuberculosis in humans.
Cliff, J. M. et al. Distinct phases of blood gene expression pattern through tuberculosis treatment reflect modulation of the humoral immune response. J. Infect. Dis. 207, 18–29 (2013).
Maertzdorf, J. et al. Human gene expression profiles of susceptibility and resistance in tuberculosis. Genes Immun. 12, 15–22 (2011).
Ottenhoff, T. H. et al. Genome-wide expression profiling identifies type 1 interferon response pathways in active tuberculosis. PLoS ONE 7, e45839 (2012).
Antonelli, L. R. et al. Intranasal poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J. Clin. Invest. 120, 1674–1682 (2010).
Mayer-Barber, K. D. et al. Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 35, 1023–1034 (2011).
McNab, F. W. et al. TPL-2-ERK1/2 signaling promotes host resistance against intracellular bacterial infection by negative regulation of type I IFN production. J. Immunol. 191, 1732–1743 (2013).
Redford, P. S. et al. Influenza A virus impairs control of Mycobacterium tuberculosis coinfection through a type I interferon receptor-dependent pathway. J. Infect. Dis. 209, 270–274 (2014).
Mayer-Barber, K. D. et al. Caspase-1 independent IL-1β production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. J. Immunol. 184, 3326–3330 (2010).
de Paus, R. A. et al. Inhibition of the type I immune responses of human monocytes by IFN-α and IFN-β. Cytokine 61, 645–655 (2013).
Novikov, A. et al. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1β production in human macrophages. J. Immunol. 187, 2540–2547 (2011).
McNab, F. W. et al. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-γ for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J. Immunol. 193, 3600–3612 (2014). This key study demonstrates the mechanisms underlying the adverse effects of type I IFNs in tuberculosis, including blocking of the protective type II IFN action, as well as blocking of IL-12, IL-1 and TNF production, in part through IL-10 induction.
Guarda, G. et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34, 213–223 (2011). This was the first study to show inhibition of the inflammasome by type I IFNs.
Mayer-Barber, K. D. et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 511, 99–103 (2014). This seminal study shows the counter-regulatory function of IL-1 and type I IFNs in controlling the outcome of M. tuberculosis infection via eicosanoids.
Xu, X. J., Reichner, J. S., Mastrofrancesco, B., Henry, W. L. Jr & Albina, J. E. Prostaglandin E2 suppresses lipopolysaccharide-stimulated IFN-β production. J. Immunol. 180, 2125–2131 (2008). This study provided the first demonstration that prostaglandin E2 suppresses lipopolysaccharide-stimulated IFNβ production.
Coulombe, F. et al. Targeted prostaglandin E2 inhibition enhances antiviral immunity through induction of type I interferon and apoptosis in macrophages. Immunity 40, 554–568 (2014).
Teles, R. M. et al. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science 339, 1448–1453 (2013). In this key study, a mechanism is reported for type I IFN-mediated blocking of the protective role of type II IFN in tuberculosis.
Desvignes, L., Wolf, A. J. & Ernst, J. D. Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. J. Immunol. 188, 6205–6215 (2012). This important study shows that type I IFNs contribute to protection against M. tuberculosis when type II IFN-mediated signalling is aberrant.
Bogunovic, D. et al. Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science 337, 1684–1688 (2012).
Mariotti, S. et al. Mycobacterium tuberculosis diverts α interferon-induced monocyte differentiation from dendritic cells into immunoprivileged macrophage-like host cells. Infect. Immun. 72, 4385–4392 (2004).
Fernandes-Alnemri, T. et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nature Immunol. 11, 385–393 (2010).
Henry, T., Brotcke, A., Weiss, D. S., Thompson, L. J. & Monack, D. M. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 204, 987–994 (2007).
Henry, T. et al. Type I IFN signaling constrains IL-17A/F secretion by γδ T cells during bacterial infections. J. Immunol. 184, 3755–3767 (2010).
Shah, S. et al. Mycobacterium tuberculosis but not nonvirulent mycobacteria inhibits IFN-β and AIM2 inflammasome-dependent IL-1β production via its ESX-1 secretion system. J. Immunol. 191, 3514–3518 (2013).
Al Moussawi, K. et al. Type I interferon induction is detrimental during infection with the Whipple's disease bacterium, Tropheryma whipplei. PLoS Pathog. 6, e1000722 (2010).
de Almeida, L. A. et al. MyD88 and STING signaling pathways are required for IRF3-mediated IFN-β induction in response to Brucella abortus infection. PLoS ONE 6, e23135 (2011).
Patel, A. A., Lee-Lewis, H., Hughes-Hanks, J., Lewis, C. A. & Anderson, D. M. Opposing roles for interferon regulatory factor-3 (IRF-3) and type I interferon signaling during plague. PLoS Pathog. 8, e1002817 (2012).
Robinson, N. et al. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nature Immunol. 13, 954–962 (2012).
Rathinam, V. A. et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by Gram-negative bacteria. Cell 150, 606–619 (2012).
Broz, P. et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291 (2012).
Martin, F. J. et al. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J. Clin. Invest. 119, 1931–1939 (2009).
Diefenbach, A. et al. Type 1 interferon (IFNα/β) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity 8, 77–87 (1998).
Mattner, J. et al. Regulation of type 2 nitric oxide synthase by type 1 interferons in macrophages infected with Leishmania major. Eur. J. Immunol. 30, 2257–2267 (2000).
Mattner, J. et al. Protection against progressive leishmaniasis by IFN-β. J. Immunol. 172, 7574–7582 (2004).
Khouri, R. et al. IFN-β impairs superoxide-dependent parasite killing in human macrophages: evidence for a deleterious role of SOD1 in cutaneous leishmaniasis. J. Immunol. 182, 2525–2531 (2009).
Xin, L. et al. Type I IFN receptor regulates neutrophil functions and innate immunity to Leishmania parasites. J. Immunol. 184, 7047–7056 (2010).
Haque, A. et al. Type I interferons suppress CD4+ T-cell-dependent parasite control during blood-stage Plasmodium infection. Eur. J. Immunol. 41, 2688–2698 (2011).
Vigario, A. M. et al. Inhibition of Plasmodium yoelii blood-stage malaria by interferon α through the inhibition of the production of its target cell, the reticulocyte. Blood 97, 3966–3971 (2001).
Vigario, A. M. et al. Recombinant human IFN-α inhibits cerebral malaria and reduces parasite burden in mice. J. Immunol. 178, 6416–6425 (2007).
Voisine, C., Mastelic, B., Sponaas, A. M. & Langhorne, J. Classical CD11c+ dendritic cells, not plasmacytoid dendritic cells, induce T cell responses to Plasmodium chabaudi malaria. Int. J. Parasitol. 40, 711–719 (2010).
Liehl, P. et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nature Med. 20, 47–53 (2014).
Costa, V. M. et al. Type I IFNs stimulate nitric oxide production and resistance to Trypanosoma cruzi infection. J. Immunol. 177, 3193–3200 (2006).
Koga, R. et al. TLR-dependent induction of IFN-β mediates host defense against Trypanosoma cruzi. J. Immunol. 177, 7059–7066 (2006).
Lopez, R., Demick, K. P., Mansfield, J. M. & Paulnock, D. M. Type I IFNs play a role in early resistance, but subsequent susceptibility, to the African trypanosomes. J. Immunol. 181, 4908–4917 (2008).
Chessler, A. D., Caradonna, K. L., Da'dara, A. & Burleigh, B. A. Type I interferons increase host susceptibility to Trypanosoma cruzi infection. Infect. Immun. 79, 2112–2119 (2011).
Une, C., Andersson, J. & Orn, A. Role of IFN-α/β and IL-12 in the activation of natural killer cells and interferon-γ production during experimental infection with Trypanosoma cruzi. Clin. Exp. Immunol. 134, 195–201 (2003).
Biondo, C. et al. IFN-α/β signaling is required for polarization of cytokine responses toward a protective type 1 pattern during experimental cryptococcosis. J. Immunol. 181, 566–573 (2008).
Biondo, C. et al. Recognition of yeast nucleic acids triggers a host-protective type I interferon response. Eur. J. Immunol. 41, 1969–1979 (2011).
del Fresno, C. et al. Interferon-β production via Dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity 38, 1176–1186 (2013).
Majer, O. et al. Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections. PLoS Pathog. 8, e1002811 (2012).
Bourgeois, C. et al. Conventional dendritic cells mount a type I IFN response against Candida spp. requiring novel phagosomal TLR7-mediated IFN-β signaling. J. Immunol. 186, 3104–3112 (2011).
Inglis, D. O., Berkes, C. A., Hocking Murray, D. R. & Sil, A. Conidia but not yeast cells of the fungal pathogen Histoplasma capsulatum trigger a type I interferon innate immune response in murine macrophages. Infect. Immun. 78, 3871–3882 (2010).
Liu, L. et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 208, 1635–1648 (2011).
van de Veerdonk, F. L. et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N. Engl. J. Med. 365, 54–61 (2011).
Morens, D. M., Taubenberger, J. K. & Fauci, A. S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J. Infect. Dis. 198, 962–970 (2008).
Li, W., Moltedo, B. & Moran, T. M. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of γδ T cells. J. Virol. 86, 12304–12312 (2012).
Nakamura, S., Davis, K. M. & Weiser, J. N. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J. Clin. Invest. 121, 3657–3665 (2011).
Shahangian, A. et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest. 119, 1910–1920 (2009).
Tian, X. et al. Poly I:C enhances susceptibility to secondary pulmonary infections by Gram-positive bacteria. PLoS ONE 7, e41879 (2012).
Navarini, A. A. et al. Increased susceptibility to bacterial superinfection as a consequence of innate antiviral responses. Proc. Natl Acad. Sci. USA 103, 15535–15539 (2006).
Kim, Y. G. et al. Viral infection augments Nod1/2 signaling to potentiate lethality associated with secondary bacterial infections. Cell Host Microbe 9, 496–507 (2011).
Belkaid, Y. & Hand, T. W. Role of the microbiota in immunity and inflammation. Cell 157, 121–141 (2014).
Ganal, S. C. et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 37, 171–186 (2012).
Abt, M. C. et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37, 158–170 (2012).
Tschurtschenthaler, M. et al. Type I interferon signalling in the intestinal epithelium affects Paneth cells, microbial ecology and epithelial regeneration. Gut 63, 1921–1931 (2014).
Kawashima, T. et al. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-β. Immunity 38, 1187–1197 (2013). This study shows that the microbiota contributes to the initial production of protective type I IFNs. References 235 and 236 collectively demonstrate a novel interplay between the microbiota, type I IFNs and consequent protection against pathogens.
Gough, D. J., Messina, N. L., Clarke, C. J., Johnstone, R. W. & Levy, D. E. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity 36, 166–174 (2012).
Acknowledgements
The authors' work was supported by the Medical Research Council, UK (grants U117565642 to A.O.G. and U117597139 to A.W.), the European Research Council (grant 294682-TB-PATH to A.O.G.) and the National Institute of Allergy and Infectious Diseases, US National Institutes of Health (grants to K.M.-B. and A.S.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- Cytosolic GAMP synthase
-
(cGAS). A cytosolic DNA sensor that catalyses the production of the second messenger cyclic di-GMP-AMP (cGAMP) in response to DNA, which is then recognized by the sensor and signalling intermediate STING (stimulator of interferon genes), triggering type I interferon production.
- Plasmacytoid dendritic cells
-
(pDCs). Immature dendritic cells with a morphology that resembles that of plasma cells. On a per-cell basis, pDCs are the main producers of type I interferons in response to viral infections or Toll-like receptor stimulation.
- Ribavirin
-
A drug that interferes with RNA metabolism and blocks viral replication. Ribavirin is used in combination with interferon-α to treat hepatitis C virus infection.
- M1 macrophage
-
A pro-inflammatory, or 'classically activated', subset of macrophages that are characterized by phagocytic activity and the expression of particular pro-inflammatory cytokines (such as tumour necrosis factor) and inflammatory mediators (such as inducible nitric oxide synthase).
Rights and permissions
About this article

Cite this article
McNab, F., Mayer-Barber, K., Sher, A. et al. Type I interferons in infectious disease. Nat Rev Immunol 15, 87–103 (2015). https://doi.org/10.1038/nri3787
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri3787
This article is cited by
-
Control of complement-induced inflammatory responses to SARS-CoV-2 infection by anti-SARS-CoV-2 antibodies
The EMBO Journal (2024)
-
Longitudinal analysis of influenza vaccination implicates regulation of RIG-I signaling by DNA methylation
Scientific Reports (2024)
-
LUBAC is required for RIG-I sensing of RNA viruses
Cell Death & Differentiation (2024)
-
Deeper look into viruses: replication intermediates do code!
Plant Cell Reports (2024)
-
MDA5 Enhances Invasive Candida albicans Infection by Regulating Macrophage Apoptosis and Phagocytosis/Killing Functions
Inflammation (2024)
