
Key Points
-
Influenza virus infection is detected by multiple pattern recognition receptors. Within the infected cells, retinoic acid-inducible gene I (RIG-I) detects the 5′-triphosphorylated RNA of replicating viral genomes, whereas in plasmacytoid dendritic cells and B cells Toll-like receptor 7 (TLR7) detects RNA that is associated with incoming virions. TLR3 is expressed in airway epithelial cells and macrophages and detects RNA that is associated with infected cells.
-
The activation of nucleic acid sensors leads to the expression of type I and type III interferons, which in turn stimulate the expression of hundreds of interferon-stimulated genes in neighbouring cells that induce antiviral state.
-
In myeloid cells, matrix 2 (M2) ion channel activity of influenza virus stimulates the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome, resulting in the activation of caspase 1 and the cleavage and release of interleukin-1β (IL-1β) and IL-18. Phagocytosis of influenza virus-infected cells containing PB1-F2 fibrils also triggers activation of the NLRP3 inflammasome. The NLRP3 inflammasome and IL-1β have an important role in host tolerance to influenza virus infection in response to high-dose viral challenge. IL-1β and IL-1R signalling promotes antiviral B cell and T cell responses after low-dose viral challenge.
-
Commensal bacteria provide signals that set the activation threshold for the induction of adaptive immune responses to influenza virus. Bacterial components drive the expression of the genes encoding pro-IL-1β and NLRP3, as well as interferon-stimulated genes, to promote the robust stimulation of B cell and T cell responses upon influenza virus infection.
-
Innate immune responses confer protection either by stimulating antiviral defence genes or by promoting disease tolerance of host tissues. Therapeutic approaches to combating influenza virus-initiated diseases should consider promoting both of these protective strategies.
Abstract
Influenza viruses are a major pathogen of both humans and animals. Recent studies using gene-knockout mice have led to an in-depth understanding of the innate sensors that detect influenza virus infection in a variety of cell types. Signalling downstream of these sensors induces distinct sets of effector mechanisms that block virus replication and promote viral clearance by inducing innate and adaptive immune responses. In this Review, we discuss the various ways in which the innate immune system uses pattern recognition receptors to detect and respond to influenza virus infection. We consider whether the outcome of innate sensor stimulation promotes antiviral resistance or disease tolerance, and propose rational treatment strategies for the acute respiratory disease that is caused by influenza virus infection.
Similar content being viewed by others

Main
Influenza virus is a member of the family Orthomyxoviridae, and is an enveloped virus that contains a genome composed of eight segments of negative-sense single-stranded RNA (ssRNA) tightly surrounded by nucleoprotein (NP). Haemagglutinin (HA) and neuraminidase (NA) are the major viral glycoproteins that are detected by antibodies, and that define the subtype of the virus. Influenza viruses are classified as seasonal or pandemic depending on the genetic changes that are incorporated from year to year that dictate the severity of disease outcome (Box 1). Influenza viruses can infect diverse host species, including pigs, birds and humans. Human infection by influenza viruses initiates in the respiratory tract and in most cases infection is contained within this organ. Influenza virus that enters the host through the oral or nasal cavities is first countered by the mucus that covers the respiratory epithelium. If the virus is successful in getting through the mucous layer, it must next attach to and invade the respiratory epithelial cells. From there, the virus can spread to both non-immune and immune cells (such as macrophages and dendritic cells (DCs)) in the respiratory tract1,2.
The defence mechanisms that are provided by the innate immune system are a formidable barrier to influenza virus. A specialized immune system exists at distinct mucosal surfaces to combat invasion by pathogens. The viral RNA that is present within infected cells is recognized as foreign by various pattern recognition receptors (PRRs), which leads to the secretion of type I interferons (IFNs), pro-inflammatory cytokines, eicosanoids and chemokines. Type I IFNs — produced by macrophages, pneumocytes, DCs and plasmacytoid DCs (pDCs)3,4,5 — stimulate the expression of hundreds of genes that are collectively known as IFN-stimulated genes (ISGs) in neighbouring cells, which induce an antiviral state. Pro-inflammatory cytokines and eicosanoids cause local and systemic inflammation, induce fever and anorexia, and instruct the adaptive immune response to influenza virus. Chemokines that are produced at the site of infection recruit additional immune cells, including neutrophils, monocytes and natural killer (NK) cells, to the airways. Virally infected epithelial cells become the targets of NK cells, which mediate viral clearance6. Monocytes and neutrophils are rapidly recruited to the influenza virus-infected lung and help to clear infected dead cells. Together with alveolar macrophages, phagocytic clearance of virus-infected cells by recruited phagocytes provides an important mechanism of viral clearance7. If the virus is successful in establishing infection despite these defences, the ultimate clearance of the virus requires adaptive immunity. The regulation of adaptive immunity to influenza virus has been recently reviewed elsewhere8 and is therefore not discussed here.
Over the past few decades, many fundamental discoveries have been made regarding the mechanisms by which influenza virus is recognized and contained by the innate immune system. The understanding of host protection at the organismal level must take into account factors that are beyond cell-intrinsic viral control. The health of the host organism can be restored following infection either by reducing the pathogen burden (antiviral resistance) or by reducing the negative impact of infection on host fitness (disease tolerance) (Fig. 1). The ability of a host to tolerate the presence of a pathogen is a distinct host defence strategy, which has mostly been ignored in animal and human studies9,10,11. Protection of a given host from infectious disease is dictated by these two fundamentally distinct strategies. Most acute infection requires resistance for host survival, however disease tolerance is a mechanism that protects the host against some acute and chronic infections: for example, infection of the African green monkey and sooty mangabey with simian immunodeficiency virus (SIV) results in high viral burden but is non-pathogenic12.

There are two distinct host strategies that deal with an infection: elimination of the pathogen or reduction of the negative impact of infection. A resistant host regains fitness by recognizing and eliminating the pathogen. A tolerant host regains fitness by reducing the immunopathology and tissue damage that is inflicted by pathogens. A host becomes susceptible if they are unable to reduce pathogen burden or unable to tolerate the negative consequences of the immune response to infection.
In this Review, we highlight the mechanisms of innate immune recognition and defence against influenza virus infection, and consider the role of innate immune responses to influenza virus in controlling disease through their effects on antiviral resistance and disease tolerance.
Innate recognition of influenza virus infection
The innate immune system detects viral infections through the recognition of pathogen-associated molecular patterns (PAMPs) by PRRs; these PAMPs are specifically present in the pathogen or are generated during infection13,14. Influenza virus is recognized by the innate immune system by members of at least three distinct classes of PRRs (with their respective ligands indicated); the Toll-like receptors TLR3 (double-stranded RNA (dsRNA)), TLR7 (ssRNA) and TLR8 (ssRNA), retinoic acid-inducible gene I (RIG-I) (5′-triphosphate RNA) and the NOD-like receptor family member NOD-, LRR- and pyrin domain-containing 3 (NLRP3) (various stimuli; see below)15 (Fig. 2). RIG-I and NLRP3 detect virus that is present within the cytosol of infected cells (cell-intrinsic recognition), whereas TLR3 detects virus-infected cells, and TLR7 (and TLR8 in humans) detect viral RNA that has been taken up into the endosomes of sentinel cells (cell-extrinsic recognition).

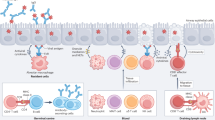
Influenza virus infection is detected by multiple host sensors that recognize unique features that are associated with the infection. a | Infected cells are phagocytosed by macrophages (left panel) for recognition of double-stranded RNA (dsRNA) by Toll-like receptor 3 (TLR3), which leads to the induction of the expression of nuclear factor-κB (NF-κB)-dependent pro-inflammatory cytokines and of type I interferon (IFN) and IFN-stimulated genes (ISGs) downstream of IFN-regulatory factor 3 (IRF3). Incoming genomic single-stranded RNA (ssRNA) that is contained within the virion is released via the degradation of the viral membrane and capsid within acidified endosomes, and ssRNA is recognized by TLR7 in plasmacytoid dendritic cells (pDCs; right panel). TLR7 signalling induces NF-κB-dependent genes from the NF-κB endosome, and IRF7 activation from the IRF7 endosome. b | Within infected cells, viral RNA in the cytosol (possibly in stress granules; not shown) is recognized by retinoic acid-inducible gene I (RIG-I), which, through the activation of mitochondrial antiviral signalling protein (MAVS), leads to the induction of pro-inflammatory cytokines and type I IFN. Matrix 2 (M2) ion channel activity in the Golgi stimulates formation of the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome, which results in caspase 1 activation and the release of the cytokines interleukin-1β (IL-1β) and IL-18. PB1-F2 fibrils accumulate in the phagosome, which results in the activation of NLRP3 and the release of IL-1β and IL-18. ER, endoplasmic reticulum; HA, haemagglutinin; NA, neuraminidase; TRIF, TIR-domain-containing adaptor protein inducing IFNβ.
Studies using high or lethal doses of influenza A viruses in mice that are deficient in innate sensors and signalling pathways have revealed the roles of these sensors and pathways in both innate resistance and host tolerance, as the host often succumbs to the infection before they have had a chance to generate robust adaptive immune responses. In parallel, challenge with sublethal doses of influenza A virus or with inactive virus enables the host to survive the infection by mounting a protective adaptive immune response, and such studies have provided insights into which viral sensors link innate recognition to adaptive immunity.
TLR-mediated recognition of influenza virus. TLR3 recognizes dsRNA in endosomes16. As influenza virus-infected cells do not generate dsRNA17, due to the activity of the cellular RNA helicase UAP56 (also known as spliceosome RNA helicase DDX39B)18, it is likely that TLR3 recognizes currently unidentified RNA structures that are present in dying influenza virus-infected cells that have been phagocytosed19 (Fig. 2a). Human respiratory epithelial cells constitutively express TLR3, the activation of which induces the production of pro-inflammatory cytokines upon influenza virus infection20 but can lead to pathology20,21,22. Indeed, Tlr3−/− mice survive longer than wild-type mice following lethal influenza virus infection, despite sustaining higher viral loads in the lungs22. In the absence of TLR3, chemokine expression in the lungs, as well as the infiltration of leukocytes and CD8+ T cells, is reduced after influenza virus challenge. Tlr3−/− mice generate normal antibody, CD4+ T cell and CD8+ T cell responses to sublethal doses of influenza virus infection23, which indicates that this sensor is dispensable for generating T cell immunity. Thus, although TLR3 induces signals that restrict viral replication, it simultaneously promotes the recruitment of innate and adaptive immune cells that cause damage to the host.
In pDCs, TLR7 recognizes the ssRNA genomes contained within the influenza virion that are taken up into the endosome24,25 (Fig. 2a). This mode of recognition does not require viral replication. TLR7 signalling via the adaptor MYD88 from distinct endosomes26,27,28 results in the activation of either nuclear factor-κB (NF-κB) or IFN-regulatory factor 7 (IRF7), which are transcription factors that are responsible for stimulating the expression of pro-inflammatory cytokines and type I IFNs, respectively.
Although one study has shown that mice deficient in TLR3 and TLR7 or MYD88 that are challenged with high doses of influenza virus are unable to control virus replication and succumb to the infection29, another study did not find a requirement for TLR7 or MYD88 in immune defence against high-dose influenza virus challenge22. Thus, the role of TLR7 in innate defence remains unclear, at least in inbred mice. However, TLR7 has a key role in innate defence in the context of mice that express IFN-induced GTP-binding protein MX1 (also known as myxovirus resistance protein 1) (Box 2). By contrast, infection with sublethal doses of influenza virus has demonstrated an important role of TLR7 in eliciting robust antibody responses to influenza virus, but not T cell responses23,30,31. Thus, TLR7 has an important role in antiviral resistance through the instruction of B cells to elicit appropriate antibody production. TLR8 is expressed by human monocytes and macrophages, and stimulation by its ligand ssRNA results in the production of IL-12 but not IFNα32. However, the relevance of TLR8 in influenza virus infection is unknown.
In summary, endosomal TLRs have distinct roles in influenza virus infection. TLR3 recognizes infected cells and induces an antiviral state but with the detrimental effect of recruiting damage-inducing inflammatory cells, whereas TLR7 induces IFN responses to block viral replication and to promote antibody responses (Table 1).
RIG-I detects replicating viral RNA within infected cells. RIG-I is crucial for viral detection and type I IFN production in infected epithelial cells, conventional DCs and alveolar macrophages33. Within the cytosol of influenza virus-infected cells, RIG-I recognizes the 5′-triphosphate viral ssRNA that is generated after viral replication17,34,35,36 (Fig. 2c). The intact genomic ssRNA containing 5′-triphosphate35 and shorter genomic segments, as well as subgenomic defective interfering particles bearing 5′-triphosphate36, have been identified as ligands for RIG-I in influenza virus-infected cells. Upon recognition of viral RNA, the helicase domain of RIG-I binds to ATP, which facilitates conformational changes that enable its caspase-recruitment domains to bind to the signalling adaptor mitochondrial antiviral signalling protein (MAVS)37,38,39.
Given that influenza virus replicates within the nucleus, the precise nature and the location of the RNA that is detected by RIG-I had previously remained a mystery. However, a recent study40 demonstrated that RIG-I enters antiviral stress granules, where viral RNA and ISG products — such as the serine/threonine kinase protein kinase R (PKR; also known as EIF2AK2) — colocalize. Disruption of antiviral stress granules was shown to reduce RIG-I-mediated IFN responses. PKR is required to generate antiviral stress granules, and the viral protein NS1 blocks antiviral stress granule formation by interfering with PKR40. This study suggests that the components that are needed for RIG-I signalling (such as viral RNA, RIG-I and PKR) are directed to stress granules to mediate the efficient induction of the type I IFN response.
MAVS signalling results in the production of pro-inflammatory cytokines downstream of NF-κB activation and in the production of type I IFNs and ISGs via IRF3 activation. Mice that are deficient in MAVS have similar viral loads and survival rates to wild-type mice when challenged with a lethal dose of influenza virus29. Furthermore, low-dose viral challenge revealed that MAVS-deficient mice have normal adaptive immune responses to influenza virus infection29,31,41. Therefore, in experimental mouse models, MAVS has a minimal role in innate and adaptive immunity to influenza virus infection in inbred mice that lack MX1 (Box 2). However, the role of RIG-I-like receptor (RLR) signalling in innate defence has not been tested in MX1-sufficient mice. Furthermore, the fact that the NS1 protein of influenza virus has evolved to block RIG-I signalling17,42 indicates that RIG-I-mediated recognition is a key antiviral determinant in naturally infected hosts.
NLRP3 inflammasome senses cellular damage. NLRPs form multiprotein inflammasome complexes consisting of an NLRP (or other PRRs), the adaptor ASC and pro-caspase 1. Activation of inflammasomes results in the autocatalytic processing of pro-caspase 1 into its active form, which then cleaves pro-IL-1β and pro-IL-18 into IL-1β and IL-18, respectively, which can then be secreted (Fig. 2b). In addition, inflammasome activation can elicit pyroptosis of infected cells43. Formation of the NLRP3 inflammasome is triggered by numerous stimuli including host cell of membrane damage, infection or stress44.
NLRP3 is expressed by myeloid cell types, such as monocytes, DCs, neutrophils and macrophages45, and also by human bronchial epithelial cells46. Two signals are required to trigger cytokine production by the inflammasome. Signal 1 activates the transcription and translation of the genes encoding pro-IL-1β, pro-IL-18 and NLRP3, and is mediated by TLR, IL-1 receptor (IL-1R) and tumour necrosis factor receptor (TNFR) signalling44. Signal 2 is induced by host damage that results in the activation and cleavage of caspase 1, and the secretion of mature IL-1β and IL-18. In vivo, signal 1 is maintained in large part by commensal bacteria (Box 3). Signal 2 is reported to be provided by at least three influenza virus-associated stimuli. First, ssRNA from influenza virus was shown to be sufficient to activate the release of IL-1β from the THP1 cell line upon transfection47. Second, proton flux through the influenza virus-encoded matrix 2 (M2) ion channel in the trans-Golgi network triggers NLRP3 activation, formation of the inflammasome and cleavage of pro-IL-1β and pro-IL-18 (Ref. 48). Proton flux through the viral M2 ion channel neutralizes the acidic pH of the trans-Golgi in order to prevent activation of HA to its fusogenic form, and this activity somehow triggers the activation of NLRP3 inflammasome. Third, in lipopolysaccharide-primed macrophages, high-molecular-weight aggregates of the influenza virus virulence protein PB1-F2 in lysosomes stimulate the activation of the NLRP3 inflammasome, presumably in cells that have taken up dying infected cells that contain the amyloid fibre-like structures of PB1-F2 (Ref. 49) (Fig. 2b).
In macrophages, influenza virus-induced NLRP3 activation requires a change in mitochondrial membrane potential and mitofusin 2, which associates with NLRP3 to form the inflammasome complex50. Therefore, mitochondria have an important role in NLRP3 activation in influenza virus-infected cells51. In human bronchial epithelial cells, the priming of signal 1 (expression of IL1B and NLRP3 mRNA) requires RIG-I-dependent IFN production followed by signalling through the type I IFN receptor (IFNAR). In addition, RIG-I directly binds to ASC to form the RIG-I–ASC inflammasome in airway epithelial cells46. Thus, influenza virus infection stimulates the activation of distinct inflammasomes — NLRP3 inflammasomes in myeloid cells and RIG-I inflammasomes in airway epithelial cells.
The importance of NLRP3 inflammasomes in innate defence against influenza virus has been demonstrated by two studies47,52. After a high-dose viral challenge with the A/PR8 strain of influenza A virus, NLRP3 was found to be essential for the recruitment of leukocytes into the lungs47. In addition, mice that are deficient in NLRP3, caspase 1 or IL-1R all suffer from increased pulmonary necrosis and reduced respiratory function, which leads to increased mortality compared with wild-type controls47. Unlike type I IFNs, IL-1β and IL-18 do not induce direct antiviral resistance53,54, which indicates that the susceptibility to influenza virus challenge in inflammasome-deficient mice is independent of a failure to contain viral replication. Consistent with this, both studies found no difference in viral load between NLRP3-deficient and wild-type mice47,52 until 7 days post infection52. This suggests that NLRP3-mediated host protection involves an increase in disease tolerance and not antiviral resistance. Specifically, NLRP3 inflammasome activation increases the tolerance of tissues to high-dose influenza virus challenge through cellular recruitment47,52 and the induction of tissue repair47 in the respiratory tract. Furthermore, the use of low-dose influenza virus challenge has revealed a crucial role for inflammasomes and IL-1R signalling in the activation of B cell and T cell responses41,55.
ISGs against influenza viruses
Genetic studies in mice have demonstrated the non-redundant role of several ISGs — including MX proteins, IFN-inducible transmembrane (IFITM) proteins and PKR — in limiting influenza virus infection and spread (Table 1).
MX proteins were among the first ISGs to be identified that restrict influenza virus infection56. In mice, the MX1 protein is expressed in the nucleus, and is a potent blocker of influenza virus infection, whereas MX2 protein is present in the cytosol and has no anti-influenza virus activity57. Humans express the MX proteins MXA and MXB when stimulated with IFNs. MXA protein is cytosolic, and has potent antiviral activity towards influenza A virus, as well as many other viruses58,59,60. By contrast, MXB protein is present at the cytoplasmic face of nuclear pore complexes and has recently been shown to inhibit HIV-1 infection but not influenza A virus infection61,62. Caution must be exercised in translating mouse data into a human setting as almost all inbred mouse strains lack functional MX1 and MX2 (Box 2).
IFITM proteins restrict the replication of a large number of viruses, including influenza A virus63, by blocking virus–host cell membrane fusion following viral attachment and endocytosis. IFITM3-deficient mice are highly susceptible to influenza virus infection64,65. IFITM3 is constitutively expressed by respiratory epithelial cells, macrophages and endothelial cells. IFITM3-deficient mice are as susceptible to influenza virus infection as mice lacking the entire Ifitm locus (that is, lacking the Ifitm1, Ifitm2, Ifitm3, Ifitm5 and Ifitm6 genes), which highlights a key requirement for IFITM3 in innate resistance to influenza virus infection64. Indeed, although IFITM3-deficient mice can initially control viral replication, they succumbed on day 4 post infection with the A/X-31 or A/H1N1/09 influenza virus strains65. Of note, IFITM3-deficient mice develop severe oedema and haemorrhagic pleural effusion65, which suggests that IFITM3 may have a role in limiting host damage, in addition to mediating resistance to influenza virus infection.
The 2′-5′-oligoadenylate synthase (OAS) family and ribonuclease L (RNase L) act together to degrade viral RNA in the cytosol. Upon binding of dsRNA, OAS becomes enzymatically active, converting ATP into 2′-5′-oligoadenylate, which functions as a second messenger to activate latent RNase L. There are four OAS genes and multiple splice variants of each gene in humans and mice66. Activated RNase L degrades viral and cellular ssRNAs, which inhibits protein synthesis and viral growth (Table 1). Mice that are deficient in RNase L suffer from increased susceptibility to RNA viruses including influenza virus67.
PKR is a serine/threonine kinase that phosphorylates the α-subunit of eukaryotic translation initiation factor 2α (EIF2α). PKR binds viral dsRNA and inhibits translation, which results in a decrease in total cellular and viral protein synthesis, thereby effectively reducing viral replication. In addition, PKR has a role in signal transduction and transcriptional control through activation of the inhibitor of NF-κB (IκB)–NF-κB pathway68. Activation of PKR in infected cells results in apoptosis, cell growth arrest and autophagy, all of which serve to curb viral replication and spread in the host66. In addition to the virus-restricting function of PKR, it also stabilizes IFNA and IFNB mRNA, thereby ensuring robust IFN protein production69. Mice that are genetically deficient in PKR are susceptible to various viral infections, including influenza virus70. Given the fact that influenza virus infection does not generate dsRNA17,18, it is likely that the panhandle secondary structure that forms between the termini of the viral RNA genome stimulates PKR during influenza virus infection71.
In addition to the well-known ISGs described above, new investigative approaches have revealed that many other ISGs are capable of blocking influenza virus infection (Table 1). For example, the overexpression of ISGs including monocarboxylate transporter 1a (Slc16a1), Fam46a, serine/threonine protein kinase D2 (Prkd2), Mx1, Irf1, Ifitm2 and Ifitm3 suppressed influenza A virus infectivity in mouse fibroblasts72. However, the mechanism by which SLC16A1, FAM46A and PRKD2 block viral replication is unknown. In addition, overexpression of the gene encoding viperin (also known as RSAD2) blocks influenza virus release by interfering with the formation of the lipid raft from which the virus buds73. The antiviral ISG cholesterol 25-hydroxylase (Ch25h) is expressed in response to IFN signalling, and the enzyme converts cholesterol to soluble 25-hydroxycholesterol (25HC), which is broadly antiviral for enveloped viruses including influenza virus74,75 by blocking viral fusion. Tripartite motif-containing protein 22 (TRIM22) blocks viral genome encapsidation, and suppresses influenza virus infection76. ISG15 is a ubiquitin-like protein that targets newly translated viral proteins77, and ISG15-deficient mice are more susceptible to influenza virus infection, as well as infection with other viruses78.
Thus, studies in knockout mice have revealed a non-redundant role for multiple ISGs in restricting influenza virus replication in vivo, prompting the question of why none of these individual ISGs is sufficient to promote innate immune protection in the host. One possible explanation is that different cell types express and use distinct sets of ISGs.
Disease tolerance versus pathology
Host protection against any infection can be conferred either by increasing innate resistance or by increasing host tolerance to infection. However, highly pathogenic influenza viruses induce excessive inflammatory responses that impair host fitness. Here we discuss some of the key innate cytokines and other soluble factors that mediate protective versus pathogenic innate immune responses. Protective innate immune responses lower the burden of disease in the infected host by increasing antiviral resistance and/or by increasing disease tolerance, whereas pathogenic innate immune responses have a negative impact on host health status by inflicting damage to host tissues as a result of trying to reduce pathogen burden.
Innate immune signals that enhance resistance. The most well-studied innate immune mechanisms are mediated by an increase in antiviral resistance (Fig. 1). As discussed above, type I IFNs represent the prototypical resistance mechanism as they induce hundreds of ISGs that serve as effectors to limit viral replication. Type I IFNs also enhance the cytolytic activity of CD8+ T cells by inducing granzyme B expression79. In addition to type I IFNs, IFNγ (the type II IFN) and IFNλ (a member of the type III IFN family) contribute to innate antiviral defence. IFNγ is secreted by NK cells and T cells, and signals through the IFNγ receptor (IFNGR).
Signalling through IFNAR and IFNGR induces overlapping but distinct sets of ISGs. For instance, MX and OAS proteins are primarily induced by type I IFNs but not type II IFN, whereas IRF1 is preferentially induced by IFNγ80. Expression of the IFNλ receptor (IFNLR) is restricted to epithelial cells and hepatocytes. Although MX1-sufficient mice (Box 2) that lacked IFNLR were only slightly more susceptible to influenza virus than wild-type mice, MX1-sufficient mice that lacked both IFNAR and IFNLR failed to restrict a usually non-pathogenic mutant of influenza virus that lacks the IFN-antagonistic factor NS1 (Ref. 81). This indicates a non-redundant role for type I and type III IFNs in resistance to influence virus infection. Mice that are deficient in signal transducer and activator of transcription 1 (STAT1), which fail to signal through all three IFN receptors, generate CD8+ T cell responses to influenza virus infection but fail to generate an IgG2a response and have exaggerated T helper 2 (TH2) cell and IgE responses82. Therefore, all three IFN types contribute to innate antiviral resistance and are required to elicit a robust adaptive immune response that promotes the clearance of the virus.
In addition to IFNs that induce antiviral effector molecules, other cytokines are known to increase resistance to influenza virus infection by recruiting other leukocytes to the affected tissue. Granulocyte colony-stimulating factor (G-CSF) is secreted in the lungs of influenza virus-infected mice and promotes the recruitment of granulocytes, which promote viral clearance. Indeed, G-CSF-deficient mice are highly susceptible to low doses of influenza virus infection owing to impaired viral clearance83.
Innate immune cytokines, such as IL-1 (Refs 41, 54, 55), IL-6 (Ref. 84) and IL-18 but not IL-12 (Ref. 85), also protect the host by promoting adaptive immune responses. IL-1 induces the migration of DCs from the lung to the mediastinal lymph nodes41, and IL-18 promotes IFNγ production by CD8+ T cells85. By contrast, IL-10 is produced in influenza virus-infected lungs and impairs antiviral defence. In one study, enhanced viral clearance in Il10−/− mice was not correlated with innate resistance or with T cell responses but was correlated with increased pulmonary anti-influenza virus antibody loads86. In another study, Il10−/− mice developed an exaggerated TH17 cell response that mediated viral clearance87. However, under some circumstances, IL-10 that is secreted by effector T cells has a key role in blocking inflammation and cell death following influenza virus infection88. Therefore, host resistance to influenza virus is induced by pro-inflammatory cytokines that instruct adaptive immune mechanisms to reduce viral burden, but this effect is antagonized by anti-inflammatory cytokines such as IL-10.
Innate immune responses that regulate disease tolerance. Innate host protection can also occur at the level of increased disease tolerance (Fig. 3). In response to IL-33, innate lymphoid cells (ILCs) that are resident in the lung secrete amphiregulin, which is a member of the epidermal growth factor family that promotes tissue repair responses89. Depletion of ILCs did not affect viral loads following infection with the A/PR8 strain of influenza virus but resulted in impaired lung function and increased tissue damage89. Of note, ILCs in the lung also secrete IL-13 in response to H3N1 influenza virus challenge, which promotes airway hyperresponsiveness90. Therefore, although ILCs have the capacity to promote tissue healing by inducing epithelial repair, under certain circumstances ILC-derived cytokines promote mucus overproduction and airway remodelling, which leads to pathogenic inflammatory conditions in the lungs.

Innate resistance (left) is conferred by type I and type III interferons (IFNs) that are secreted upon stimulation of retinoic acid-inducible gene I (RIG-I) in infected cells and Toll-like receptor 7 (TLR7) in plasmacytoid dendritic cells (pDCs). Type I IFNs act on most cells, whereas type III IFN (IFNλ) acts on epithelial cells to block virus replication. DCs and macrophages that are infected with influenza virus release interleukin-1β (IL-1β), which enables bystander DCs to upregulate CC-chemokine receptor 7 (CCR7) expression and migrate to the draining lymph nodes to stimulate T cells. NLRP3 inflammasomes also increase disease tolerance by promoting tissue repair. T cells and natural killer (NK) cells secrete IFNγ to induce an antiviral state or induce the granzyme B-mediated lysis of virus-infected cells, whereas B cells secrete antibodies to viral antigens to mediate adaptive immune protection of the host. Inflammatory cytokines (such as IL-1, tumour necrosis factor (TNF) and IL-17) that are induced as a result of innate signalling can also lead to pathology when the duration and extent of cytokine release is increased. Negative regulators of cytokines and inflammatory cells — such as IL-10 and CD200–CD200R — suppress inflammatory consequences, while positive regulators of tissue repair — such as amphiregulin and transforming growth factor-β (TGFβ) — promote a return to homeostasis. The balance between these negative and positive regulators determines whether the host succumbs to disease or can enter a state of tolerance (right). ILC2, type 2 innate lymphoid cell; NLRP3, NOD-, LRR- and pyrin domain-containing 3; ssRNA, single-stranded RNA.
The classic immune regulator transforming growth factor-β (TGFβ) is expressed in a latent inactive form by almost all cell types. Influenza virus NA can cleave latent TGFβ into its active form, and in vivo blockade of TGFβ increases the mortality of mice that have been infected with the 2009 H1N1 or H5N1 influenza virus strains with a minimal effect on viral loads91. Expression of TGFβ from plasmid DNA that was injected into the lungs of influenza virus-infected mice resulted in reduced inflammatory responses but impaired viral clearance92. Therefore, TGFβ increases disease tolerance but impairs antiviral resistance, and an improved understanding of the balance of these processes would be required to determine the potential of TGFβ as a therapeutic target. Another mechanism that promotes tissue resistance against damage during influenza virus infection is mediated by CD200R, which is expressed by alveolar macrophages and suppresses the inflammatory function of macrophages through its interaction with CD200, which is expressed by lung epithelial cells. Mice that are deficient in CD200 succumb to A/X-31 H3N2 influenza virus infection and show increased nitric oxide production and inflammatory cellular infiltrates in the lungs, despite reduced viral loads93. Thus, both TGFβ and CD200 increase host tolerance to the immune damage that occurs following influenza virus infection, but this comes at the cost of an increase in viral burden.
In contrast to these molecules that promote host tolerance, other molecules can lead to a breakdown of tolerance. IL-17A and IL-17F are rapidly secreted by γδ T cells in the lungs of BALB/c mice in response to infection with the A/PR8 strain of influenza virus94. Mice that are deficient in IL-17R lose less weight, show reduced recruitment of neutrophils and have less tissue injury than wild-type mice, without affecting viral loads or on the activation of CD8+ T cells94. Similarly, TNFR1-deficient mice exhibit greatly reduced morbidity compared with wild-type mice following challenge with the H5N1 influenza virus strain, without any difference in viral replication and spread95. Thus, IL-17 and TNF promote untoward inflammation and the loss of tolerance to disease with no obvious contribution to resistance, thus providing ideal targets for clinical intervention.
In summary, pro-inflammatory cytokines can be classified as those that promote antiviral resistance, those that promote host tolerance or those that inflict tissue damage without an apparent benefit to the host (Table 2). Understanding the relative impact of a given cytokine in resistance versus tolerance will help to better define opportunities to block or promote the effect of such a cytokine for improving host fitness in the clinic.
Innate signals that instruct adaptive immunity
Although innate immune responses control virus replication during the early phase of influenza virus infection, clearance and recovery from the infection requires adaptive immune responses. In particular, immunoglobulins that are specific for the HA and NA influenza proteins bind and block viral spread, and cytotoxic T cells and TH1 cells promote the elimination of influenza virus-infected cells. The innate sensors that are expressed by antigen-presenting cells and B cells send key signals to promote these immune responses.
Redundancy, cooperation and hierarchy of innate sensors. PAMPs, such as viral genomic RNA that is contained within the virion, signal the presence of a pathogen to the host but do not necessarily indicate active infection. However, viability-associated PAMPs (vita-PAMPs)96 are distinct patterns that signify to the host that the pathogen is alive. A marker of viability within the context of viral infection is viral replication, whereby replicating viral RNA could serve as vita-PAMPs.
Is there cooperation, complementation or redundancy in innate sensing through PRRs97? Deficiency in MYD88 (downstream of TLRs), MAVS (downstream of RIG-I) or both has showed that these pathways compensate for the loss of the other upon high-dose influenza virus challenge in mice31,98, which suggests that there is redundancy between TLRs and RIG-I. However, infection with a sublethal dose revealed a paradoxical role for TLR7 and MAVS signalling in the enhancement of viral replication through the recruitment of monocytes (viral target cells) to the respiratory tract98, which indicates that the virus utilizes redundancy between TLR7 and RIG-I to promote its replication. A systematic comparison of the genes that are activated by TLRs compared with RLRs showed that a small proportion of genes are uniquely turned on by TLRs but not RLRs, and vice versa99, revealing a non-redundant set of effector molecules. Therefore, the redundancy between PRRs for a given response depends on the selective cellular expression of PRRs, whether the transcriptomes engaged by these receptors overlap, and the extent of viral burden. By contrast, it is likely that the NLRP pathway has a non-redundant role, as the cytokines and effector molecules that are induced by this pathway are distinct from those that are induced by TLR7 and RIG-I.
Is there a hierarchy of these viral signatures in the instruction of adaptive antiviral immune responses to influenza virus? We speculate that the degree of threat that is associated with a given viral signature correlates with immunogenicity. The detection of cellular damage by inflammasomes outweighs the contributions of signals derived from TLR7 or RIG-I for CD8+ T cell induction; CD8+ T cells are generated normally in the absence of TLR7 or RIG-I signalling but are not generated in IL-1R-deficient mice upon infection with the A/PR8 influenza virus strain41. The source of IL-1 is probably inflammasome-dependent, as mice that are deficient in ASC and caspase 1 share the same phenotype of defective CD8+ T cell responses to influenza virus infection55 that is seen in IL-1R-deficient mice.
Why do IL-1R-deficient mice fail to generate CD8+ T cell immunity if PAMPs and vita-PAMPs are still robustly generated? A probable explanation is that IL-1R activation enables uninfected DCs to present viral peptides to CD8+ T cells, whereas RIG-I-stimulated DCs are by definition infected with the virus, which often renders them incapable of their antigen-presenting function. Furthermore, TLR7 is not expressed by the DCs that mediate cross-priming in vivo100. Thus, it is possible that if the antigen-presenting capabilities of DCs can be preserved by infection with an attenuated influenza virus, the importance of RIG-I in the generation of CD8+ T cell immunity might be revealed. Furthermore, infection with a whole inactivated influenza virus — that contains PAMPs but that is not capable of generating vita-PAMPs — has been used to identify a role of TLR7 in promoting the CD8+ T cell response101.
In addition, antibody responses mainly rely on TLR7 (Refs 31, 101). B cells express TLR7, and the detection of pathogen by the B cell receptor and TLR7 within the same endosome promotes robust antibody responses102. The physical connection between the PAMP and antigen that is necessary to elicit influenza virus-specific antibody responses indicates that the 'surrogate' detection of infection via the IL-1R that is used for T cell priming does not apply to B cell priming. The requirement of IL-1R for the induction of antibody responses54,55 may reflect the need to generate good T helper cell responses to support B cell function. The use of knockout mice that lack key innate sensors in specific immune cell subsets will reveal the cell type-specific requirements for these sensors in instructing various aspects of adaptive immunity.
Sensors triggered by influenza virus infection and vaccines. The concept of hierarchical immunogenicity associated with the extent of threat has bearings on our understanding of immune responses to influenza virus infection, as well as on the better design of influenza vaccines. Although a seasonal influenza virus strain would probably elicit all three pathways of innate recognition, a pandemic influenza virus infection might elicit higher levels of TLR7, RIG-I103,104,105,106 and inflammasome105 activation. Live attenuated vaccines, such as FluMist (MedImmune), are cold temperature-adapted and attenuated for replication. Although a live-attenuated influenza vaccine generates PAMPs and vita-PAMPs, the extent to which it triggers inflammasome activation is unknown. Whole inactivated vaccines contain viral genomic RNA for TLR7 engagement and rely solely on this pathway to elicit adaptive immunity101. However, the most common influenza vaccine given to adults — the trivalent or quadrivalent influenza split virion inactivated vaccine — consists of recombinant membrane HA and NA that are derived from three circulating strains and internal virion proteins that are derived from the A/PR8/34 influenza virus strain30. Although the split vaccine is not administered with any adjuvants, the viral RNA that contaminates vaccine preparation is thought to induce TLR7-dependent antibody responses30.
These studies collectively indicate that distinct effector molecules are induced downstream of PRRs, and that depending on the viral burden and cell types infected, different PRRs have a non-redundant role in protecting the host from influenza virus infection. The degree of threat that is associated with the molecular signatures of infection seems to set the hierarchy for T cell priming. Understanding the principles of immune priming by the signatures that are associated with the top of the hierarchy could be used to improve vaccine design in stimulating more robust B cell and T cell immunity.
Therapies for influenza-induced acute lung injury
Severe lethality associated with influenza virus infection is a product of both intrinsic viral virulence and the overt innate immune response to the infection. Viral determinants of pathogenicity that depend on host innate immune responses have been intensively studied107. The common feature of highly pathogenic influenza virus strains — such as the 1918 H1N1 and H5N1 strains — is the overt pro-inflammatory gene signature in the respiratory tract of infected hosts, which results from a combination of intrinsic viral virulence, viral replication capacity and aberrant host innate immune responses to the virus. In addition to virus-associated molecular signatures, a host-derived oxidized phospholipid (termed OxPAPC) contributes to inflammatory pathology in acute lung injury after H5N1 infection108. The oxidized phospholipid is produced in the lungs of mice after respiratory challenge with an inactivated H5N1 avian influenza virus and in the lungs of humans who have been infected with H5N1 virus108. The oxidized phospholipid stimulates TLR4 and induces inflammation and lung damage. A comprehensive lipidomic analysis of influenza virus-infected mice showed that 5-lipoxygenase metabolites predominated in pathogenic influenza virus infection, whereas the protective metabolites generated by 2,15-lipoxygenase were markedly reduced109.
Current general immunotherapeutic strategies, including aspirin, statins, cyclooxygenase inhibitors and corticosteroids, have not proved to be effective in treating acute respiratory disease following influenza virus infection110. Recent efforts to treat acute lung injury caused by influenza virus can be classified into those intended to increase antiviral resistance and those that reduce the tissue damage that is mediated by the virus or by the immune system. Agents that aim to increase resistance must be administered in the early stages of infection and must be without inflammatory side effects. For example, recombinant type I IFNs cause 'flu-like symptoms' (Ref. 111), which precludes them from being used to block such symptoms. A recent lipidomic screen has identified an omega-3 polyunsaturated fatty acid-derived lipid mediator, protectin D1, that inhibits influenza virus replication through the blockade of RNA nuclear export machinery109. Treatment of mice with protectin D1 was shown to protect the host against challenge with a lethal dose of influenza virus by reducing viral loads.
There are a number of approaches to reduce inflammation and immunopathology. Adenoviral delivery of active TGFβ was shown to delay mortality and reduce viral loads following H5N1 infection in mice91 but in some cases, TGFβ leads to impaired viral resistance and death in mice92. A more promising approach is treatment with TNF-specific antibodies, which reduced pulmonary recruitment of inflammatory cells and the severity of illness without preventing viral clearance112. Another approach is to block the infiltration of leukocytes to the lung, such as the infiltration of neutrophils, which mainly accounts for gene signatures that are associated with fatal influenza virus infection113. Sphingosine 1-phosphate receptor 1 (S1PR1; also known as S1P1) is expressed by lymphocytes and endothelial cells, and is known to control lymphocyte egress from lymph nodes114. S1PR1 agonists administered shortly after influenza virus challenge block chemokine expression in lung endothelial cells and protect mice from lethality without any effect on adaptive immune responses or viral loads115. In addition, attempts to block the TLR4-dependent inflammatory effect of oxidized phospholipids that are generated during influenza virus infection show promise in an animal model; blockade of TLR4 with eritoran (E5564; Eisai), a TLR4 antagonist, protected mice from disease and death following infection with the A/PR8 strain of influenza virus116.
A different approach to treating the acute respiratory damage caused by influenza viruses could be to increase host tolerance. Because the tolerance capacity of each cell type differs considerably, and the vascular and respiratory systems are most vulnerable to the damage sequelae9,10,11, protecting endothelial cells and epithelial cells of the respiratory tract from influenza virus-induced damage is a priority. The administration of agents that increase tissue protective and reparatory factors — such as vascular endothelial growth factors, epidermal growth factors and antioxidants — to limit the damage caused by reactive oxygen species may be beneficial. In this regard, the administration of recombinant human catalase117 (an enzyme that converts hydrogen peroxide into molecular oxygen and water), or apocynin118 (an inhibitor of NOX2 NADPH oxidase) to the lung of influenza virus-infected mice was shown to reduce the levels of reactive oxygen and to confer some level of protection.
Conclusions and perspectives
The past decade has brought a great expansion in our understanding of innate sensing and responses to influenza virus infection. The field is now ripe for translating these findings into the clinic to prevent and treat diseases that are caused by influenza viruses. In this regard, it is important to keep in mind that a given influenza virus causes very different disease outcomes in humans depending on age, comorbidities, pregnancy and host genetics. The risk of influenza virus-related death increases exponentially after the age of 65, with over 90% of the overall annual influenza virus-related mortality in the United States being from this age group119. What makes the elderly more susceptible to influenza virus-related illness and fatality? The age-dependent increase in comorbidities that are associated with a higher risk of influenza virus-related complications — such as chronic respiratory conditions, hepatic, renal, cardiovascular, haematological, neurological and metabolic disorders — has a role. In older humans, complications from secondary bacterial infection are a leading cause of the morbidity and mortality that is associated with influenza virus infection120,121. Therefore, efforts to minimize the damage that is caused by secondary bacterial infection are key to reducing mortality in this vulnerable population. In addition to these physiological factors, socioeconomic status dictates the outcome of influenza virus infection, as lower income populations with comorbidities are tenfold more likely to be hospitalized due to influenza virus-associated illness in the United States across all age groups122. Therefore, influenza virus disease should be treated as a complex disease, and like many other such diseases clinical outcomes could be improved by universal access to healthcare.
References
Manicassamy, B. et al. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc. Natl Acad. Sci. 107, 11531–11536 (2010).
Perrone, L. A., Plowden, J. K., García-Sastre, A., Katz, J. M. & Tumpey, T. M. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 4, e1000115 (2008).
Hogner, K. et al. Macrophage-expressed IFN-β contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog. 9, e1003188 (2013).
Kallfass, C., Lienenklaus, S., Weiss, S. & Staeheli, P. Visualizing the beta interferon response in mice during infection with influenza A viruses expressing or lacking nonstructural protein 1. J. Virol. 87, 6925–6930 (2013).
Jewell, N. A. et al. Differential type I interferon induction by respiratory syncytial virus and influenza a virus in vivo. J. Virol. 81, 9790–9800 (2007).
Gazit, R. et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nature Immunol. 7, 517–523 (2006).
Hashimoto, Y., Moki, T., Takizawa, T., Shiratsuchi, A. & Nakanishi, Y. Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J. Immunol. 178, 2448–2457 (2007).
Braciale, T. J., Sun, J. & Kim, T. S. Regulating the adaptive immune response to respiratory virus infection. Nature Rev. Immunol. 12, 295–305 (2012).
Medzhitov, R., Schneider, D. S. & Soares, M. P. Disease tolerance as a defense strategy. Science 335, 936–941 (2012).
Raberg, L., Sim, D. & Read, A. F. Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science 318, 812–814 (2007).
Ayres, J. S. & Schneider, D. S. Tolerance of infections. Annu. Rev. Immunol. 30, 271–294 (2012).
Paiardini, M., Pandrea, I., Apetrei, C. & Silvestri, G. Lessons learned from the natural hosts of HIV-related viruses. Annu. Rev. Med. 60, 485–495 (2009).
Janeway, C. A. Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 54, 1–13 (1989).
Medzhitov, R. Toll-like receptors and innate immunity. Nature Rev. Immunol. 1, 135–145 (2001).
Pang, I. K. & Iwasaki, A. Control of antiviral immunity by pattern recognition and the microbiome. Immunol. Rev. 245, 209–226 (2012).
Alexopoulou, L., Holt, A. C., Medzhitov, R. & Flavell, R. A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413, 732–738 (2001).
Pichlmair, A. et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314, 997–1001 (2006).
Wisskirchen, C., Ludersdorfer, T. H., Muller, D. A., Moritz, E. & Pavlovic, J. The cellular RNA helicase UAP56 is required for prevention of double-stranded RNA formation during influenza A virus infection. J. Virol. 85, 8646–8655 (2011).
Schulz, O. et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 433, 887–892 (2005).
Le Goffic, R. et al. Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J. Immunol. 178, 3368–3372 (2007).
Guillot, L. et al. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J. Biol. Chem. 280, 5571–5580 (2005).
Le Goffic, R. et al. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2, 0526 (2006). This study demonstrates a paradoxical role of TLR3 in promoting both antiviral resistance and inflammation, but despite an increase in viral load, Tlr3−/− mice had a survival advantage owing to reduced inflammation.
Heer, A. K. et al. TLR signaling fine-tunes anti-influenza B cell responses without regulating effector T cell responses. J. Immunol. 178, 2182–2191 (2007).
Lund, J. M. et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl Acad. Sci. USA 101, 5598–5603 (2004).
Diebold, S. S., Kaisho, T., Hemmi, H., Akira, S. & Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531 (2004).
Honda, K. et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 434, 1035–1040 (2005).
Sasai, M., Linehan, M. M. & Iwasaki, A. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science 329, 1530–1534 (2010).
Lande, R. et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449, 564–569 (2007).
Seo, S. U. et al. MyD88 signaling is indispensable for primary influenza A virus infection but dispensable for secondary infection. J. Virol. 84, 12713–12722 (2010).
Jeisy-Scott, V. et al. J. Virol. 86, 10988–10998 (2012).
Koyama, S. et al. Differential role of TLR- and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J. Immunol. 179, 4711–4720 (2007).
Ablasser, A. et al. Selection of molecular structure and delivery of RNA oligonucleotides to activate TLR7 versus TLR8 and to induce high amounts of IL-12p70 in primary human monocytes. J. Immunol. 182, 6824–6833 (2009).
Kato, H. et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23, 19–28 (2005).
Hornung, V. et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 (2006).
Rehwinkel, J. et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140, 397–408 (2010).
Baum, A., Sachidanandam, R. & Garcia-Sastre, A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl Acad. Sci. USA 107, 16303–16308 (2010).
Luo, D. et al. Structural insights into RNA recognition by RIG-I. Cell 147, 409–422 (2011).
Jiang, F. et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 479, 423–427 (2011).
Kowalinski, E. et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147, 423–435 (2011).
Onomoto, K. et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS ONE 7, e43031 (2012). This study demonstrates that the stress granules that form in infected cells serve as a hub for viral RNA accumulation and RIG-I signalling.
Pang, I. K., Ichinohe, T. & Iwasaki, A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8+ T cell responses to influenza A virus. 14, 246–253 (2013).
Gack, M. U. et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5, 439–449 (2009).
Bergsbaken, T., Fink, S. L. & Cookson, B. T. Pyroptosis: host cell death and inflammation. Nature Rev. Microbiol. 7, 99–109 (2009).
Martinon, F., Mayor, A. & Tschopp, J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265 (2009).
Guarda, G. et al. Differential expression of NLRP3 among hematopoietic cells. J. Immunol. 186, 2529–2534 (2011).
Pothlichet, J. et al. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 9, e1003256 (2013).
Thomas, P. G. et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30, 566–575 (2009).
Ichinohe, T., Pang, I. K. & Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nature Immunol. 11, 404–410 (2010).
McAuley, J. L. et al. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 9, e1003392 (2013).
Ichinohe, T., Yamazaki, T., Koshiba, T. & Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl Acad. Sci. USA 110, 17963–17968 (2013).
Tal, M. C. & Iwasaki, A. Mitoxosome: a mitochondrial platform for cross-talk between cellular stress and antiviral signaling. Immunol. Rev. 243, 215–234 (2011).
Allen, I. C. et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30, 556–565 (2009).
Van Der Sluijs, K. F. et al. Enhanced viral clearance in interleukin-18 gene-deficient mice after pulmonary infection with influenza A virus. Immunology 114, 112–120 (2005).
Schmitz, N., Kurrer, M., Bachmann, M. F. & Kopf, M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J. Virol. 79, 6441–6448 (2005).
Ichinohe, T., Lee, H. K., Ogura, Y., Flavell, R. & Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 206, 79–87 (2009).
Staeheli, P., Haller, O., Boll, W., Lindenmann, J. & Weissmann, C. Mx protein: constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell 44, 147–158 (1986).
Zurcher, T., Pavlovic, J. & Staeheli, P. Mouse Mx2 protein inhibits vesicular stomatitis virus but not influenza virus. Virology 187, 796–800 (1992).
Hefti, H. P. et al. Human MxA protein protects mice lacking a functional alpha/beta interferon system against La Crosse virus and other lethal viral infections. J. Virol. 73, 6984–6991 (1999).
Turan, K. et al. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res. 32, 643–652 (2004).
Pavlovic, J. et al. Enhanced virus resistance of transgenic mice expressing the human MxA protein. J. Virol. 69, 4506–4510 (1995).
Goujon, C. et al. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502, 559–562 (2013).
Liu, Z. et al. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 14, 398–410 (2013).
Brass, A. L. et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139, 1243–1254 (2009). This study uses a functional genomic screen and shows that IFITM1, IFITM2 and IFITM3 restrict an early step in the replication of influenza A virus.
Bailey, C. C., Huang, I. C., Kam, C. & Farzan, M. Ifitm3 limits the severity of acute influenza in mice. PLoS Pathog. 8, e1002909 (2012).
Everitt, A. R. et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484, 519–523 (2012). References 64 and 65 show that despite the presence of other IFITM proteins, Ifitm3−/− mice succumb to influenza virus infection owing to impaired viral control and severe pathology.
Sadler, A. J. & Williams, B. R. Interferon-inducible antiviral effectors. Nature Rev. Immunol. 8, 559–568 (2008).
Silverman, R. H. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 81, 12720–12729 (2007).
Kumar, A., Haque, J., Lacoste, J., Hiscott, J. & Williams, B. R. Double-stranded RNA-dependent protein kinase activates transcription factor NF-κB by phosphorylating IκB. Proc. Natl Acad. Sci. USA 91, 6288–6292 (1994).
Schulz, O. et al. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 7, 354–361 (2010).
Balachandran, S. et al. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 13, 129–141 (2000).
Dauber, B. et al. Influenza B virus ribonucleoprotein is a potent activator of the antiviral kinase PKR. PLoS Pathog. 5, e1000473 (2009).
Schoggins, J. W. et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695 (2013). This study uses an ectopic expression assay to screen a library of more than 350 human ISGs for their effects on 14 viruses representing seven families and 11 genera. It reveals several new ISGs that are capable of restricting influenza virus replication. It is also a great resource paper.
Wang, X., Hinson, E. R. & Cresswell, P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2, 96–105 (2007).
Blanc, M. et al. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38, 106–118 (2013).
Liu, S. Y. et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38, 92–105 (2013).
Di Pietro, A. et al. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 87, 4523–4533 (2013).
Durfee, L. A., Lyon, N., Seo, K. & Huibregtse, J. M. The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol. Cell 38, 722–732 (2010).
Lenschow, D. J. et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl Acad. Sci. USA 104, 1371–1376 (2007).
Kohlmeier, J. E., Cookenham, T., Roberts, A. D., Miller, S. C. & Woodland, D. L. Type I interferons regulate cytolytic activity of memory CD8+ T cells in the lung airways during respiratory virus challenge. Immunity 33, 96–105 (2010).
Der, S. D., Zhou, A., Williams, B. R. & Silverman, R. H. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc. Natl Acad. Sci. USA 95, 15623–15628 (1998).
Mordstein, M. et al. Interferon-λ contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 4, e1000151 (2008).
Durbin, J. E. et al. Type I IFN modulates innate and specific antiviral immunity. J. Immunol. 164, 4220–4228 (2000).
Hermesh, T., Moran, T. M., Jain, D. & Lopez, C. B. Granulocyte colony-stimulating factor protects mice during respiratory virus infections. PLoS ONE 7, e37334 (2012).
Lauder, S. N. et al. Interleukin-6 limits influenza-induced inflammation and protects against fatal lung pathology. Eur. J. Immunol. 43, 2613–2625 (2013).
Denton, A. E., Doherty, P. C., Turner, S. J. & La Gruta, N. L. IL-18, but not IL-12, is required for optimal cytokine production by influenza virus-specific CD8+ T cells. Eur. J. Immunol. 37, 368–375 (2007).
Sun, K., Torres, L. & Metzger, D. W. A detrimental effect of interleukin-10 on protective pulmonary humoral immunity during primary influenza A virus infection. J. Virol. 84, 5007–5014 (2010).
McKinstry, K. K. et al. IL-10 deficiency unleashes an influenza-specific TH17 response and enhances survival against high-dose challenge. J. Immunol. 182, 7353–7363 (2009).
Sun, J., Madan, R., Karp, C. L. & Braciale, T. J. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nature Med. 15, 277–284 (2009).
Monticelli, L. A. et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nature Immunol. 12, 1045–1054 (2011).
Chang, Y. J. et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nature Immunol. 12, 631–638 (2011).
Carlson, C. M. et al. Transforming growth factor-β: activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog. 6, e1001136 (2010).
Williams, A. E. et al. TGF-β prevents eosinophilic lung disease but impairs pathogen clearance. Microbes Infect. 7, 365–374 (2005).
Snelgrove, R. J. et al. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nature Immunol. 9, 1074–1083 (2008).
Crowe, C. R. et al. Critical role of IL-17RA in immunopathology of influenza infection. J. Immunol. 183, 5301–5310 (2009).
Szretter, K. J. et al. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J. Virol. 81, 2736–2744 (2007).
Blander, J. M. & Sander, L. E. Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nature Rev. Immunol. 12, 215–225 (2012).
Nish, S. & Medzhitov, R. Host defense pathways: role of redundancy and compensation in infectious disease phenotypes. Immunity 34, 629–636 (2011).
Pang, I. K., Pillai, P. S. & Iwasaki, A. Efficient influenza A virus replication in the respiratory tract requires signals from TLR7 and RIG-I. Proc. Natl Acad. Sci. USA 110, 13910–13915 (2013).
Negishi, H. et al. Cross-interference of RLR and TLR signaling pathways modulates antibacterial T cell responses. Nature Immunol. 13, 659–666 (2012).
Edwards, A. D. et al. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8α+ DC correlates with unresponsiveness to imidazoquinolines. Eur. J. Immunol. 33, 827–833 (2003).
Geeraedts, F. et al. Superior immunogenicity of inactivated whole virus H5N1 influenza vaccine is primarily controlled by Toll-like receptor signalling. PLoS Pathog. 4, e1000138 (2008). This study provides definitive evidence for the role of TLR7 in generating protective antibody responses to whole killed influenza vaccine, and demonstrates that split vaccines are inferior owing to the loss of viral RNA during vaccine preparation.
Clingan, J. M. & Matloubian, M. B. Cell-intrinsic TLR7 signaling is required for optimal B cell responses during chronic viral infection. J. Immunol. 191, 810–818 (2013).
Kash, J. C. et al. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443, 578–581 (2006).
Kobasa, D. et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445, 319–323 (2007).
Cilloniz, C. et al. Lethal influenza virus infection in macaques is associated with early dysregulation of inflammatory related genes. PLoS Pathog. 5, e1000604 (2009).
Baskin, C. R. et al. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc. Natl Acad. Sci. USA 106, 3455–3460 (2009).
Palese, P. Influenza: old and new threats. Nature Med. 10, S82–87 (2004).
Imai, Y. et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133, 235–249 (2008).
Morita, M. et al. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell 153, 112–125 (2013). This study uses a mediator lipidomics and bioactive lipid screen and demonstrates the regulation of lipid metabolites that are generated during influenza virus infection. The study also identifies protectin D1 as a candidate antiviral molecule that can reduce viral load in vivo.
Tisoncik, J. R. et al. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 76, 16–32 (2012).
Calvaruso, V., Mazza, M. & Almasio, P. L. Pegylated-interferon-α2a in clinical practice: how to manage patients suffering from side effects. Expert Opin. Drug Saf 10, 429–435 (2011).
Hussell, T., Pennycook, A. & Openshaw, P. J. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur. J. Immunol. 31, 2566–2573 (2001).
Brandes, M., Klauschen, F., Kuchen, S. & Germain, R. N. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 154, 197–212 (2013).
Cyster, J. G. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu. Rev. Immunol. 23, 127–159 (2005).
Teijaro, J. R. et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 146, 980–991 (2011).
Shirey, K. A. et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 497, 498–502 (2013).
Shi, X. et al. PEGylated human catalase elicits potent therapeutic effects on H1N1 influenza-induced pneumonia in mice. Appl. Microbiol. Biotechnol. 97, 10025–10033 (2013).
Vlahos, R. et al. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog. 7, e1001271 (2011).
Thompson, W.W. et al. Influenza-associated hospitalizations in the United States. JAMA 292, 1333–1340 (2004).
Beadling, C. & Slifka, M. K. How do viral infections predispose patients to bacterial infections? Curr. Opin. Infecti. Diseases 17, 185–191 (2004).
McCullers, J. A. Insights into the interaction between influenza virus and pneumococcus. Clin. Microbiol. Rev. 19, 571–582 (2006).
Glezen, W. P., Greenberg, S. B., Atmar, R. L., Piedra, P. A. & Couch, R. B. Impact of respiratory virus infections on persons with chronic underlying conditions. JAMA 283, 499–505 (2000).
Horimoto, T. & Kawaoka, Y. Pandemic threat posed by avian influenza A viruses. Clin. Microbiol. Rev. 14, 129–149 (2001).
Tscherne, D. M. & Garcia-Sastre, A. Virulence determinants of pandemic influenza viruses. J. Clin. Invest. 121, 6–13 (2011).
Ungchusak, K. et al. Probable person-to-person transmission of avian influenza A (H5N1). New Engl. J. Med. 352, 333–340 (2005).
World Health Organization Global Influenza Program Surveillance Network. Evolution of H5N1 avian influenza viruses in Asia. Emerg. Infect. Diseases 11, 1515–1521 (2005).
Herfst, S. et al. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336, 1534–1541 (2012).
Imai, M. et al. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 486, 420–428 (2012).
Staeheli, P., Grob, R., Meier, E., Sutcliffe, J. G. & Haller, O. Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol. Cell. Biol. 8, 4518–4523 (1988).
Koerner, I., Kochs, G., Kalinke, U., Weiss, S. & Staeheli, P. Protective role of beta interferon in host defense against influenza A virus. J. Virol. 81, 2025–2030 (2007).
Kaminski, M. M., Ohnemus, A., Cornitescu, M. & Staeheli, P. Plasmacytoid dendritic cells and Toll-like receptor 7-dependent signalling promote efficient protection of mice against highly virulent influenza A virus. J. General Virol. 93, 555–559 (2012). The authors demonstrate a key role of TLR7 and pDCs in antiviral defence, which has not previously been possible to study in inbred mice that lack MX1. This study indicates the importance of studying immune defence in the context of MX1-sufficient mice.
GeurtsvanKessel, C. H. et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b− but not plasmacytoid dendritic cells. J. Exp. Med. 205, 1621–1634 (2008).
Wolf, A. I. et al. Plasmacytoid dendritic cells are dispensable during primary influenza virus infection. J. Immunol. 182, 871–879 (2009).
Ichinohe, T. et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl Acad. Sci. USA 108, 5354–5359 (2011).
Shaw, M. H., Kamada, N., Kim, Y. G. & Nunez, G. Microbiota-induced IL-1β, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J. Exp. Med. 209, 251–258 (2012).
Hoshi, N. et al. MyD88 signalling in colonic mononuclear phagocytes drives colitis in IL-10-deficient mice. Nature Commun. 3, 1120 (2012).
Helft, J. et al. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J. Clin. Invest. 122, 4037–4047 (2012).
Langlois, R. A., Varble, A., Chua, M. A., Garcia-Sastre, A. & tenOever, B. R. Hematopoietic-specific targeting of influenza A virus reveals replication requirements for induction of antiviral immune responses. Proc. Natl Acad. Sci. USA 109, 12117–12122 (2012).
Abt, M. C. et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37, 158–170 (2012).
Zurcher, T., Pavlovic, J. & Staeheli, P. Nuclear localization of mouse Mx1 protein is necessary for inhibition of influenza virus. J. Virol. 66, 5059–5066 (1992).
Cilloniz, C. et al. Molecular signatures associated with Mx1-mediated resistance to highly pathogenic influenza virus infection: mechanisms of survival. J. Virol. 86, 2437–2446 (2012).
Garber, E. A., Hreniuk, D. L., Scheidel, L. M. & van der Ploeg, L. H. Mutations in murine Mx1: effects on localization and antiviral activity. Virology 194, 715–723 (1993).
Sadler, A. J. & Williams, B. R. Structure and function of the protein kinase R. Curr. Top. Microbiol. Immunol. 316, 253–292 (2007).
Jia, R. et al. Identification of an endocytic signal essential for the antiviral action of IFITM3. Cell. Microbiol. http://dx.doi.org/10.1111/cmi.12262 (2014).
Hinson, E. R. & Cresswell, P. The N-terminal amphipathic α-helix of viperin mediates localization to the cytosolic face of the endoplasmic reticulum and inhibits protein secretion. J. Biol. Chem. 284, 4705–4712 (2009).
Hinson, E. R. & Cresswell, P. The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic α-helix. Proc. Natl Acad. Sci. USA 106, 20452–20457 (2009).
Hidaka, F. et al. A missense mutation of the Toll-like receptor 3 gene in a patient with influenza-associated encephalopathy. Clin. Immunol. 119, 188–194 (2006).
Esposito, S. et al. Toll-like receptor 3 gene polymorphisms and severity of pandemic A/H1N1/2009 influenza in otherwise healthy children. Virol. J. 9, 270 (2012).
Goodman, A. G. et al. The alpha/beta interferon receptor provides protection against influenza virus replication but is dispensable for inflammatory response signaling. J. Virol. 84, 2027–2037 (2010).
Price, G. E., Gaszewska-Mastarlarz, A. & Moskophidis, D. The role of alpha/beta and gamma interferons in development of immunity to influenza A virus in mice. J. Virol. 74, 3996–4003 (2000).
Garcia-Sastre, A. et al. The role of interferon in influenza virus tissue tropism. J. Virol. 72, 8550–8558 (1998).
Woods, A. et al. Influenza virus-induced type I interferon leads to polyclonal B-cell activation but does not break down B-cell tolerance. J. Virol. 81, 12525–12534 (2007).
Acknowledgements
The authors would like to thank R. Medzhitov for critical discussion and the US National Institutes of Health (NIH) for its support of research in the laboratory (grant numbers AI081884, AI054359, AI062428 and AI064705). Research on influenza virus in the laboratory was supported in part by the National Institute of Allergy and Infectious Diseases, NIH, USA, under award number U54AI057160 to the Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (MRCE), USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- Pattern recognition receptors
-
(PRRs). Germ-line encoded host receptors that are membrane bound (such as Toll-like receptors) or soluble and present in the cytoplasm (such as RIG-I, MDA5 and NLRs) and sense pathogen-associated molecular patterns and initiate signalling cascades that lead to innate immune responses.
- Type I interferons
-
(Type I IFNs). Cytokines that are rapidly induced by virus infection, as well as by some bacterial infections. They act on the IFN receptor to limit viral replication and to enhance antigen-specific immune responses.
- Antigenic drift
-
A process by which circulating influenza virus genomes are constantly changing, which allows the virus to cause annual epidemics. Antigenic drift occurs when mutations accumulate in the haemagglutinin and neuraminidase genes that alter the antigenicity of these proteins such that the 'drifted' strains are no longer neutralized by antibodies that were specific for previously circulating strains.
- Antigenic shift
-
A process by which a new influenza A virus haemagglutinin subtype (with or without an accompanying new neuraminidase subtype) is introduced into the human population, which often lacks prior experience of and immunity to the subtype. Antigenic shift can occur as a result of the direct introduction of an influenza virus from an animal or avian host into humans or by the exchange or reassortment of gene segments between human and non-human influenza viruses when they co-infect animals or humans.
- Eicosanoids
-
Fatty acid derivatives, mainly derived from arachidonic acid precursors, that have a wide variety of biological activities. There are four main classes of eicosanoid — the prostaglandins, prostacyclins, thromboxanes and leukotrienes — that are derived from the activities of cyclooxygenases and lipoxygenases on membrane-associated fatty acid precursors.
- IFN-stimulated genes
-
(ISGs). These genes contain interferon (IFN)-responsive promoters and are responsible for the antiviral, antiproliferative and immunomodulatory properties of IFN. More than 400 such genes have been identified by microarray analysis. Some — such as protein kinase R, ribonuclease L, IFN-induced GTP-binding protein MX1 and ISG15 — have well-documented antiviral activities, but the precise biological function of the majority of these genes is unknown.
- Antiviral resistance
-
A host strategy to reduce viral burden by recognizing and eliminating viruses and virus-infected cells.
- Disease tolerance
-
A host strategy to reduce tissue damage inflicted by pathogens or the immune response to pathogens.
- Antiviral stress granules
-
Dense cytoplasmic aggregates of RNA and proteins that appear when a cell is under stress. Stress granules contain stalled translation initiation complexes and processing bodies, and are a site for the storage of transiently repressed mRNA.
- Inflammasome
-
A molecular complex of several proteins that upon assembly activates caspase 1, which in turn cleaves substrates including pro-interleukin-1 (pro-IL-1), thereby producing active IL-1.
- Pyroptosis
-
A form of programmed cell death that requires caspase 1 downstream of inflammasome activation. Pyroptosis is accompanied by pore formation and cell swelling, and is an inflammatory form of death.
- Mitofusin 2
-
A mitochondrial GTPase that is embedded in the outer membrane that participates in mitochondrial fusion.
- Innate lymphoid cells
-
(ILCs). A group of innate immune cells that are lymphoid in morphology and developmental origin, but lack properties of adaptive B cells and T cells such as recombined antigen-specific receptors. They function in the regulation of immunity, tissue homeostasis and inflammation in response to cytokine stimulation.
- Amphiregulin
-
An epidermal growth factor (EGF)-like growth factor that stimulates tissue remodelling and the growth of epithelial cells through binding to its receptor, EGFR. Amphiregulin has been shown to restore lung function after influenza virus infection.
- Airway hyperresponsiveness
-
A hyperreactivity of the airways that is initiated by exposure to a defined stimulus and is usually tolerated by normal individuals but that causes bronchoconstriction and inflammatory cell infiltration in allergic individuals.
- γδ T cells
-
T cells that express a T cell receptor that consists of a γ-chain and a δ-chain and that are involved in innate immune responses. γδ T cells are present in the skin, digestive tract and reproductive mucosa.
- Viability-associated PAMPs
-
(Vita-PAMPs). Members of a special class of pathogen-associated molecular patterns (PAMPs) that are recognized by the innate immune system to signify microbial life. These patterns differentiate dead and living microorganisms to allow for the scaling of appropriate immune responses based on the level of threat that the microbial signals represents.
- Cross-priming
-
The initiation of a CD8+ T cell response to an antigen that has been taken up by antigen-presenting cells and re-routed to the MHC class I presentation pathway.
- NADPH oxidase
-
An enzyme system that consists of multiple cytosolic and membrane-bound subunits. The complex is assembled in activated neutrophils mainly on the phagolysosomal membrane. NADPH oxidase uses electrons from NADPH to reduce molecular oxygen to form superoxide anions. Superoxide anions are enzymatically converted into hydrogen peroxide, which is converted by myeloperoxidase to hypochloric acid, a highly toxic and microbicidal agent.
Rights and permissions
About this article

Cite this article
Iwasaki, A., Pillai, P. Innate immunity to influenza virus infection. Nat Rev Immunol 14, 315–328 (2014). https://doi.org/10.1038/nri3665
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri3665
This article is cited by
-
Cullin5 drives experimental asthma exacerbations by modulating alveolar macrophage antiviral immunity
Nature Communications (2024)
-
TRIM38 Induced in Respiratory Syncytial Virus-infected Cells Downregulates Type I Interferon Expression by Competing with TRIM25 to Bind RIG-I
Inflammation (2024)
-
Double-edged sword of JAK/STAT signaling pathway in viral infections: novel insights into virotherapy
Cell Communication and Signaling (2023)
-
TRAF3 activates STING-mediated suppression of EV-A71 and target of viral evasion
Signal Transduction and Targeted Therapy (2023)
-
A quantitative systems pharmacology model of the pathophysiology and treatment of COVID-19 predicts optimal timing of pharmacological interventions
npj Systems Biology and Applications (2023)
