
Key Points
-
Epigenetic regulation of gene expression relies on covalent, reversible and sequence-specific modifications of histone proteins that package DNA. Deregulation of this control mechanism can lead to multiple diseases.
-
Acetylation and methylation of lysine and arginine residues are the most abundant modifications of the known histone marks. Histone acetyl marks are written by histone acetyltranferases, read by bromodomain-containing proteins and erased by histone deacetylases.
-
Histone methyl marks are written by protein methyltransferases and read by proteins containing chromodomains, Tudor domains, malignant brain tumour domains, PWWP domains and plant homeodomain (PHD) fingers. Histone methyl marks are erased by lysine demethylases.
-
Domains that write, read or erase histone marks are often grouped within a single gene, and these proteins are often combined within multiprotein complexes that mediate crosstalk between specific marks.
-
When they are mutated, aberrantly expressed or driven by aberrant upstream signals, writers, readers and erasers of histone marks can affect the expression patterns of genes that lead to — or even drive — and maintain disease states. Direct associations have been observed in cancer, neuropsychiatric disorders, inflammation and metabolic diseases.
-
Several histone deacetylase inhibitors have reached the clinic and two are approved in oncology. Small-molecule antagonists of bromodomain-containing proteins have demonstrated efficacy in animal models of cancer and inflammation. The chemical tractability of histone acetyltransferases, however, remains unclear.
-
Potent protein methyltransferase inhibitors have been reported, and one compound was active against tumour xenografts in mice. Lysine demethylase inhibitors and antagonists of methyl-lysine readers are at an earlier development stage.
Abstract
Epigenetic regulation of gene expression is a dynamic and reversible process that establishes normal cellular phenotypes but also contributes to human diseases. At the molecular level, epigenetic regulation involves hierarchical covalent modification of DNA and the proteins that package DNA, such as histones. Here, we review the key protein families that mediate epigenetic signalling through the acetylation and methylation of histones, including histone deacetylases, protein methyltransferases, lysine demethylases, bromodomain-containing proteins and proteins that bind to methylated histones. These protein families are emerging as druggable classes of enzymes and druggable classes of protein–protein interaction domains. In this article, we discuss the known links with disease, basic molecular mechanisms of action and recent progress in the pharmacological modulation of each class of proteins.
Similar content being viewed by others
Main
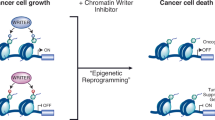
Although all cells in an organism inherit the same genetic material, the ability of cells to maintain the unique physical characteristics and biological functions of specific tissues and organs is due to heritable differences in the packaging of DNA and chromatin. These differences dictate distinct cellular gene expression programmes but do not involve changes in the underlying DNA sequence of the organism. Thus, epigenetics (which literally means 'above genetics') underpins the fundamental basis of human physiology. Importantly, the epigenetic state of a cell is malleable; it evolves in an ordered manner during the cellular differentiation and development of an organism, and epigenetic changes are responsible for cellular plasticity that enables cellular reprogramming and response to the environment. Because epigenetic mechanisms are responsible for the integration of environmental cues at the cellular level, they have an important role in diseases related to diet, lifestyle, early life experience and environmental exposure to toxins1. Thus, epigenetics is of therapeutic relevance in multiple diseases such as cancer, inflammation, metabolic disease and neuropsychiatric disorders, as well as in regenerative medicine2,3,4.
The dynamic nature of epigenetics means that it may be possible to alter disease-associated epigenetic states through direct manipulation of the molecular factors involved in this process. Several interrelated molecular mechanisms contribute to epigenetic gene regulation, including chromatin remodelling via ATP-dependent processes and exchange of histone variants, regulation by non-coding RNAs, methylation and related modifications of cytosines on DNA, as well as covalent modification of histones5 (Fig. 1). Inhibitors of DNA methylation and histone deacetylase (HDAC) inhibitors are approved for clinical use in haematological malignancies, thus providing proof of concept for epigenetic therapies6. Over the past decade, knowledge of the proteins involved in the post-translational modification of histones has grown tremendously. These proteins comprise several families of related enzymes and chromatin-interacting proteins, and are a rich source of potential therapeutic targets. Here, we review the proteins involved in depositing, removing or binding to acetyl and methyl groups — the two most abundant histone post-translational modifications (which are commonly referred to as histone marks). We focus on the mediators of acetyl and methyl histone marks because of their prominent role in several diseases, as well as the emerging realization that many of these proteins are susceptible to inhibition by small molecules.

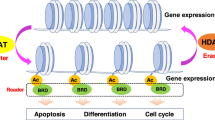
DNA is packaged into chromatin by wrapping around histone proteins (two copies each of histones H2A, H2B, H3 and H4) to form a nucleosome. Nucleosomes are further compacted by additional protein factors to form chromatin, with the degree of compactness dependent on the types of post-translational modification present on the histones, especially on their terminal residues, which protrude from the nucleosome particle. Acetylated histones tend to be less compact and more accessible to RNA polymerase and the transcriptional machinery, thereby enabling transcription of nearby genes. Methylated histones can be either repressive or activating, depending on the site and degree of methylation. The combination of modifications on each histone and/or nucleosome establishes a code that relates to the transcriptional properties of the nearby genes. The primary protein families that mediate histone post-translational modifications are illustrated in the inset. Proteins that covalently attach acetyl or methyl groups produce (or 'write') the code (these include histone acetyltransferases and histone methyltransferases) and are termed 'writers'. Proteins that recognize and bind to histone modifications are termed 'readers' of the code (these include bromodomains, plant homeodomains (PHDs) and members of the royal family of methyl-lysine-binding domains). Enzymes that remove histone marks are termed 'erasers' (these include histone deacetylases and lysine demethylases).
Defining the druggable epigenome
Acetylation and methylation networks define a large component of the human epigenome. Although several histone post-translational modifications — including phosphorylation and ubiquitylation — are important components of the epigenome, acetyl and methyl marks are the most abundant and among the most widely studied, and have a large number of druggable proteins that mediate their dynamic activity. A feature of epigenetic regulation that is mediated by histone marks is the collaboration among combinations of marks to affect specific cellular outcomes — often referred to as the histone code hypothesis7,8,9,10 (Fig. 1). For example, the recent mapping of nine acetyl and methyl histone marks across the genomes of nine different cell types showed that combinations of marks defined 15 chromatin states related to the transcriptional activity of surrounding genes11.
Individual marks and combinations of marks are recognized by several classes of conserved protein domains, usually within the context of larger multiprotein complexes. Thus, histone marks and the multiprotein complexes that bind to them contribute to the physical make-up of chromatin and to the recruitment of specific proteins to genomic loci that contain specific histone marks. For example, most of the enzymes that are 'writers' of methyl or acetyl histone marks are large proteins that, in addition to their catalytic domain, contain other domains or regions that 'read' histone marks and/or interact with DNA or other proteins. Together, these proteins form complexes that integrate upstream cellular and environmental signals to establish and maintain cellular identity and contribute to the genesis and/or maintenance of disease states10. Owing to remarkable progress over the past decade, we now know the basic complement of regulatory proteins that 'read', 'write' and 'erase' the major histone marks. These are summarized in Table 1, and further delineated in Fig. 2 as phylogenetic trees of structurally and evolutionarily related families of proteins.

Proteins are clustered on branches on the basis of the similarity of their amino acid sequences. The phylogenetic representation tends to cluster structurally (and sometimes functionally) related proteins. Drugs targeting a specific protein are more likely to be active against other proteins on the same branch. Distinct phylogenetic branches are highlighted with distinct colours (in the case of the malignant brain tumour (MBT) family, where only a few MBT domains are actually binding methyl-lysines, the red colour coding indicates the branch where all known methyl-lysine-binding domains are clustered). We assembled protein families by looking for domains associated with 'writing', 'reading' and 'erasing' acetyl and methyl marks in the Human Protein Reference Database, and by complementing the list with data from the literature, as well as data from the Pfam protein family database and the SMART (Simple Modular Architecture Research Tool) database. The phylogeny outlined in the trees is derived from multiple sequence alignments of the domain after which the family was named (full-length sequences were used for acetyltransferases as the catalytic domain is not always clearly defined for this family). If a domain is present multiple times in a protein, the protein is shown multiple times in the corresponding tree, followed by the sequential iteration of the domain in parenthesis: for example, L3MBTL(2) corresponds to the second MBT domain of the protein L3MBTL. If multiple variants with insertions or deletions were reported for a gene, the variant number according to Swiss-Prot nomenclature is indicated after a hyphen: for example, TRIM33-2 in the tree of bromodomain-containing proteins corresponds to the second Swiss-Prot variant of the TRIM33 (tripartite motif-containing protein 33) bromodomain. For each tree, a seed alignment was derived from available protein structures by aligning residues that were superimposed in the three-dimensional space. Additional sequences were appended by aligning them to the closest seed sequence. A larger version of the protein methyltransferase family was reported that includes numerous putative arginine methyltransferases; these are not depicted here as the authors of that work stated that they did not want to imply that these proteins are protein arginine methyltransferases per se173. For further trees, as well as details on sequence and domain boundaries, see Supplementary information S1–S10 (tables) and Supplementary information S11–S14 (figures). Small variations in domain boundaries or alignment methods can result in minor changes in the phylogeny15,173. ASH1L, ASH1-like protein; ATAD2, ATPase family AAA domain-containing protein 2; ATAT1, α-tubulin acetyltransferase 1; BAZ2A, bromodomain adjacent to zinc finger domain protein 2A; BPTF, bromodomain PHD finger transcription factor; BRD1, bromodomain containing protein 1; BRDT, bromodomain testis-specific protein; BRPF1, bromodomain and PHD finger-containing protein 1; BRWD1, bromodomain and WD repeat-containing protein 1; CECR2, cat eye syndrome chromosome region candidate protein 2; CLOCK, circadian locomoter output cycles kaput protein; CREBBP, CREB binding protein; DOT1L, DOT1-like protein; EHMT1, euchromatic histone lysine N-methyltransferase 1; ELP3, elongator complex protein 3; EP300, E1A binding protein p300; EZH1, histone lysine N-methyltransferase EZH1; GTF3C4, general transcription factor 3C polypeptide 4; HAT, histone acetylase; HDAC, histone deacetylase; JARID2, Jumonji/ARID domain-containing protein 2; JMJD1C, Jumonji domain-containing protein 1C; KAT2A, lysine acetyltransferase 2A; KDM, lysine demethylase; KDM1A, lysine-specific histone demethylase 1A; L3MBTL, lethal 3 MBT-like protein 1; MBTD1, MBT domain-containing protein 1; MDS1, myelodysplasia syndrome 1; MINA, MYC-induced nuclear antigen; MLL, mixed lineage leukaemia; MYST1, histone acetyltransferase MYST1; NCOA1, nuclear receptor co-activator 1; NO66, nucleolar protein 66; NSD1, nuclear receptor binding SET domain protein 1; PBRM1, protein polybromo 1; PHF2, PHD finger protein 2; PHIP, pleckstrin homology domain interacting protein; PMT, protein methyltransferase; PRDM1, PR domain-containing protein 1; PRMT1, protein arginine methyltransferase 1; SETD1A, SET domain containing protein 1A; SETD2, SET domain-containing protein 2; SETMAR, SET domain and mariner transposase fusion gene; SFMBT1, SCM-like with four MBT domains protein 1; SIRT1, sirtuin 1; SMARCA2, SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 2; SMYD1, SET and MYND domain-containing protein; SP100, nuclear antigen SP100; SP110, nuclear body protein SP110; SP140, nuclear body protein SP140; SP140L, nuclear body protein SP140-like protein; SUV39H1, suppressor of variegation 3–9 homolog 1; SUV420H1, suppressor of variegation 4–20 homolog 1; TAF1, TBP-associated factor 1; TAF1L, TAF1-like protein; UTY, ubiquitously transcribed Y chromosome tetratricopeptide repeat protein; WHSC1, Wolf–Hirschhorn syndrome candidate 1 protein; WHSC1L1, WHSC1-like protein; ZMYND8, zinc finger MYND domain-containing protein 8.
Histone acetylation. Since the first description of histone acetylation in 1964 (Ref. 12), it has been established that this is a highly dynamic process that is regulated by two families of enzymes — histone acetyltransferases (HATs) and HDACs — that operate in an opposing manner. HATs use acetyl-CoA as a cofactor and catalyse the transfer of an acetyl group to the ɛ-amino group of lysine side chains on the histone protein. This neutralizes the positive charge on lysine, thus reducing the affinity of the histone tail that protrudes from the nucleosome core of DNA. As a result, chromatin adopts a more relaxed structure, enabling the recruitment of the transcriptional machinery. HDACs oppose the effects of HATs and reverse the acetylation of lysine residues to restore their positive charge and stabilize the local chromatin architecture.
Among the various sites of histone lysine acetylation, Lys16 of histone 4 (H4K16) appears to be crucial in the regulation of chromatin folding and in the switch from heterochromatin to euchromatin13. In addition to the acetylation of histone tails, there are several lysine substrates within the globular core of the histone proteins (such as H3K56), which suggests that acetylation can also directly affect the interaction between histones and DNA14. There is evidence that histone acetylation, particularly of H4K5 and H4K12, is important for the recognition of chaperones during histone assembly and deposition into DNA.
Histone acetylation also promotes transcription by providing binding sites for proteins that are involved in gene activation. In particular, the bromodomain-containing family of proteins recognize (that is, 'read') modified lysine residues within histone proteins. Bromodomains are a common feature in a diverse set of proteins united by their importance in transcriptional co-activation, and the ability of bromodomains to identify and bind to acetylated lysine residues within histone proteins is key to their activity15,16.
Histone methylation
The significance of and the associated mechanisms of histone methylation have been gradually elucidated over the past decade. Lysine residues on histones can be monomethylated, dimethylated or trimethylated. Arginine residues are also subject to monomethylation and dimethylation. Dimethylation of arginine residues can occur in a symmetric manner (via monomethylation of both terminal guanidino nitrogens) or in an asymmetric manner (via dimethylation of one of the terminal guanidino nitrogens). As with acetylation, methylation is dynamic. Methyl marks are written by S-adenosylmethionine (SAM)-dependent methyltransferases and erased by either the Jumonji family of 2-oxoglutarate-dependent demethylases17 or the flavin-dependent enzymes lysine-specific histone demethylase 1 (LSD1; also known as KDM1A) and LSD2 (also known as KDM1B)18.
Because methylation does not change the charged state of a lysine or arginine residue, it does not appear to effect chromatin structure directly. Instead, the various methyl marks act as binding sites for other proteins that compact nucleosomes together19,20 or bring additional regulatory proteins to chromatin sites marked by methylation21,22. Each type of mark constitutes a specific signal that is recognized by highly evolved methyl-lysine-binding domains that recognize the level of methylation and, in many cases, the surrounding amino acid sequence (Table 1). Thus, trimethylated Lys4 of histone 3 (H3K4me3), H3K9me3 and H4K20me2 each interact with a distinct set of reader domains.
Histone lysine methylation can be associated with either transcriptional activation or repression. For example, H3K4me3 is a hallmark of transcriptionally active genes, whereas H3K9me3 and H3K27me3 (Refs 23, 24) are associated with silenced genes. Although protein arginine methylation is abundant and has been known for a long time, histone arginine methylation has only recently become recognized as an important transcriptional regulatory mechanism25. Arginine methylation of histones can promote or antagonize the interaction of nuclear factors with other nearby histone marks, thereby increasing the complexity of the histone code26,27.
Disease association
The readers, writers and erasers of epigenetic marks can contribute to or drive disease via two primary mechanisms. First, aberrant activity due to mutation or altered expression of epigenetic factors can alter subsequent cellular gene expression patterns that lead to or even drive and maintain disease states. Second, because the readers, writers and erasers are general factors that work in concert with many other cellular proteins, especially tissue-specific and environmentally responsive DNA-binding transcription factors, they can mediate altered gene expression patterns driven by upstream signals10. Importantly, the latter case offers the opportunity to target disease pathways whose primary drivers (for example, certain transcription factors or external stimuli) may not be druggable.
Cancer. Epigenetic mechanisms have long been known to be involved in cancer, beginning with the observation that levels of DNA methylation were dramatically altered in most cancers. Although cancer is fundamentally a genetic disease that is driven by irreversible genomic mutations that subsequently activate oncogenes or inactivate tumour suppressor genes, there is increasing evidence that many epigenetic regulatory proteins are among those dysregulated in cancer, and that histone marks are globally and locally altered within cancer epigenomes28.
This knowledge stimulated the development of inhibitors of DNA methyltransferases and HDACs that are clinically effective in several cancers, attesting to the value of epigenetic therapies in oncology28. However, these agents are non-selective within their target protein families and have substantial side effects. Although it remains to be demonstrated in the clinic, agents that target specific HDACs with greater selectivity may be beneficial in certain cancers. For example, treatment of neuroblastoma cell lines with a selective inhibitor of HDAC8 mimicked genetic knockdown of HDAC8 as well as inhibiting cellular proliferation and triggering differentiation29,30. Second-generation HDAC inhibitors — several of which are more selective — are currently in clinical trials for multiple types of cancer (Table 2).
Deregulation of epigenetic regulatory proteins and their signalling networks can occur via several mechanisms, including direct inactivating or activating mutations, gene amplification, indirect upregulation or inactivation of enzymes, and translocations that lead to the expression of gain-of-function fusion proteins that contain reader domains31. Well-known examples include overexpression of the key developmental histone lysine N-methyltransferase EZH2 in several types of leukaemia and in various solid tumours32.
The gene encoding the protein methyltransferase MLL is also subject to many chromosomal translocations that lead to the expression of chimeric fusion proteins and inappropriate recruitment of other epigenetic factors such as the methyltransferase DOT1-like protein (DOT1L)33. Inhibition of DOT1L was recently shown to selectively kill cells and tumour xenografts that contained MLL translocations34. EZH2 can be aberrantly upregulated by the overexpression of dominant mutations that increase its trimethylation activity, offering the possibility of selective therapy targeting the mutant protein35. A recent example of a potential epigenetic targeted therapy was shown in a model of midline carcinoma. In this cancer, carcinogenesis is driven by chromosomal translocation, which results in the expression of a fusion protein containing the bromodomain of bromodomain-containing protein 4 (BRD4) or BRD3 and a testis-specific transcription factor (NUT) that drives carcinogenesis. A selective antagonist of the BET family of bromodomains (which includes BRD2, BRD3, BRD4 and bromodomain testis-specific protein (BRDT)) resulted in the selective killing of BRD4–NUT-positive midline carcinoma xenografts36.
Modulation of epigenetic mechanisms also offers the potential for overcoming the genetic changes that drive cancer — especially oncoproteins that may not be druggable. For example, with the exception of nuclear hormone receptors, it is recognized that it is extremely challenging to inhibit most sequence-specific transcription factors using small molecules37. This includes the transcription factor MYC, whose pathological activation is among the most common genetic events observed in cancer genomes38. Although MYC was one of the first known and most common oncoproteins39, over 30 years of research have failed to identify compounds that can directly inhibit the activity of the MYC protein. However, several recent exciting reports indicate that MYC may be effectively inhibited in several haematological malignancies through pharmacological inhibition of one of its regulatory partners, BRD4. BRD4 binds acetylated histones via its bromodomain and mediates chromatin-dependent signalling and transcription at MYC target loci40. Inhibition of the interaction between BRD4 and acetylated histones results in reduced levels of MYC target genes and inhibition of transcription of the MYC gene itself41,42.
Similarly, overexpression of the bromodomain-containing nuclear cofactor ATPase AAA domain-containing protein 2 (ATAD2) is crucial for the proliferation and survival of triple-negative/basal-like breast cancer cells and controls the expression of the oncogene MYB43. The bromodomain of ATAD2 has a key role in tumorigenesis44. These results highlight the potential for targeting 'undruggable' oncogenic transcription factors by inhibiting the catalytic or chromatin-interaction activities of druggable epigenetic cofactors that drive the expression of oncogenic transcription factors.
There are numerous other cancer-linked alterations in the genes coding for (and the activity of) readers, writers and erasers of histone marks. Many of these alterations occur in key developmental genes and are associated with cancers that derive from stem cell-like early progenitors of a given tissue type, such as many haematological malignancies45,46,47,48 and medulloblastoma49,50. Thus, these self-renewing cells may be locked in an epigenetic state that prevents them from undergoing differentiation. Inhibition of mutated epigenetic proteins or inhibition of the transcriptional programme of other oncogenic signalling factors could be an attractive strategy for overcoming the block to differentiation in these types of cancers. Similarly, the oxygen-independent glycolytic metabolism that is observed in rapidly proliferating cancer cells (known as the Warburg effect) may be orchestrated and maintained by epigenetic signalling networks51.
Genomic instability is also a hallmark of cancer, and inactivation of epigenetic proteins that contribute to DNA damage checkpoints (such as the HAT 60 kDa Tat-interactive protein (TIP60; also known as KAT5)52 or the tumour protein p53 binding protein 1 (TP53BP1; a Tudor domain-containing protein)53 appears to contribute to oncogenesis. Although TIP60 and TP53BP1 act as tumour suppressors and are not likely to be therapeutic targets, the actions of these proteins underscore the extensive role of epigenetic proteins in oncogenesis, both positive (driving tumour growth) and negative (suppressing tumour growth). This dichotomy also raises important safety-related issues for potential epigenetic therapy (see below).
Neuropsychiatric disorders. Several studies have shown that levels of epigenetic proteins are altered in clinical neurodisease states, especially in intellectual disability syndromes. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, developmental delays and behavioural problems54. Moreover, haploinsufficiency of the HAT CREB binding protein (CREBBP) causes Rubinstein–Taybi syndrome, a genetic disorder that results in cognitive dysfunction. In a mouse model of this disorder — neonatal Crebbp+/− mice — the mice exhibit behavioural impairments, and this phenotype can be reversed by inhibition of histone deacetylation55. CREBBP might also be a key target for presenilins in the regulation of memory formation and neuronal survival56. In addition, mutations in epigenetic proteins can result in neuropsychiatric disorders: for example, mutations in the gene encoding the euchromatic histone-lysine N-methyltransferase 1 (EHMT1; also known as G9A-like protein 1 (GLP1)) result in a complex intellectual disability syndrome that is mirrored following deletion of this gene in the adult mouse brain57,58,59.
X-linked mental retardation (XLMR) is an inherited disorder mostly affecting males, and is caused by genetic abnormalities of the X chromosome, including many transcriptional co-activator proteins60. For example, the XLMR protein PHF8 (PHD finger protein 8) catalyses the demethylation of H3K9me2 and H3K9me1 (Ref. 61). The PHD of PHF8 binds to H3K4me3, and colocalizes with H3K4me3 at transcription initiation sites. Furthermore, PHF8 interacts with another XLMR protein, zinc finger protein 711 (ZNF711), which binds to a subset of PHF8-regulated proteins including the histone demethylase lysine-specific demethylase 5C (KDM5C; also known as JARID1C). These results functionally connect the XLMR-linked gene PHF8 to two other XLMR-linked genes, ZNF711 and JARID1C, indicating that genes linked to intellectual disability may be genetically associated within pathways that cause the complex phenotypes that are observed in patients who develop intellectual disability61.
Sirtuin 1 (SIRT1) is ubiquitously expressed in areas of the brain that are especially susceptible to age-related neurodegenerative states in rats and humans. Therefore, activation of endogenous sirtuin pathways may offer a therapeutic approach to delay and/or treat human age-related diseases62. Reduced levels of HDAC11 mRNA and increased levels of HDAC2 mRNA are observed in the brain and spinal cord of patients with amyotrophic lateral sclerosis63. The functional and therapeutic implications of these findings will be realized once more selective inhibitors of HDAC2 are available. Despite the lack of such tools, studies with currently available, partially selective HDAC inhibitors such as vorinostat (Zolinza; Merck)64 and MS-275 (Ref. 65) are revealing great insights into the role of these HDACs in central nervous system pathologies.
Schizophrenia is another disorder in which there are altered levels of epigenetic proteins. The gene encoding SMARCA2 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 2) expresses BRM, which is a protein component of the SWI/SNF chromatin-remodelling complex; this complex has been associated with schizophrenia in genome-wide association studies. Polymorphisms in SMARCA2 that are linked to the disease produce changes in the expression of the gene and/or in the encoded amino acid sequence66. In addition, a polymorphism in BRD1 has been shown to be associated with schizophrenia and bipolar affective disorder67.
Inflammation. The adaptive immune response exhibits hallmarks of a system that is subject to epigenetic regulation. The adaptive immune system is composed of multipotent precursor cells that undergo differentiation and clonal expansion upon exposure to an appropriate stimulus (for example, an antigen) to become activated lymphocytes, which then retain a memory against future exposure. It is therefore unsurprising that non-selective HDAC inhibitors have demonstrated preclinical efficacy in several rodent models of inflammatory conditions, both in rodent disease models68 and in clinical samples taken from patients with autoimmune disease69. These studies have revealed that several specific HDACs are implicated in various aspects of the immune response, including the innate and adaptive system70. For example, HDAC6 and HDAC9 enhance the activity of the transcription factor forkhead box P3 to promote the activity of anti-inflammatory regulatory T lymphocytes71, whereas HDAC6 has recently been implicated in the differentiation and maturation of antigen-presenting dendritic cells72.
In addition, several sirtuins have been shown to regulate the immune response by modulating the activity of key transcription factors. For example, SIRT1 and SIRT6 modulate nuclear factor-κB (NF-κB) activity via post-translational modification of the NF-κB p65 subunit and by altering the accessibility of the promoters to p65, respectively73. Accordingly, SIRT1 activators have anti-inflammatory effects in in vitro and in vivo models of inflammation74.
Several HATs also regulate the inflammatory response, both through acetylation of histones and through transcription factors such as NF-κB75. These findings clearly indicate an important role for acetylation in the regulation of the immune response; this is further supported by recent findings indicating that the BET family of bromodomain-containing proteins is pivotal in the systemic global inflammatory response to endotoxin76.
In addition, there is growing evidence of a role for histone lysine methylation in the regulation of immune processes. In particular, the protein methyltransferase G9A (also known as EHMT2) is important in mediating the silencing of specific genes during endotoxin shock via H3K9 dimethylation77. The flavin-dependent amine oxidase LSD2 mediates NF-κB demethylation and, in doing so, it has been implicated in a regulatory circuit that controls the expression of pro-inflammatory genes in dendritic cells78. Moreover, the histone demethylase Jumonji domain-containing protein 3 (JMJD3; also known as KDM6B) has been implicated in the response of macrophages to lipopolysaccharides and in the activation and maintenance of so-called 'alternatively activated' macrophages, which are thought to be involved in the host response to parasites, tissue remodelling and angiogenesis79,80.
In summary, there is a growing body of molecular and pharmacological evidence that epigenetic machinery is involved in the regulation of the immune system via mechanisms that involve modulation of transcription factors and modification of histones. In addition, there is clinical evidence to suggest that these mechanisms may be deregulated in autoimmune diseases81,82; targeting epigenetic regulators may therefore represent powerful new approaches for the amelioration of these conditions.
Metabolic disorders. The sirtuins, which deacetylate both histone and non-histone substrates, are major regulators of metabolism83. Two common variants in SIRT1 have been associated with a lower body mass index in two independent Dutch populations. Carriers of these variants have a reported 13–18% decreased risk of obesity84. Reduced levels or reduced activity of SIRT1 has been associated with complications of type 2 diabetes in humans85 and mice86. Thus, activation of one or more sirtuins can have favourable physiological effects. Resveratrol, through the indirect activation of SIRT1, stimulates insulin release in insulinoma INS-1E cells and human islets87. In a separate study, long-term intracerebroventricular infusion of resveratrol to diet-induced obese diabetic mice normalized hyperglycaemia and improved hyperinsulinaemia88. These effects were also reported to be mediated via SIRT1, as demonstrated by knockdown of the protein using short hairpin RNA89.
Histone methylation contributes to hyperglycaemic memory in models of transient hyperglycaemia90. The methyltransferases SET domain-containing protein 7 (SETD7) and suppressor of variegation 3–9 homolog 1 (SUV39H1), as well as the demethylase LSD1, contribute to sustained upregulation of the gene encoding the p65 subunit of NF-κB in response to glucose. Knockdown of SETD7 reverses effects that are associated with diabetic vascular injury, suggesting that this protein lysine methyltransferase (PKMT) is a potential target for the treatment of diabetes91.
Regenerative medicine: role in embryonic stem cell differentiation and reprogramming. Modulation of epigenetic proteins has shown utility in regenerative medicine, particularly in the directed differentiation of embryonic stem cells towards a committed lineage, and in the formation of induced pluripotent stem cells by reprogramming somatic cells92. Important opportunities associated with HDAC proteins in regenerative medicine are in the treatment of diabetes and neurodegenerative disorders such as Parkinson's disease and Alzheimer's disease. Stem cells treated with putative HDAC inhibitors demonstrated lineage progression towards insulin-producing β-cells with the generation of definitive endoderms, as well as the efficient production of pancreatic progenitors that expressed key transcription factors (for example, pancreatic and duodenal homeobox 1 (PDX1)) that are necessary for pancreatic development and β-cell maturation93. In addition, the class I HDAC inhibitor valproic acid promotes neuronal differentiation of multipotent adult rat neuroprogenitor cells in vitro94 and neurogenesis in the rat brain in vivo95.
The generation of a plentiful supply of stem cells whereby lineage specification could be orchestrated would substantially advance regenerative cell therapies. Yamanaka96 first demonstrated the propensity for four genetic transcription factors (octamer-binding protein 3 (OCT3; also known as OCT4), SOX2, MYC and Krüppel-like factor 4 (KLF4)) to induce pluripotency in murine somatic cells, and considerable recent efforts have focused on discovering small-molecule substitutes for these transcription factors to make the overall process more efficient and avoid eventual carcinogenesis.
When combined with two or more of these specific genetic factors, small-molecule inhibitors of HDACs, PKMTs or lysine demethylases improve the reprogramming efficiency to a level that is comparable to transduction with all four factors. This demonstrates the key role of epigenetic regulation in cellular reprogramming. For example, valproic acid enables the reprogramming of primary human fibroblasts with two factors, OCT4 and SOX2, without the need for the oncogenes MYC or KLF4. Induced pluripotent stem cells created under these conditions resemble human embyronic stem cells in pluripotency, global gene expression profiles and epigenetic states97. Similarly, the G9A inhibitor BIX-01294 improved reprogramming efficiency in neural progenitor cells transduced with only OCT3/OCT4 and KLF4 (Ref. 98). These studies suggest that the generation of induced pluripotent stem cells using only small molecules may soon become feasible.
Drugging the epigenome
There is experimental evidence for small-molecule inhibition of each of the major classes of acetyl and methyl readers, writers and erasers (Figs 3, 5, although only HDAC inhibitors are currently in the clinic (Table 2). The past decade has seen a large increase in the amount of knowledge related to the biochemistry, substrate selectivity and three-dimensional structures of these classes of proteins, revealing common structural and mechanistic features of their active sites. This knowledge has enabled the recent reports of new inhibitors for HATs, histone methyltransferases, lysine methyltransferases, bromodomains and malignant brain tumour domains (MBT domains), which are discussed below.

Compound C646 inhibits the histone acetyltransferase EP300 (E1A-associated protein p300) and has an IC50 (half-maximal inhibitory concentration) value of 1,600 nM133. Bromodomain antagonists have recently proved to be efficacious in vivo: JQ1 and IBET, two potent antagonists of bromodomain-containing protein 2 (BRD2), BRD3 and BRD4, are in preclinical development in cancer and inflammation, respectively36,76. Compound 6a is at a less advanced developmental stage but provides a new chemical scaffold174. Several histone deacetylase (HDAC) inhibitors have reached the clinic. Vorinostat (an inhibitor of HDAC1, HDAC2, HDAC3 and HDAC6) and romidepsin (an inhibitor of HDAC1, HDAC2, HDAC3 and HDAC8) are both approved for oncology indications111; other compounds in the clinic for oncology indications include: panobinostat (which is in Phase III development, and targets HDAC1, HDAC2, HDAC3 and HDAC6)111; entinostat (which is in Phase II development, and targets HDAC1 and HDAC2)111; and mocetinostat (which is in Phase II development, and targets HDAC1 and HDAC2)111. The sirtuin 1 inhibitor EX-527 is in Phase II clinical trials for Huntington's disease127. Compound 6J inhibits sirtuin 2 with an IC50 of 1μm175.

Potent inhibitors of lysine and arginine methyltransferases have recently been reported. EPZ004777, the first published protein methyltransferase inhibitor with in vivo efficacy, targets DOT1-like protein with an IC50 (half-maximal inhibitory concentration) in the picomolar range, and is active in a mouse tumour xenograft model34. UNC638 is a 10 nM inhibitor of lysine methyltransferase G9A (also known as EHMT2) and G9A-like protein 1 (GLP1; also known as EHMT1) that reduces the abundance of the histone mark H3K9me2 (dimethylated Lys9 of histone 3) in cells140. AZ505 inhibits SET and MYND domain-containing protein 2 with an IC50 of 120 nM142, and Compound 2 inhibits co-activator-associated arginine methyltransferase 1 with an IC50 of 30 nM146. A 100 nM inhibitor of lysine-specific histone demethylase 1 has been reported (Compound 10)154. Compound 15c has an IC50 of 110 nM against lysine-specific demethylase 4D-like protein (KDM4DL)157, and the smaller compounds SID 85736331 and 2,4-pyridine-dicarboxylate (2,4-PDCA) exploit the metal centre of KDM4A and KDM4DL (IC50 values range from 600 nM to 2.4 μM)158. The first chemical antagonist of a methyl mark reader is UNC669, which specifically targets lethal 3 malignant brain tumour-like protein (L3MBTL) with an IC50 of 5 μM165.
HDACs. HDACs are divided into five phylogenetic classes99 (Fig. 2): class I comprises HDAC1, HDAC2, HDAC3 and HDAC8; class IIa comprises HDAC4, HDAC5, HDAC7 and HDAC9; class IIb comprises HDAC6 and HDAC10; class III comprises the sirtuins SIRT1–SIRT7; and class IV contains HDAC11. Enzymes from classes I, II and IV require a divalent metal ion for catalysis100. Sirtuins are NAD+-dependent enzymes with protein deacetylase and ADP-ribosylase activity, and are structurally and biochemically unrelated to the other classes101,102.
Reflecting the ubiquitous distribution of acetyl marks within the cell103, HDACs deacetylate both histone and non-histone substrates. For example, HDAC6 is not involved in epigenetic signalling but it deacetylates microtubules and heat shock protein 90 (Refs 104, 105). Several metal-dependent HDAC inhibitors are in the clinic (Fig. 3); most of these target haematological malignancies, and two drugs, vorinostat and romidepsin (Istodax; Celgene), were first approved for the treatment of cutaneous T cell lymphoma in 2006 and 2009, respectively106,107. Increased acetylation of both histone and non-histone substrates mediated by these drugs and related agents is linked to the arrest of tumour cell growth, apoptosis and anti-angiogenesis108,109.
All HDAC inhibitors occupy the canonical acetyl-lysine channel of HDACs (Fig. 4). Interactions at the surface-accessible rim and at a 'foot pocket' next to the catalytic site mediate selectivity110, and chelation of a zinc ion at the metal-dependent catalytic site drives both potency and selectivity111. Several types of HDAC inhibitors — such as hydroxamates, cyclic peptides, benzamides and fatty acids — differentially satisfy these pharmacophoric rules. The hydroxamic acid group of vorinostat and the sulfhydryl group of romidepsin chelate the catalytic zinc ion with little specificity between different HDACs, but it has been reported that vorinostat preferentially inhibits HDAC1, HDAC2, HDAC3 and HDA6, whereas romidepsin preferentially targets HDAC1, HDAC2, HDAC3 and HDAC8 (Ref. 111).

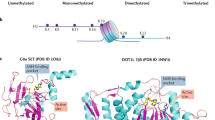
a | Vorinostat is shown in complex with the acetyl mark eraser histone deacetylase 8 (HDAC8) (Protein Data Bank (PDB) ID code: 1T69). b | UNC638 is shown in complex with the methyl mark writer lysine methyltransferase G9A (PDB ID code: 3RJW). c | 2,4-pyridine-dicarboxylate is shown in complex with the methyl mark eraser Jumonji domain-containing protein 2A (JMJD2A) (PDB ID code: 2VD7). d | JQ1 is shown in complex with the acetyl lysine reader bromodomain-containing protein 2 (BRD2) (PDB ID code: 3ONI). e | UNC669 is shown in complex with the methyl mark reader lethal 3 malignant brain tumour-like protein 1 (L3MBTL1). (PDB ID code: 3UWN) Most compounds compete with the substrate lysine (shown in magenta), whereas 2,4-pyridine-dicarboxylate competes with the cofactor of JMJD2A (shown in orange)34. Similarly, the methyltransferase inhibitor EPZ-04777 competes with the cofactor S-adenosylmethionine (not shown). Binding sites are shaded in grey, nitrogen atoms are dark blue and oxygen atoms are red. A catalytic zinc or nickel ion (shown as a yellow sphere) is co-crystallized with HDAC8 or JMJD2A, respectively.
The deacetylase domain of class I, II and IV enzymes is highly conserved but a catalytic residue is absent in class IIa enzymes, which results in minimal deacetylase activity and raises the possibility of an undiscovered substrate112 or allosteric stimulation of activity113. Alternatively, class IIa enzymes may act as scaffolding proteins that help recruit catalytically active HDACs within multiprotein complexes. This mechanism was recently confirmed for the HDAC4/HDAC5-mediated deacetylation of a non-histone substrate involved in glucose homeostasis114. Additionally, class IIa HDACs bind acetylated peptides with an affinity that is comparable to that of other metal-dependent HDACs, and may — like bromodomains — act as readers of acetyl marks111. The use of an improved, non-natural trifluoroacetylated class IIa substrate revealed that most HDAC inhibitors are inactive against class IIa HDACs at pharmacologically relevant concentrations but HDAC1, HDAC2 and HDAC3 were inhibited by most of the compounds that were tested111.
Importantly, HDACs are components of larger complexes in cells, and the selectivity of inhibitors or substrates observed against purified proteins may be altered in the context of multiprotein complexes115, adding a further layer of complexity to the design of selective HDAC inhibitors. A more detailed understanding of the cellular and cell-type specific HDAC complexes and their substrates is required to better design selective inhibition. This issue is also likely to be important for the development of inhibitors of the other epigenetic protein families. Whether increased selectivity (achieved by exploiting structural diversity at the rim and foot of the pocket) can translate into better targeted therapy or an improved therapeutic window has yet to be confirmed, but this is currently under investigation in the laboratory and the clinic using next-generation compounds108 (Fig. 3).
Among sirtuins, deacetylase activity has been reported for SIRT1, SIRT2, SIRT3 and SIRT6 (Refs 116,117,118,119). Other sirtuins can hydrolyse different marks such as succinyl, malonyl or propionyl marks120,121, or they can exclusively act as ADP-ribosylases. SIRT1 can be shuttled from the nucleus to the cytoplasm and deacetylate an array of substrates, including histones and the tumour suppressor p53 (Ref. 122). SIRT1 has been associated with enhancement of lifespan and memory, and has shown beneficial effects in neurodegeneration, metabolic syndrome and cancer, thus raising considerable interest in the drug discovery community123. A report presenting the first SIRT1 activators, including the natural product resveratrol and various synthetic molecules124, has been at the centre of an extensive controversy. Mounting evidence now suggests that the observed SIRT1 activation was a biochemical artefact, and that cellular activity was mediated by unrelated targets125,126. The need for potent and selective SIRT1 activators remains unfulfilled. The most advanced compound is the SIRT1 inhibitor selisistat (also known as EX-527 or SEN196), which has reached Phase II clinical trials for Huntington's disease127 (see the Siena Biotech website).
HATs. Despite having very low sequence homology, the catalytic domains of all HATs solved to date are organized around a conserved central fold where the cofactor acetyl-CoA binds and catalysis takes place. The only solved crystal structure of human HAT bound to a peptide substrate reveals a shallow peptide-binding site where only the acetylated lysine is inserted in a solvent-accessible groove, which suggests that this site may be difficult to efficiently target with drugs. Several HATs have been co-crystallized with acetyl-CoA: in all HATs except for E1A-associated protein p300 (EP300), the cofactor lies in an open but structurally diverse pocket. Whether this cofactor pocket is druggable is unclear. By contrast, EP300 contains a unique loop that folds onto the cofactor, which becomes buried in an enclosed and probably chemically tractable pocket128.
An array of HAT inhibitors have been identified and reviewed129,130. However, most of these compounds are either promiscuous natural substances that bind multiple classes of proteins129 or they are covalently modifying isothiazolones131. A very large bi-substrate inhibitor, Lys-CoA, was shown to be a submicromolar EP300 inhibitor with surprising selectivity but it does not have drug-like properties132. Another more recently described EP300 inhibitor, C646, may be the only potent, selective and drug-like HAT inhibitor published to date133 (Fig. 3). The compound binds at the predicted druggable pocket of EP300 and acts as a cofactor competitor. Moreover, C646 can mimic the caspase-dependent pro-apoptotic effect of short interfering RNA-mediated EP300 knockdown, which involves both extrinsic and intrinsic cell death pathways, in androgen-dependent and castration-resistant prostate cancer cells134.
The chemical tractability of other HATs remains unclear, but the lack of obvious druggable sites in available structures and the lack of convincing inhibitors reported to date suggest that screening chemical libraries against the isolated enzymes is not an adequate approach. HATs function in cells as part of large multiprotein complexes, and the formation of these complexes may be necessary for the discovery of inhibitors. This would require biochemical screening against the reconstituted complexes, or phenotypic screening in cells.
Protein methyltransferases. The structure of protein methyltransferases, which comprise two distinct but adjacent binding sites, offers two locations where small molecules can bind and inhibit enzyme function. Indeed, both the peptide substrate channel and the binding site for the cofactor SAM have been exploited to produce potent inhibitors of protein methyltransferases135,136. Currently, the successful identification of selective and cell-active inhibitors of histone lysine methyltransferases (HKMTs) has been restricted to those targeting the closely related enzymes G9A and GLP1, as well as DOT1L.
BIX-01294 was the first selective inhibitor of a PKMT. BIX-01294 binds at the protein substrate channel of G9A and GLP1, but its modest affinity and cytotoxicity limit its use to cell-based assays137. Second-generation inhibitors such as E72 (Ref. 138) and UNC321 (Ref. 139), both of which incorporate a 7-alkoxyamine tethered to the quinazoline core as a key structural modification, showed significantly improved enzyme affinity. UNC638 (Ref. 140) is a potent and selective inhibitor of G9A and GLP1, and was further optimized for improved cellular potency and low toxicity. UNC638 also retains the 7-alkoxyamine group, indicating that the incorporation of this group may represent a viable strategy for designing compounds that target this HKMT family.
Structure-based studies — which have been insightful in aiding the design of novel compounds — have shown that the conserved quinazoline core of UNC638 (Ref. 140) occupies the peptide groove (as previously seen with BIX-01294)141 and that the new alkoxyamine substituents bind inside the lysine channel in a similar manner to the lysine of the histone substrate140 (Fig. 4). Other small-molecule inhibitors of HKMTs that bind to the peptide-binding groove include AZ505, which is a potent and selective inhibitor of SET and MYND domain-containing protein 2 (SMYD2)142. The oncogenic protein SMYD2 represses the functional activities of p53 and retinoblastoma protein, making it an attractive drug target for the development of small-molecule inhibitors.
Compounds that bind to the SAM binding site include the DOT1L inhibitor EPZ004777, which has activity against mixed lineage leukaemia fusions that cause aberrant localization of DOT1L34. Although EPZ004777 was designed as a SAM analogue (it retains the nucleoside core), it displays remarkable selectivity (>1,000-fold) for inhibition of DOT1L over other histone methyltransferases. Other compounds that bind to the SAM binding site include: the fungal metabolite chaetocin, which is an inhibitor of SUV39H1 and G9A143; and sinefungin, which is a promiscuous natural product and an analogue of SAM144. Thus, in analogy to kinase inhibitors that bind at the ATP site, it appears that targeting the cofactor binding site of protein methyltransferases could be a general strategy for this target class. This is supported by computational analysis of the structural diversity observed within the SAM binding site across all human SAM-dependent methyltransferases, indicating that selectivity should be achievable145. Potent inhibitors of histone arginine methyltransferases such as protein arginine methyltransferase 1 (PRMT1) and PRMT4 have also been identified146,147,148, providing further evidence that protein methyltransferases can be inhibited by small molecules.
Lysine demethylases. First-generation mechanism-based inhibitors of the flavin-dependent lysine-specific demethylases LSD1 and LSD2, such as tranylcypromine, lacked potency and selectivity over their historical targets — the monoamine oxidases149,150. Structure–activity relationships subsequently demonstrated that extension of the chemical structure further into the lysine substrate pocket resulted in more potent and selective inhibitors151,152,153 (for example, compound 10; Fig. 5)154. The LSD class of demethylases are structurally and mechanistically distinct from the Jumonji domain-containing histone demethylases and appear to primarily target H3K4. Thus, LSDs may offer the possibility of developing selective H3K4 demethylase antagonists more readily than by selectively targeting the subset of Jumonjidomain-containing H3K4 demethylases.
All current inhibitors of Jumonji domain-containing lysine demethlyases compete with the cofactor 2-oxoglutarate and bind to the catalytic iron in the active site. The highly polar compound 2,4-pyridine-dicarboxylate inhibits the Jumonji domain-containing demethylases as well as other 2-oxoglutarate-dependent oxygenases such as HIF prolyl hydroxylase 1 (HPH1; also known as EGLN2) and HPH2 (also known as EGLN1)155. As observed with the LSD1 inhibitors, extending the chemical structure of the Jumonji domain-containing demethylase inhibitor template so that the compound binds directly to the iron in the substrate binding pocket increases potency, as seen with metal-chelating hydroxamic acids156. These compounds are selective for the Jumonji domain-containing demethylases over other 2-oxoglutarate-dependent oxygenases, but the molecular and physicochemical properties of the compound may limit bioavailability156.
Two new series of Jumonji domain-containing demethylase inhibitors, 8-hydroxyquinolines (for example, SID 85736331) and 2,2′-bipyridines (for example, compound 15c) (Fig. 5), are potent inhibitors with subtype selectivity and more drug-like properties157,158. These new lead compounds have smaller and more compact chemical structures, and represent good lead compounds for further optimization. The compounds gain their potency and selectivity through favourable inhibitor–protein interactions in the active site closer to the metal centre. Thus, as potent and selective inhibitors of histone demethylases have now been identified, the next challenge will be to identify compounds that have improved cell permeability, which will be better suited to investigate activity in whole-cell assays.
Bromodomain-containing proteins. The bromodomain-containing family of proteins represents an important class of histone modification reader proteins that recognize acetylated lysine residues. The bromodomain was first described in 1992 as a domain of ∼110 amino acids that was conserved in several transcriptionally important genes from humans, fruitflies and yeast16. The human genome encodes 42 bromodomain-containing proteins, each of which contains between one and six bromodomains, encompassing a total of 61 unique human bromodomains15. Interestingly, bromodomains are commonly found in proteins that also contain enzymatic domains (for example, HATs) or other reader domains (for example, PHDs)159 in configurations that contribute to specific combinatorial recognition of multiple histone marks160. To date, the structures of 23 of the 61 human bromodomains have been experimentally determined, demonstrating a conserved hydrophobic pocket that accommodates one (and sometimes two) acetyl-lysine side chains15,161.
Bromodomains adopt a left-handed, four-helix bundle comprising amphipathic helices known as alphaZ, alphaA, alphaB and alphaC. At one end of the helical bundle, the amino- and carboxyl termini come together, emphasizing the modular architecture of this domain and underscoring the idea that the bromodomain could act as an independent functional unit that interacts with other proteins. Based on these findings, Zhou and colleagues162 conducted a nuclear magnetic resonance (NMR)-based chemical screen to identify compounds that bound to the bromodomain of the HAT P300/CBP-associated factor with an affinity comparable to that of the Tat peptideacetylated on Lys50 (IC50 (half-maximal inhibitory concentration) ∼5 μM). The lead compound did not bind to the structurally related bromodomains of CREBBP and TIF1β (transcriptional intermediary factor 1β), indicating that it is possible to identify small-molecule inhibitors that have specificity within the bromodomain family162.
Building on this observation, Zhou et al.163 described the rational design of cyclic peptide modulators of the bromodomain-containing transcriptional co-activator CREBBP. The affinity of the cyclic peptides for the CREBBP bromodomain was significantly higher than the affinity of the bromodomain for its biological ligands, which included lysine-acetylated histones and tumour suppressor p53. The best cyclic peptide exhibited a Kd (dissociation constant) of 8.0 μM, representing a 24-fold improvement in affinity over that of the linear Lys382-acetylated p53 peptide. This lead peptide was highly selective for the bromodomain of CREBBP compared with bromodomains from other transcriptional proteins163.
Recently, two independent groups reported the first selective inhibitors with low nanomolar affinity for the tandem bromodomain-containing family of transcriptional regulators known as the BET proteins (BRD2, BRD3, BRD4 and BRDT)36,76,164. The compounds JQ1 and IBET represent novel chemical templates that are distinct from the previously reported simple acetyl-containing templates, and they have a clear mode of action. These studies demonstrate that it is feasible to produce inhibitors that have a high affinity (in the nanomolar range), specificity and cell permeability (Fig. 3). The development of these inhibitors has revealed novel insights into the physiological role and therapeutic potential of inhibiting BET function. Indeed, beneficial effects of these inhibitors have been observed in several cancers and systemic inflammatory conditions36,40,41,76.
Methyl-lysine readers. As is the case with bromodomains, the ability to inhibit the interaction between a methyl-lysine reader domain and its target methyl-lysine mark appears to be possible. A common theme among all methyl-lysine reader domains is the presence of a conserved 'aromatic cage' that comprises the binding cleft for the methyl-lysine side chain and provides π electron interactions with the positively charged methylammonium moiety160. The geometry of the aromatic cage as well as the presence and configuration of a countercharge or hydrogen-bond acceptor determines the degree of methylation that is optimal for binding. These structural features are attractive for drug discovery.
Of all the known structures of methyl-lysine binding pockets, the deep narrow clefts that bind mono- and di-methyl-lysine (such as those found in MBT domains), may be the most attractive targets for the design of small molecules. The first example of such a small-molecule antagonist, UNC669, was recently developed for the MBT domain-containing protein L3MBTL1 (lethal 3 MBT-like protein 1), using a structure-guided approach165. Interestingly, UNC669 uses the same pyrrolidine moiety as the protein methyltransferase inhibitor UNC638 (which is an inhibitor of the dimethylase G9A) to mimic dimethyl-lysine, suggesting that pyrrolidine may be used as a universal 'warhead' in compounds that target dimethyl-lysine binding pockets in proteins. At present, there are no reported antagonists of trimethyl-lysine readers. Because some trimethyl-lysine binding pockets tend to be more open and shallow compared with those of MBT domains, they may be more challenging to target.
Safety of drugging epigenetic modifiers
As with all new potential drug targets, there will be a need to demonstrate that epigenetic modifiers havea clear benefit in the treatment of diseases that can be achieved with an acceptable safety and tolerability profile. This is especially important when evaluating epigenetic protein targets, owing to their fundamental role as general factors in the regulation of global gene expression patterns.
The first wave of epigenetic drugs — HDAC inhibitors — have been beneficial in the treatment of cutaneous T cell lymphoma, with acceptable adverse event profiles; additional clinical studies are underway to determine their utility in treating other cancers166. There are ongoing studies to determine the potential therapeutic utility of HDAC inhibitors for non-oncology indications in which the adverse event profile requirements may be more stringent167. For non-oncology indications, key safety issues include the long-term effects of the drug on stem cells and germ cells, especially potential transgenerational effects168. For example, embryonic exposure to environmental endocrine disruptors169 or nutrient restriction170 during gonadal development and sex determination is capable of inducing adult-onset disease states that can be perpetuated across multiple generations.
Because modulation of the epigenome has the potential to reprogramme all cells, adverse effects on stem cells (or on germ cells before conception or embryonic development) may only become apparent over longer periods of time. Strategies to investigate and avoid such effects will need to be developed, and may include tools such as epigenomic profiling of histone and DNA marks in the appropriate cell types. Identification of inhibitors that are truly subtype- or target-selective for their epigenetic protein will also help to tease apart the balance between efficacy and safety for a given target.
Conclusion
There is ample and growing evidence for an association of epigenetic factors with disease — especially in chronic conditions such as cancer, inflammation, diabetes and neuropsychiatric disorders. In these types of pathologies, there is support for cellular memory that is linked to precursors of the disease state or environmental interactions that lead to the disease state. For example, the well-known link between inflammation and cancer has a strong epigenetic component171. Inflammation-specific gene expression patterns mediated by an epigenetic mechanism, not mutations, are preserved in cancers that arise from chronically inflamed tissue in a lung carcinogenesis model172. Similarly, situations such as hyperglycaemic memory in diabetes, epigenomic states that maintain and perpetuate stem-like tumour-initiating cells and the Warburg effect in cancer are potentially reversible cellular states that may be unlocked by epigenetic therapies. With the ability to reprogramme normal somatic cells into different cell types (assisted by small molecules), it is conceivable that disease states of cells could eventually be selectively reprogrammed into either normal tissue or an apoptotic state using small molecules. In order to achieve this goal, there is an urgent need for well-characterized tool compounds that will enable the identification of the targets and disease states that are selectively vulnerable to epigenetic therapy.
The protein families highlighted in this Review, which together contain several hundreds of targets, represent a new frontier in drug discovery that has huge potential for the development of future therapeutics. For most epigenetic protein families, there is experimental evidence — using selective small-molecule inhibitors — that these targets are likely to be druggable (Figs 3, 4, 5). Because of the multidomain nature of these proteins, and their participation in large protein complexes, there are probably several possibilities to target a single gene or multifunctional complex. For example, many of the enzymes or complexes that write histone marks also have reader domains for the same mark. This is thought to aid the enzyme in spreading the mark along chromatin by binding to the first written mark via the reader domain, thereby allowing the writing of a subsequent mark on a neighbouring nucleosome, and so on. Thus, targeting a reader domain involved in histone binding may result in cellular effects that are distinctly different from inhibition of enzymatic activity by changing the localization of enzymes or their complexes, or disrupting the positive feedback and spreading of the mark. Indeed, the recent expansion of exciting activities reported for bromodomain antagonists are examples of successful inhibition of protein–protein interactions and point to an exciting new frontier in drug discovery36,40,41,76.
References
Meaney, M. J. Epigenetics and the biological definition of gene × environment interactions. Child Dev. 81, 41–79 (2010).
Kelly, T. K., De Carvalho, D. D. & Jones, P. A. Epigenetic modifications as therapeutic targets. Nature Biotech. 28, 1069–1078 (2010).
Meaney, M. J. & Ferguson-Smith, A. C. Epigenetic regulation of the neural transcriptome: the meaning of the marks. Nature Neurosci. 13, 1313–1318 (2010).
Portela, A. & Esteller, M. Epigenetic modifications and human disease. Nature Biotech. 28, 1057–1068 (2010).
Allis, C. D., Jenuwein, T. & Reinberg, D. Epigenetics (Cold Spring Harbor Laboratory Press, New York, 2007).
Taberlay, P. C. & Jones, P. A. DNA methylation and cancer. Prog. Drug Res. 67, 1–23 (2011).
Strahl, B. D. & Allis, C. D. The language of covalent histone modifications. Nature 403, 41–45 (2000).
Turner, B. M. Histone acetylation and an epigenetic code. Bioessays 22, 836–845 (2000).
Turner, B. M. Cellular memory and the histone code. Cell 111, 285–291 (2002).
Schreiber, S. L. & Bernstein, B. E. Signaling network model of chromatin. Cell 111, 771–778 (2002).
Ernst, J. et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 (2011).
Allfrey, V. G., Faulkner, R. & Mirsky, A. E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl Acad. Sci. USA 51, 786–794 (1964). References 7, 8 and 12 highlight the role of histone modifications in gene regulation, and the concept of an epigenetic code.
Shahbazian, M. D. & Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76, 75–100 (2007).
Das, C., Lucia, M. S., Hansen, K. C. & Tyler, J. K. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459, 113–117 (2009).
Filippakopoulos, P. et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 30 Mar 2012 (doi: 10.1016/j.cell.2012.02.013). This is a full structural analysis of the entire human bromodomain-containing family of proteins, which established the size and details of this target class.
Haynes, S. R. et al. The bromodomain: a conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 20, 2603 (1992).
Tsukada, Y. et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816 (2006).
Shi, Y. et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004). References 17 and 18 describe the discovery of the two classes of lysine demethylases, both of which belong to larger druggable enzyme classes.
Trojer, P. et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell 129, 915–928 (2007).
Nielsen, A. L. et al. Heterochromatin formation in mammalian cells: interaction between histones and HP1 proteins. Mol. Cell 7, 729–739 (2001).
Lee, M. G. et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318, 447–450 (2007).
Kuzmichev, A., Nishioka, K., Erdjument-Bromage, H., Tempst, P. & Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 16, 2893–2905 (2002).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011).
Kouzarides, T. Chromatin modifications and their function. Cell 128, 693–705 (2007).
Di Lorenzo, A. & Bedford, M. T. Histone arginine methylation. FEBS Lett. 585, 2024–2031 (2011).
Kirmizis, A. et al. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature 449, 928–932 (2007).
Guccione, E. et al. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 449, 933–937 (2007).
Jones, P. A. & Baylin, S. B. The epigenomics of cancer. Cell 128, 683–692 (2007).
Krennhrubec, K., Marshall, B. L., Hedglin, M., Verdin, E. & Ulrich, S. M. Design and evaluation of 'Linkerless' hydroxamic acids as selective HDAC8 inhibitors. Bioorg. Med. Chem. Lett. 17, 2874–2878 (2007).
Oehme, I. et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 15, 91–99 (2009).
Chi, P., Allis, C. D. & Wang, G. G. Covalent histone modifications — miswritten, misinterpreted and mis-erased in human cancers. Nature Rev. Cancer 10, 457–469 (2010). This is a summary of the extensive number and types of 'errors' in epigenetic proteins implicated in cancer.
Simon, J. A. & Lange, C. A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 647, 21–29 (2008).
Okada, Y. et al. hDOT1L links histone methylation to leukemogenesis. Cell 121, 167–178 (2005).
Daigle, S. R. et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65 (2011). This is the first example of selective inhibition of a histone methyltransferase that has efficacy in a cancer model.
Yap, D. B. et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117, 2451–2459 (2010).
Filippakopoulos, P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2011).
Darnell, J. E. Jr. Transcription factors as targets for cancer therapy. Nature Rev. Cancer 2, 740–749 (2002).
Beroukhim, R. et al. The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 (2010).
Meyer, N. & Penn, L. Z. Reflecting on 25 years with MYC. Nature Rev. Cancer 8, 976–990 (2008).
Delmore, J. E. et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 (2011). References 36 and 40 provide the first examples of bromodomain inhibition that has apparent therapeutic benefit.
Dawson, M. A. et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478, 529–533 (2011).
Zuber, J. et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528 (2011).
Kalashnikova, E. V. et al. ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple-negative cells through control of B-Myb and EZH2. Cancer Res. 70, 9402–9412 (2010).
Caron, C. et al. Functional characterization of ATAD2 as a new cancer/testis factor and a predictor of poor prognosis in breast and lung cancers. Oncogene 29, 5171–5181 (2010).
Zhang, Y. et al. PR-domain-containing Mds1–Evi1 is critical for long-term hematopoietic stem cell function. Blood 118, 3853–3861 (2011).
Velichutina, I. et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood 116, 5247–5255 (2010).
Trubia, M. et al. Characterization of a recurrent translocation t(2;3)(p15–22;q26) occurring in acute myeloid leukaemia. Leukemia 20, 48–54 (2006).
Wang, G. G. et al. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 459, 847–851 (2009).
Parsons, D. W. et al. The genetic landscape of the childhood cancer medulloblastoma. Science 331, 435–439 (2011).
Northcott, P. A. et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nature Genet. 41, 465–472 (2009).
Wang, X. & Jin, H. The epigenetic basis of the Warburg effect. Epigenetics 5, 566–568 (2010).
Gorrini, C. et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 448, 1063–1067 (2007).
Bouwman, P. et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nature Struct. Mol. Biol. 17, 688–695 (2010).
Williams, S. R. et al. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am. J. Hum. Genet. 87, 219–228 (2010).
Wang, J. et al. CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein–Taybi syndrome brain. Dev. Cell 18, 114–125 (2010).
Chen, G., Zou, X., Watanabe, H., van Deursen, J. M. & Shen, J. CREB binding protein is required for both short-term and long-term memory formation. J. Neurosci. 30, 13066–13077 (2010).
Schaefer, A. et al. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron 64, 678–691 (2009).
Kleefstra, T. et al. Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J. Med. Genet. 46, 598–606 (2009).
Kramer, J. M. & van Bokhoven, H. Genetic and epigenetic defects in mental retardation. Int. J. Biochem. Cell Biol. 41, 96–107 (2009).
Chiurazzi P., Schwartz C. E., Gecz, J. & Neri G. XLMR genes: update 2007. Eur. J. Hum. Genet. 16, 422–434 (2008).
Kleine-Kohlbrecher, D. et al. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol. Cell 38, 165–178 (2010).
Zakhary, S. M. et al. Distribution analysis of deacetylase SIRT1 in rodent and human nervous systems. Anat. Rec. (Hoboken) 293, 1024–1032 (2010).
Janssen, C. et al. Differential histone deacetylase mRNA expression patterns in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 69, 573–581 (2010).
Riessland, M. et al. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum. Mol. Genet. 19, 1492–1506 (2010).
Covington, H. E. et al. Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 29, 11451–11460 (2009).
Loe-Mie, Y. et al. SMARCA2 and other genome-wide supported schizophrenia-associated genes: regulation by REST/NRSF, network organization and primate-specific evolution. Hum. Mol. Genet. 19, 2841–2857 (2010).
Severinsen, J. E. et al. Evidence implicating BRD1 with brain development and susceptibility to both schizophrenia and bipolar affective disorder. Mol. Psychiatry 11, 1126–1138 (2006).
Lin, H. S. et al. Anti-rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen-induced arthritis in rodents. Br. J. Pharmacol. 150, 862–872 (2007).
Grabiec, A. M., Korchynskyi, O., Tak, P. P. & Reedquist, K. A. Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Ann. Rheum. Dis. 71, 424–431 (2011).
Shakespear, M. R., Halili, M. A., Irvine, K. M., Fairlie, D. P. & Sweet, M. J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 32, 335–343 (2011).
Beier, U. H., Akimova, T., Liu, Y., Wang, L. & Hancock, W. W. Histone/protein deacetylases control Foxp3 expression and the heat shock response of T-regulatory cells. Curr. Opin. Immunol. 23, 670–678 (2011).
de Zoeten, E. F. et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3+ T-regulatory cells. Mol. Cell Biol. 31, 2066–2078 (2011).
Galli, M., Van Gool, F. & Leo, O. Sirtuins and inflammation: friends or foes? Biochem. Pharmacol. 81, 569–576 (2011).
Yoshizaki, T. et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 298, e419–e428 (2010).
Ghizzoni, M., Haisma, H. J., Maarsingh, H. & Dekker, F. J. Histone acetyltransferases are crucial regulators in NF-κB mediated inflammation. Drug Discov. Today 16, 504–511 (2011).
Nicodeme, E. et al. Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123 (2010).
Chen, X., El Gazzar, M., Yoza, B. K. & McCall, C. E. The NF-κB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J. Biol. Chem. 284, 27857–27865 (2009).
van Essen, D., Zhu, Y. & Saccani, S. A feed-forward circuit controlling inducible NF-κB target gene activation by promoter histone demethylation. Mol. Cell 39, 750–760 (2010).
De Santa, F. et al. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J. 28, 3341–3352 (2009).
Ishii, M. et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 114, 3244–3254 (2009).
Ciavatta, D. J. et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J. Clin. Invest. 120, 3209–3219 (2010).
Hu, N. et al. Abnormal histone modification patterns in lupus CD4+ T cells. J. Rheumatol. 35, 804–810 (2008).
Zhong, L. & Mostoslavsky, R. Fine tuning our cellular factories: sirtuins in mitochondrial biology. Cell Metab. 13, 621–626 (2011).
Zillikens, M. C. et al. SIRT1 genetic variation is related to BMI and risk of obesity. Diabetes 58, 2828–2834 (2009).
Cardellini, M. et al. TIMP3 is reduced in atherosclerotic plaques from subjects with type 2 diabetes and increased by SirT1. Diabetes 58, 2396–2401 (2009).
Orimo, M. et al. Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler. Thromb. Vasc. Biol. 29, 889–894 (2009).
Vetterli, L., Brun, T., Giovannoni, L., Bosco, D. & Maechler, P. Resveratrol potentiates glucose-stimulated insulin secretion in INS-1E β-cells and human islets through a SIRT1-dependent mechanism. J. Biol. Chem. 286, 6049–6060 (2011).
Ramadori, G. et al. Central administration of resveratrol improves diet-induced diabetes. Endocrinology 150, 5326–5333 (2009).
Fischer-Posovszky, P. et al. Resveratrol regulates human adipocyte number and function in a Sirt1-dependent manner. Am. J. Clin. Nutr. 92, 5–15 (2010).
Brasacchio, D. et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 58, 1229–1236 (2009).
Siebel, A. L., Fernandez, A. Z. & El-Osta, A. Glycemic memory associated epigenetic changes. Biochem. Pharmacol. 80, 1853–1859 (2010).
Meissner, A. Epigenetic modifications in pluripotent and differentiated cells. Nature Biotech. 28, 1079–1088 (2010).
Borowiak, M. et al. Small molecules efficiently direct endodermal differentiation of mouse and human embryonic stem cells. Cell Stem Cell 4, 348–358 (2009).
Hsieh, J., Nakashima, K., Kuwabara, T., Mejia, E. & Gage, F. H. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc. Natl Acad. Sci. USA 101, 16659–16664 (2004).
Hao, Y. et al. Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. J. Neurosci. 24, 6590–6599 (2004).
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Huangfu, D. et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nature Biotech. 26, 1269–1275 (2008).
Shi, Y. et al. A combined chemical and genetic approach for the generation of induced pluripotent stem cells. Cell Stem Cell 2, 525–528 (2008).
de Ruijter, A. J., van Gennip, A. H., Caron, H. N., Kemp, S. & van Kuilenburg, A. B. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370, 737–749 (2003).
Finnin, M. S. et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188–193 (1999).
Sauve, A. A. Sirtuin chemical mechanisms. Biochim. Biophys. Acta 1804, 1591–1603 (2010).
Sauve, A. A., Wolberger, C., Schramm, V. L. & Boeke, J. D. The biochemistry of sirtuins. Annu. Rev. Biochem. 75, 435–465 (2006).
Choudhary, C. et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 (2009).
Kovacs, J. J. et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 18, 601–607 (2005).
Hubbert, C. et al. HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458 (2002).
Prince, H. M., Bishton, M. J. & Harrison, S. J. Clinical studies of histone deacetylase inhibitors. Clin. Cancer Res. 15, 3958–3969 (2009).
Grant, S., Easley, C. & Kirkpatrick, P. Vorinostat. Nature Rev. Drug Discov. 6, 21–22 (2007).
Marks, P. A. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin. Investig. Drugs 19, 1049–1066 (2010).
Butler, L. M. et al. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 60, 5165–5170 (2000). This was the first evidence that an HDAC inhibitor has anticancer activity in an animal model.
Schapira, M. Structural biology of human metal-dependent histone deacetylases. Handb. Exp. Pharmacol. 206, 225–240 (2011).
Bradner, J. E. et al. Chemical phylogenetics of histone deacetylases. Nature Chem. Biol. 6, 238–243 (2010).
Lahm, A. et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl Acad. Sci. USA 104, 17335–17340 (2007).
Schuetz, A. et al. Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J. Biol. Chem. 283, 11355–11363 (2008).
Mihaylova, M. M. et al. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 145, 607–621 (2011).
Bantscheff, M. et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nature Biotech. 29, 255–265 (2011).
Vaquero, A. et al. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 20, 1256–1261 (2006).
Vaquero, A. et al. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 16, 93–105 (2004).
Vempati, R. K. et al. p300-mediated acetylation of histone H3 lysine 56 functions in DNA damage response in mammals. J. Biol. Chem. 285, 28553–28564 (2010).
Michishita, E. et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452, 492–496 (2008).
Du, J. et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 (2011).
Liu, B. et al. Identification and characterization of propionylation at histone H3 lysine 23 in mammalian cells. J. Biol. Chem. 284, 32288–32295 (2009).
Vaziri, H. et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159 (2001).
Haigis, M. C. & Sinclair, D. A. Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295 (2010).
Milne, J. C. et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450, 712–716 (2007).
Stunkel, W. & Campbell, R. M. Sirtuin 1 (SIRT1): the misunderstood HDAC. J. Biomol. Screen. 16, 1153–1169 (2011).
Pacholec, M. et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 285, 8340–8351 (2010).
Solomon, J. M. et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell Biol. 26, 28–38 (2006).
Liu, X. et al. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 451, 846–850 (2008).
Piaz, F. D. et al. Chemical biology of histone acetyltransferase natural compounds modulators. Mol. Divers. 15, 401–416 (2011).
Dekker, F. J. & Haisma, H. J. Histone acetyl transferases as emerging drug targets. Drug Discov. Today 14, 942–948 (2009).
Wisastra, R. et al. Isothiazolones; thiol-reactive inhibitors of cysteine protease cathepsin B and histone acetyltransferase PCAF. Org. Biomol. Chem. 9, 1817–1822 (2011).
Lau, O. D. et al. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol. Cell 5, 589–595 (2000).
Bowers, E. M. et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem. Biol. 17, 471–482 (2010).
Santer, F. R. et al. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol. Cancer Ther. 10, 1644–1655 (2011).
Spannhoff, A., Hauser, A. T., Heinke, R., Sippl, W. & Jung, M. The emerging therapeutic potential of histone methyltransferase and demethylase inhibitors. ChemMedChem 4, 1568–1582 (2009).
Copeland, R. A., Solomon, M. E. & Richon, V. M. Protein methyltransferases as a target class for drug discovery. Nature Rev. Drug Discov. 8, 724–732 (2009).
Kubicek, S. et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 25, 473–481 (2007). This was the first example of selective, substrate-competitive inhibition of a histone methyltransferase.
Chang, Y. et al. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J. Mol. Biol. 400, 1–7 (2010).
Liu, F. et al. Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J. Med. Chem. 53, 5844–5857 (2010).
Vedadi, M. et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nature Chem. Biol. 7, 566–574 (2011).
Chang, Y. et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nature Struct. Mol. Biol. 16, 312–317 (2009).
Ferguson, A. D. et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure 19, 1262–1273 (2011).
Greiner, D., Bonaldi, T., Eskeland, R., Roemer, E. & Imhof, A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nature Chem. Biol. 1, 143–145 (2005).
Couture, J. F., Hauk, G., Thompson, M. J., Blackburn, G. M. & Trievel, R. C. Catalytic roles for carbon-oxygen hydrogen bonding in SET domain lysine methyltransferases. J. Biol. Chem. 281, 19280–19287 (2006).
Campagna-Slater, V. et al. Structural chemistry of the histone methyltransferases cofactor binding site. J. Chem. Inf. Model 51, 612–623 (2011).
Sack, J. S. et al. Structural basis for CARM1 inhibition by indole and pyrazole inhibitors. Biochem. J. 436, 331–339 (2011).
Dowden, J., Hong, W., Parry, R. V., Pike, R. A. & Ward, S. G. Toward the development of potent and selective bisubstrate inhibitors of protein arginine methyltransferases. Bioorg. Med. Chem. Lett. 20, 2103–2105 (2010).
Cheng, D. et al. Small molecule regulators of protein arginine methyltransferases. J. Biol. Chem. 279, 23892–23899 (2004).
Culhane, J. C., Wang, D., Yen, P. M. & Cole, P. A. Comparative analysis of small molecules and histone substrate analogues as LSD1 lysine demethylase inhibitors. J. Am. Chem. Soc. 132, 3164–3176 (2010).
Schmidt, D. M. & McCafferty, D. G. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 46, 4408–4416 (2007).
Mimasu, S. et al. Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry 49, 6494–6503 (2010).
Binda, C. et al. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J. Am. Chem. Soc. 132, 6827–6833 (2010).
Ogasawara, D. et al. Synthesis and biological activity of optically active NCL-1, a lysine-specific demethylase 1 selective inhibitor. Bioorg. Med. Chem. 19, 3702–3708 (2011).
Ortega Muñoz, A., Castro- Palomino-Laria, J. & Fyfe, M. C. T. Lysine specific demethylase-1 inhibitors and their use. Patent WO2011035941A1 (2011).
Rose, N. R. et al. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. J. Med. Chem. 51, 7053–7056 (2008).
Luo, X. et al. A selective inhibitor and probe of the cellular functions of jumonji C domain-containing histone demethylases. J. Am. Chem. Soc. 133, 9451–9456 (2011).
Chang, K. H. et al. Inhibition of histone demethylases by 4-carboxy-2,2′-bipyridyl compounds. ChemMedChem 6, 759–764 (2011).
King, O. N. et al. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS ONE 5, e15535 (2010).
Ruthenburg, A. J., Li, H., Patel, D. J. & Allis, C. D. Multivalent engagement of chromatin modifications by linked binding modules. Nature Rev. Mol. Cell Biol. 8, 983–994 (2007).
Taverna, S. D., Li, H., Ruthenburg, A. J., Allis, C. D. & Patel, D. J. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nature Struct. Mol. Biol. 14, 1025–1040 (2007).
Jacobson, R. H., Ladurner, A. G., King, D. S. & Tjian, R. Structure and function of a human TAFII250 double bromodomain module. Science 288, 1422–1425 (2000).
Zeng, L. et al. Selective small molecules blocking HIV-1 Tat and coactivator PCAF association. J. Am. Chem. Soc. 127, 2376–2377 (2005).
Borah, J. C. et al. A small molecule binding to the coactivator CREB-binding protein blocks apoptosis in cardiomyocytes. Chem. Biol. 18, 531–541 (2011).
Chung, C. W. et al. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem. 54, 3827–3838 (2011).
Herold, J. M. et al. Small-molecule ligands of methyl-lysine binding proteins. J. Med. Chem. 54, 2504–2511 (2011). This was the first demonstration that methyl-lysine binding pockets can be antagonized using small molecules.
Best, J. D. & Carey, N. Epigenetic opportunities and challenges in cancer. Drug Discov. Today 15, 65–70 (2010).
Best, J. D. & Carey, N. Epigenetic therapies for non-oncology indications. Drug Discov. Today 15, 1008–1014 (2010).
Anway, M. D., Cupp, A. S., Uzumcu, M. & Skinner, M. K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308, 1466–1469 (2005).
Anway, M. D., Leathers, C. & Skinner, M. K. Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology 147, 5515–5523 (2006).
Bertram, C. et al. Transgenerational effects of prenatal nutrient restriction on cardiovascular and hypothalamic-pituitary-adrenal function. J. Physiol. 586, 2217–2229 (2008).
Brower, V. Epigenetics: unravelling the cancer code. Nature 471, S12–S13 (2011).
Blanco, D. et al. Molecular analysis of a multistep lung cancer model induced by chronic inflammation reveals epigenetic regulation of p16 and activation of the DNA damage response pathway. Neoplasia 9, 840–852 (2007).
Richon, V. M. et al. Chemogenetic analysis of human protein methyltransferases. Chem. Biol. Drug Des. 78, 199–210 (2011).
Bamborough, P. et al. Fragment-based discovery of bromodomain inhibitors part 2: optimization of phenylisoxazole sulfonamides. J. Med. Chem. 55, 587–596 (2012).
Medda, F. et al. Novel cambinol analogs as sirtuin inhibitors: synthesis, biological evaluation, and rationalization of activity. J. Med. Chem. 52, 2673–2682 (2009).
Marks, P. A. & Breslow, R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nature Biotech. 25, 84–90 (2007).
Bertino, E. M. & Otterson, G. A. Romidepsin: a novel histone deacetylase inhibitor for cancer. Expert Opin. Investig. Drugs 20, 1151–1158 (2011).
Zhou, Q., Atadja, P. & Davidson, N. E. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor α (ER) gene expression without loss of DNA hypermethylation. Cancer Biol. Ther. 6, 64–69 (2007).
Plumb, J. A. et al. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 2, 721–728 (2003).
Hu, E. et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J. Pharmacol. Exp. Ther. 307, 720–728 (2003).
Fournel, M. et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 7, 759–768 (2008).
Younes, A. et al. Mocetinostat for relapsed classical Hodgkin's lymphoma: an open-label, single-arm, Phase 2 trial. Lancet Oncol. 12, 1222–1228 (2011).
Mandl-Weber, S. et al. The novel inhibitor of histone deacetylase resminostat (RAS2410) inhibits proliferation and induces apoptosis in multiple myeloma (MM) cells. Br. J. Haematol. 149, 518–528 (2010).
Furlan, A. et al. Pharmacokinetics, safety and inducible cytokine responses during a Phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (givinostat). Mol. Med. 17, 353–362 (2011).
Wang, H. et al. Discovery of (2E)-3-{2-butyl-1-[2-(diethylamino)ethyl]-1H-benzimidazol-5-yl}-N-hydroxya crylamide (SB939), an orally active histone deacetylase inhibitor with a superior preclinical profile. J. Med. Chem. 54, 4694–4720 (2011).
Novotny-Diermayr, V. et al. SB939, a novel potent and orally active histone deacetylase inhibitor with high tumor exposure and efficacy in mouse models of colorectal cancer. Mol. Cancer Ther. 9, 642–652 (2010).
Lai, C. J. et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 70, 3647–3656 (2010).
Rivera-Del Valle, N. et al. PCI-24781, a novel hydroxamic acid HDAC inhibitor, exerts cytotoxicity and histone alterations via caspase-8 and FADD in leukemia cells. Int. J. Cell Biol. 2010, 207420 (2010).
Lucas, D. M. et al. The novel deacetylase inhibitor AR-42 demonstrates pre-clinical activity in B-cell malignancies in vitro and in vivo. PLoS ONE 5, e10941 (2010).
Hwang, J. J. et al. A novel histone deacetylase inhibitor, CG200745, potentiates anticancer effect of docetaxel in prostate cancer via decreasing Mcl-1 and Bcl-(XL). Invest. New Drugs 20 Jul 2011 (doi: 10.1007/s10637-011-9718-1).
Rosato, R. R. HDAC inhibitors — CHI's third annual conference. IDrugs 13, 13–15 (2010).
Acknowledgements
We are grateful to P. Brennan for his contribution on demethylase inhibitors, and S. Knapp for his comments on the manuscript. The Structural Genomics Consortium is a registered charity (charity number 1097737) that receives funds from the Canadian Institutes of Health Research, Eli Lilly, Genome Canada (through the Ontario Genomics Institute), GlaxoSmithKline, the Ontario Ministry for Research and Innovation, the Novartis Research Foundation, Pfizer and the Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
K.L. was an employee of GlaxoSmithKline and is now an employee of Pfizer. All other authors declare no competing financial interests.
Supplementary information
Supplementary information
BROMO_domain_fullname (XLS 150 kb)
Supplementary information
CHROMO_domain_fullname (XLS 95 kb)
Supplementary information
HAT_full_length_fullname (XLS 61 kb)
Supplementary information
HDAC_SIRT_domain_fullname (XLS 71 kb)
Supplementary information
KDM_domain_fullname (XLS 93 kb)
Supplementary information
MBT_domain_fullname (XLS 60 kb)
Supplementary information
PHD_domain_fullname (XLS 271 kb)
Supplementary information
PMT_domain_fullname (XLS 164 kb)
Supplementary information
PWWP_domain_fullname (XLS 74 kb)
Supplementary information
TUDOR_domain_fullname (XLS 119 kb)
Supplementary information
In format provided by Fish et al. (MAY 2012) (PDF 186 kb)
Supplementary information
In format provided by Fish et al. (MAY 2012) (PDF 555 kb)
Supplementary information
In format provided by Fish et al. (MAY 2012) (PDF 148 kb)
Supplementary information
In format provided by Fish et al. (MAY 2012) (PDF 211 kb)
Related links
Related links
FURTHER INFORMATION
EnVivo Pharmaceuticals website — Pipeline & Programs
RCSB Protein Data Bank website
RepliGen website (RG2833: Potential to Alter the Course of Friedreich's Ataxia)
Siena Biotech website — Development Pipeline
SMART (Simple Modular Architecture Research Tool) database
Structural Genomics Consortium (Annotated Phylogenetic Trees)
Structural Genomics Consortium (Chemical Probes)
Structural Genomics Consortium (Epigenetics: Chemical Probes for Drug Discovery)
Glossary
- Chromatin
-
The fibres in which DNA and genes are packaged in the nucleus of a cell. Chromatin consists of the DNA double helix wrapped around a complex of histone proteins — together called the nucleosome.
- Epigenetics
-
Heritable changes in gene expression or phenotype that are stable between cell divisions, and sometimes between generations, but do not involve changes in the underlying DNA sequence of the organism.
- Differentiation
-
The process by whicha stem cell, or other precursor cell, commits towards a more specialized cell type with a specific function, and represents an exit from self-renewal. Differentiation is controlled by cell signalling pathways and maintained through epigenetic mechanisms.
- Post-translational modification
-
A chemical modification of proteins that acts as a signal to other proteins that recognize the modification. In the context of epigenetic signalling, post-translational modifications are often called 'marks'.
- Epigenome
-
The combination of histone and DNA post-translational modifications and related interacting proteins that together package the genome and help define the transcriptional programme in a given cell.
- Heterochromatin
-
A tightly packed form of DNA associated with transcriptionally silent or repressed genes. It is highly correlated with di- and trimethlyated H3K9 (Lys9 of histone 3) marks.
- Euchromatin
-
A more loosely packed form of DNA that is associated with transcriptionally active genes.
- Bromodomain
-
An evolutionarily conserved, ∼110-amino-acid motif composed of four left-handed, antiparallel α-helices.
- Stem cell
-
An unspecialized precursor cell withthe capacity to self-renew (continuously produce unaltered progeny) and to differentiate into more mature specialized cell types.
- Haploinsufficiency
-
A disease mechanism in which one of two copies of a gene is mutated, resulting in insufficient activity of the gene products (typically a protein) to bring about a functional, wild-type condition.
- Brachydactyly mental retardation syndrome
-
A disorder that presents with a range of features, including intellectual disabilities, developmental delays, behavioural abnormalities, sleep disturbance, craniofacial and skeletal abnormalities, and autism spectrum disorder.
- Presenilins
-
A family of related multipass transmembrane proteins that function as part of the γ-secretase intramembrane protease complex. They were first identified in screens for mutations causing early-onset forms of familial Alzheimer's disease.
- Amyotrophic lateral sclerosis
-
A progressive neurological disease that is associated with the degeneration of central and spinal motor neurons. This neuron loss causes muscles to weaken and waste away, leading to paralysis.
- Jumonji domain
-
A conserved domain originally identified in the Jumonji family of transcription factors, now known to be histone demethylases. The Jumonji C domain comprises the catalytic domain of the 2-oxoglutarate-dependent lysine demethylases.
- Hyperglycaemic memory
-
A phenomenon in which the deleterious end-organ effects resulting from exposure to high glucose levels persist for several years after usual glycaemic control is restored.
- Malignant brain tumour domains
-
Conserved sequence motifs found in certain developmental proteins. These domains bind to mono- or dimethylated lysine-containing peptides and, when deleted in fruitflies, lead to brain tumours.
- Allosteric stimulation
-
The regulation of an enzyme or protein by binding an effector molecule at a site other than the protein's active site, thereby causing a conformational change in the protein.
- P300/CBP-associated factor
-
A trancriptional co-activator protein containing a bromodomain and a histone acetyltransferase domain.
- π electron interactions
-
A non-covalent interaction between the π-electron cloud of aromatic rings and the cationic charge of, for example, methylated lysine.
Rights and permissions
About this article
Cite this article
Arrowsmith, C., Bountra, C., Fish, P. et al. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov 11, 384–400 (2012). https://doi.org/10.1038/nrd3674
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrd3674
This article is cited by
-
3D Models of Sarcomas: The Next-generation Tool for Personalized Medicine
Phenomics (2023)
-
Combined low levels of H4K16ac and H4K20me3 predicts poor prognosis in breast cancer
International Journal of Clinical Oncology (2023)
-
Structural classification of EZH2 inhibitors and prospects for the treatment of tumor: a review
Medicinal Chemistry Research (2023)
-
The role of histone methylation in renal cell cancer: an update
Molecular Biology Reports (2023)
-
Fabrication and characterization of bilayer scaffolds made of decellularized dermis/nanofibrous collagen for healing of full-thickness wounds
Drug Delivery and Translational Research (2023)
