
Abstract
Heart failure is a pressing worldwide public-health problem with millions of patients having worsening heart failure. Despite all the available therapies, the condition carries a very poor prognosis. Existing therapies provide symptomatic and clinical benefit, but do not fully address molecular abnormalities that occur in cardiomyocytes. This shortcoming is particularly important given that most patients with heart failure have viable dysfunctional myocardium, in which an improvement or normalization of function might be possible. Although the pathophysiology of heart failure is complex, mitochondrial dysfunction seems to be an important target for therapy to improve cardiac function directly. Mitochondrial abnormalities include impaired mitochondrial electron transport chain activity, increased formation of reactive oxygen species, shifted metabolic substrate utilization, aberrant mitochondrial dynamics, and altered ion homeostasis. In this Consensus Statement, insights into the mechanisms of mitochondrial dysfunction in heart failure are presented, along with an overview of emerging treatments with the potential to improve the function of the failing heart by targeting mitochondria.
Similar content being viewed by others
Main
Heart failure (HF) is associated with substantial clinical burden and economic costs worldwide. The disease is particularly prevalent in elderly individuals, in whom the incidence and associated costs are projected to double over the next 20 years1,2. Economic costs associated with the management of patients with HF is estimated at >US$30 billion annually in the USA alone, and accounts for roughly 2–3% of total health-care spending globally3,4. Despite these enormous costs, mortality from HF remains high. Death from HF within 5 years of diagnosis is common despite current optimal medical therapy. Mortality and rehospitalization within 60–90 days after discharge from hospital can be as high as 15% and 35%, respectively5. These event rates have largely not changed over the past 15 years, despite implementation of evidence-based therapy5. HF rehospitalization rates also remain high, with care typically focused on symptomatic relief. Patients with HF are often designated as having either reduced ejection fraction (HFrEF), or preserved ejection fraction (HFpEF). Patients with HFpEF also have poor prognosis after the first diagnosis6. Regardless of the HF aetiology, novel treatments that improve intrinsic cardiac function remain elusive.
Advances in the treatment of ischaemic and valvular heart disease have clearly improved patient survival. The residual cardiac dysfunction and associated comorbidities, however, have led, in the long-term, to the development of HF with attendant poor quality of life. Commonly prescribed HF medications, although beneficial in promoting some symptom relief, often do not fully address the underlying causes of progressive left ventricular dysfunction7. Most standard-of-care pharmacological approaches to HF act by reducing workload on the failing heart and, in doing so, attempt to rebalance energy supply and energy demand, albeit to a lower level (Fig. 1). Hallmarks of current therapies include modulation of neurohormonal abnormalities, unloading the heart (that is, vasodilatation), and/or reducing the heart rate — all important determinants of reducing myocardial oxygen consumption8. β-Blockers, ivabradine, and antagonism of the renin–angiotensin–aldosterone system all act in concert to reduce myocardial energy requirements and attenuate or prevent further adverse cardiac remodelling. Although these therapies have improved survival in patients with chronic ambulatory HFrEF over the past 2–3 decades, death and poor quality of life continue to adversely affect this ever-increasing patient population. This unmet need is probably not going to be met by drugs that modulate neurohormonal abnormalities and lower heart rates, because further intervention along these axes is likely to be counterproductive as hypotension and bradycardia become limiting factors. The search for more effective and complementary therapy for this patient population must be focused on improving the intrinsic function of the viable, but dysfunctional, cardiac unit — the cardiomyocytes3,9. The novel therapy must be haemodynamically neutral (no decrease in blood pressure or heart rate) and must target the myocardium as the centrepiece of the therapeutic intervention10.

The delicate balance between cardiac demands for energy and supply of energy is tipped in heart failure, in which energy supply cannot match demand. Next-generation therapeutics can improve on existing standard-of-care therapies by bolstering mitochondrial energy production. ACE, angiotensin-converting enzyme; ARB, angiotensin II-receptor blocker; ETC, electron transport chain; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; ROS, reactive oxygen species.
The vast majority of phase III trials in patients with HF conducted in the past decade have been negative, arguably for the same reasons discussed above11,12. Furthermore, a relative underinvestment in cardiovascular drug development, as well as strategic abandonment by pharmaceutical companies of new therapies for which the risks are perceived to be higher than the rewards, have also contributed to slow development of drugs for HF13. Moreover, the development of effective therapies for HFpEF is imperative to treat this patient population, but the variability in HFpEF phenotypes (such as age, and the presence of diabetes mellitus or hypertension), and the difficulty in establishing reliable preclinical models of HFpEF, also hinder progress. Despite these obstacles, ample opportunity exists to improve HF treatments, provided the focus is directed towards cardiomyocytes and their intrinsic function.
A roundtable meeting was held in Stresa, Italy on 23 October 2015 to discuss the multifaceted problem of insufficient energy production in HF, and the role it has in progressive left ventricular dysfunction. This meeting was attended by academics, clinicians, and representatives from the pharmaceutical industry. The meeting focused on mitochondrial dysfunction as the source of energy deprivation in HF, and how correction of mitochondrial dysfunction using emerging novel therapies might lead to functional improvement of the HF phenotype. This Consensus Statement summarizes the findings from that roundtable discussion.
Bioenergetics of the beating heart
Aristotle considered the heart to be the body's furnace, radiating energy in the form of heat14. Given the astounding energetic cost of cardiac function, this concept is not so far from the truth. Humans produce and consume roughly their body weight in ATP (about 65 kg) every single day15. The heart accounts for only ∼0.5% of body weight, but is responsible for roughly 8% of ATP consumption. This high energy flux is dynamic: the heart stores only enough energy to support pumping for a few heart beats, turning over the entire metabolite pool approximately every 10 s even at resting heart rates16. As the most metabolically active organ in the body, the heart possesses the highest content of mitochondria of any tissue. Mitochondria comprise 25–30% of cell volume across mammalian species17,18, with only the myofilaments being more densely packed within cardiac myocytes. The high mitochondrial content of cardiomyocytes is needed to meet the enormous energy requirement for contraction and relaxation (which is also an active process). About 90% of cellular ATP is utilized to support the contraction–relaxation cycle within the myocardium19. ATP-dependent release of actin from myosin is required for both contraction (as myosin heads cycle through cross-bridges with actin) and relaxation. Cellular sequestration of calcium back into the sarcoplasmic reticulum during diastole also requires a tremendous amount of ATP. Cells sustain the energy requirements necessary to support cardiac function through remarkable metabolic supply–demand matching20,21 (Fig. 1). Bioenergetic homeostasis is accomplished almost exclusively through an 'energy grid' comprised of a mitochondrial network and their associated phosphate-transfer couples. Cardiac mitochondria must operate at high efficiency levels to respond instantaneously to the energetic needs of contractile units, a demand that is ever-changing and necessitated by the body's dynamic requirements for oxygen-bearing blood.
Myocardial energy requirements are more pronounced during physical activity, when demands for energy increase to maintain cardiac function commensurate with the needs of the body. However, other mitochondrial abnormalities besides energy deprivation during physical activity can contribute to the pathologies seen in patients with HF. Mitochondrial abnormalities in HF are not only a question of reduced capacity to generate ATP (even though that capacity is reduced at rest in HF compared with resting normal), but can also be directly linked to cardiomyocyte injury and death and, therefore, to disease progression. Abnormal mitochondria are a major source of reactive oxygen species (ROS) production, which can induce cellular damage. Abnormal mitochondria can promote programmed cell death through the release of cytochrome c into the cytosolic compartment and activation of caspases. Therefore, mitochondria directly influence ongoing cell injury and death. Mitochondrial abnormalities have also been implicated in aberrant cellular calcium homeostasis, vascular smooth muscle pathology, myofibrillar disruption, and altered cell differentiation, all important issues in cardiovascular disease, including HF.
Mitochondria in cardiomyocytes
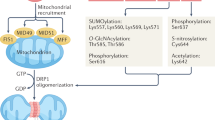
Mitochondria are primarily located within subsarcolemmal, perinuclear, and intrafibrillar regions of the cardiomyocyte. Although they are symbiotic partners with the other cellular compartments, mitochondria are in many ways discrete entities. Mitochondrial dynamics in the form of fission, fusion, and autophagy are highly regulated processes that are essential for energy production and structural integrity of the organelles22,23,24,25,26,27,28,29. Altered mitochondrial biogenesis, fragmentation, and hyperplasia have been observed in studies of human30 and animal models31,32 of HF. These effects seem to be caused by altered expression of proteins that regulate mitochondrial dynamics33. As many of these factors are 'master regulators' of mitochondrial metabolism, these changes might be directly related to the decreased capacity to oxidize fatty acid substrates often seen in HF34,35.
Mitochondria have their own DNA (mtDNA) and a genetic code that is distinct from the host-cell nuclear DNA. mtDNA is circular in shape, analogous to DNA found in lower organisms, and a primitive fingerprint leftover from bacterial origin. Evolutionary selection pressures have led to mitochondria 'outsourcing' almost all their protein-making needs to their cellular hosts. The overwhelming majority (>99%) of mitochondrial proteins come from nuclear-encoded DNA. These proteins are synthesized via cellular protein synthesis machinery, and are actively imported into mitochondria through mitochondrial membrane transporters36. mtDNA encodes 13 protein subunits found within three of the electron transport protein complexes, and a handful of ribosomal and transfer RNAs37. These proteins are made in specialized ribosomes or 'mitoribosomes', which are physically attached to the mitochondrial inner membrane38.
Many inherited familial cardiomyopathies (both adult and paediatric) are associated with mtDNA mutations39. In humans, mitochondria are maternally inherited40, owing to high mitochondrial density in the egg and the active degradation of mitochondria in the sperm during fertilization41. The proximity of mtDNA to sites of mitochondrial ROS generation, poor repair mechanisms, and a lack of protective histones combine to make mtDNA particularly susceptible to oxidative injury and mutation.
Mitochondrial genetics contribute to cardiomyopathies by expressing mutant proteins that influence energy homeostasis. With 1,000–10,000 genes per mitochondria (polyploidy), mitochondrial genetics operate on population-based (instead of Mendelian) principles37. Mutated mtDNA is found alongside nonmutated copies, leading to mitochondrial 'heteroplasmy'. The extent of heteroplasmy in mutated mtDNA influences the susceptibility to inherited mitochondrial disease42. Mutated mtDNA can be found in 1 in 200 individuals, a frequency that is 20-fold higher than the incidence of mitochondrial disease. This mismatch indicates that healthy individuals often harbour mutated mtDNA that has no observable phenotypic consequences until a certain mutation threshold is reached37. Although very early in preclinical development, various innovative approaches to reduce the extent of heteroplasmy using genome editing might ultimately lead to effective therapy for HF caused by genetic mitochondrial disease43,44,45. Given that mitochondrial abnormalities, such as increased ROS production, altered mitochondrial energetics, and impaired mitochondrial ion homeostasis, are observed in genetic mitochondrial diseases as well as HF, innovative approaches that target mitochondrial dysfunction might share efficacy across these diseases.
Heart failure is a bioenergetic disease
The 'myocardial power grid' consists of mitochondrial ATP supply that transfers energy throughout the cell along intracellular phosphotransfer buffering systems (Fig. 2). Mitochondria utilize carbon sources from food substrates, which are catabolized and passed through the Krebs cycle and are then channelled through a series of redox reactions along the inner mitochondrial membrane. The oxidation of these substrates creates a proton electrochemical gradient, predominantly in the form of mitochondrial membrane potential (ΔΨm)46. Protons that re-enter the mitochondrial matrix through complex V (mitochondrial ATP synthase) liberate energy that phosphorylates ADP, regenerating ATP. Newly synthesized ATP is rapidly transferred out of mitochondria and energy is subsequently distributed throughout the cell via reversible phosphate exchange networks, primarily catalysed by creatine kinase and adenylate kinase-associated reactions16,47.

Decreased capacity of mitochondria to generate and transfer energy within heart cells results in energy deficits, which influences all cellular processes that require energy, most notably the processes of contraction and relaxation.
The evidence that HF involves impaired cellular energy production and transfer is considerable (Table 1). Among studies that have directly examined energetics in human HF, all but three noted some form of bioenergetic impairment in the failing heart. This decrement in bioenergetics is reflected by a decrease in cellular ATP, phosphocreatine (PCr), or the PCr/ATP ratio. Impaired bioenergetics affect patients with HFrEF and those with HFpEF (Table 1).
Although it is difficult to tell from the heterogeneous patient population included in Table 1, the progression to HF is likely to be associated with a gradual decline in bioenergetic reserve capacity that ultimately reaches a critical threshold, after which endogenous mechanisms can no longer compensate for faltering energy supply48. Attempts to improve bioenergetics in HF tend to focus on mitochondrial energy production as a target, because direct augmentation of myocardial creatine with oral creatine supplementation is thwarted by a decreased capacity to transport creatine into the failing cardiomyocytes49. Skeletal muscles also show mitochondrial dysfunction in HF, contributing to the exercise intolerance that characterizes the HF state50. Abnormal mitochondrial function has also been reported in patients with renal insufficiency51, and in patients with insulin resistance52. Given that patients with HF often manifest both renal insufficiency and insulin resistance, treating mitochondrial dysfunction in HF derives benefits that go beyond improving cardiac function (Fig. 3).

Aberrant mitochondrial energy production is involved in many symptoms commonly found in patients with heart failure, including skeletal muscle dysfunction and renal pathologies. LV, left ventricular.
Several interventions are currently being tested in clinical trials to stimulate mitochondrial biogenesis in HF. These include epicatechin and resveratrol, which are naturally-occurring polyphenols found in foods such as red wine, green tea, and dark chocolate. Preclinical HF models suggest that these molecules are biologically active53,54,55, and some success in improving cardiac function has been reported in small trials of patients with myocardial infarction56. Larger trials in patients with HF are required.
Mitochondrial substrate selectivity
Substrate utilization in the failing heart has been extensively reviewed previously57,58,59,60. Overall, altered substrate metabolism seems to be centrally involved in HF, although the direction of the metabolic alterations is complex and is likely to depend on the particular stage of HF progression and differences in the availability of substrate (whether the heart is in a 'fed' or 'fasted' state)58,59.
The heart utilizes different substrates simultaneously to produce energy. Mitochondrial fatty acid oxidation (FAO) is the predominate substrate used in the healthy adult human heart, being responsible for 60–80% of cardiac ATP production, followed by lesser contributions from glucose, lactate, and ketone bodies61. However, the heart can shift the relative contribution of these substrates in an effort to adapt to varying physiological conditions. Under conditions of low oxygen content, such as ischaemia and HF, ATP content is thought to decrease by as much as 40%3. In HF, fatty acid oxidation and the oxidative capacity of the mitochondria decline, and can no longer maintain sufficient levels of ATP, especially during conditions of increased cardiac workload such as exercise. The failing heart shifts its predominant fuel source from mitochondrial FAO toward glycolytic pathways. This switch is most apparent in late and end-stage HF57, and is 30% more energetically efficient in the failing heart, because more ATP is produced per mole of oxygen during carbohydrate oxidation62. Numerous studies investigating FAO, glucose oxidation, and (to a lesser extent) ketone body oxidation have aimed to establish a metabolic phenotype, underlying molecular mechanisms, and potential therapeutic targets of the failing heart.
The reduction in fatty acid uptake and FAO that occurs during HF might be owing to dysregulated molecular mechanisms responsible for fatty acid metabolism. For example, the level of peroxisome proliferator-activated receptor-α (PPARα), a transcription factor highly expressed in the heart and responsible for fatty acid transport into the mitochondria and peroxisomes, has been reported to be downregulated in both animal models and humans with HF63,64. Similarly, tissue from animals and humans with HF has reduced activity of the transcription factor responsible for mitochondrial biogenesis, PPAR-γ co-activator (PGC)-1α64,65. Because these transcription factors have a critical role in the regulation of cardiac mitochondrial energy production, these data suggest that decreased PPARα and PGC-1α activity might be an important precursor leading to impaired FAO during HF. Therefore, further inhibition of FAO to increase glycolytic flux via PPARα and/or PGC-1α is a plausible therapeutic target. Small-molecule regulators of PGC-1α are needed, and animal models overexpressing the transcription factor are inherently problematic, ostensibly owing to increased mitochondrial biogenesis-induced cardiomyopathy66. Similarly, PPARα antagonists in animal models of HF have yielded inconclusive data67, whereas clinical PPARα ligands are reportedly safe, but their efficacy in a HF population is currently unknown61. Although the safety of PPARα ligands is promising, further evidence demonstrating their efficacy in both animal models and humans with HF is needed.
Levels of circulating free fatty acids might be higher in the failing heart than under healthy conditions owing to hormonal stimulation. The rise in serum catecholamine levels increases plasma free fatty acid concentrations, and subsequently stimulates FAO68. As a result, reducing the availability of circulating free fatty acids via transient adrenergic antagonists might be a viable therapy to inhibit FAO and increase glycolytic ATP production. Traditionally, β-adrenergic receptor antagonists are used in HF owing to their negative ionotropic effects that reduce cardiac workload and spare oxygen by decreasing sympathetic activity68. Many, such as carvedilol, have been clinically shown to lessen infarct size after ischaemia by decreasing sympathetic activity, followed by inhibition of mitochondrial fatty acid uptake and increased glucose oxidation69.
Malonyl-CoA endogenously regulates fatty acid concentrations by controlling the activity of carnitine O-palmitoyltransferase (CPT) 1, a rate-limiting enzyme in mitochondrial fatty acid uptake68. When intracellular levels of malonyl-CoA are increased, CPT1 is inhibited and mitochondrial fatty acid uptake is stopped70. The intracellular concentration of malonyl-CoA is dependent on the balance between its synthesis via acetyl-CoA carboxylase and degradation via malonyl-CoA decarboxylase. Therefore, the upregulation of acetyl-CoA carboxylase or inhibition of malonyl-CoA decarboxylase would increase intracellular malonyl-CoA levels, and prevent mitochondrial uptake of free fatty acids to reduce FAO. As expected, inhibiting malonyl-CoA decarboxylase in animal models has reportedly improved cardiac function after ischaemia, reduced cardiac FAO, and increased glycolytic flux71,72. Studies of malonyl-CoA decarboxylase inhibitors in patients with HF are needed.
Trends in glucose oxidation across the spectrum of HF are more variable, particularly among animal models of HF58. Compensatory substrate switching towards glucose use has been observed in both animal models and humans59, with a higher contribution coming from glycolysis. Stimulating mitochondrial glucose oxidation, either directly or by inhibiting fatty acid catabolism, has been suggested as a viable therapeutic strategy to compensate for the energetically 'starved' failing heart59.
Ketone body metabolism also seems to be altered in HF. Ketones are formed in the liver via fatty acid metabolism, and provide a small substrate pool for oxidation within the myocardium. In conditions such as diabetes or starvation, ketone catabolism is upregulated in response to lowered insulin availability and higher fatty acid levels57,73. Studies have reported increased ketone utilization in the severely failing heart in humans73,74. Further research is needed to understand the role of ketone oxidation in the failing myocardium, and to determine whether targeting ketone metabolism is a plausible therapy to improve energetics in HF.
Novel insights into the regulation of metabolic substrate demand in the heart have been provided through studies of microRNAs and acetylation of mitochondrial lysine residues. Alterations in microRNA levels through any number of upregulation and downregulation events can alter substrate utilization in the heart75. Alterations in protein levels modulated by microRNA expression have been proposed to have important implications for glycolysis, β-oxidation, ketone metabolism, the Krebs cycle, and the electron transport chain (ETC)75. For example, increased levels of ROS can alter calcium handling in HF by modifying microRNA that leads to inhibition of sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) 2a transcription75. Post-translational modification via lysine acetylation has been suggested to have an important role in metabolic enzyme regulation in the mitochondria59.
Overactivation of the SNS
As all substrates converge on mitochondria, understanding the specific abnormalities that occur in HF is central to the development of new treatments. ROS production increases in many aetiologies of HF, a phenomenon that might be directly related to increased sympathetic nervous system (SNS) tone76. Sustained sympathetic drive and chronically elevated circulating catecholamines — processes that are normally transient to mediate acute increases in cardiac output — are commonly observed in patients with HF (particularly HFrEF)77,78. Chronic stimulation of β-adrenergic receptors has been directly linked to mitochondrial ROS production through adrenergic receptor-mediated second messenger signalling79,80. ROS-mediated initiation of mitochondria-dependent cell death cascades has been repeatedly observed after chronic sympathetic activation, leading to overall declines in mitochondrial function81,82,83,84,85,86. These processes can be amplified by the formation of aminochromes, catecholamine metabolites known to impair mitochondrial redox balance87. Attenuation of HF pathology with β-blockers and renin–angiotensin–aldosterone antagonism has resulted in substantial clinical improvements88, and is likely to relieve some of the mitochondrial dysfunction that accompanies increased sympathetic tone. The capacity to complement these existing background therapies with compounds that directly target mitochondrial dysfunction is a potentially promising novel paradigm (Fig. 1).
Increased ROS production
Cellular ROS production occurs when ROS formation outpaces or exhausts compensatory signals and overwhelms endogenous scavenging systems89,90,91. ROS are produced at several different sites within cells, both within and outside of mitochondria (reviewed in detail previously92,93,94,95). Mitochondrial ROS production occurs at various sites along the inner mitochondrial membrane as well as in the mitochondrial matrix by components of the ETC and the Krebs cycle, respectively96 (Fig. 4). ROS production is typically low under normal physiological conditions93, and is kept in check by intracellular and intramitochondrial scavenging systems. Pathological ROS levels in the heart typically occur when ROS production outpaces endogenous scavenging capacity. ROS (and other associated reactive intermediates) can damage proteins and lipids, trigger cell-death cascades, and evoke synchronized collapses in the cellular energy grid97,98. Heightened mitochondrial ROS production and downstream ROS-mediated damage has been reported in patients with HF as well as in preclinical models of the disease31,99,100,101.

Enzyme complexes responsible for energy production are packed into the mitochondrial inner membrane, often with the help of phospholipids such as cardiolipin. Failing mitochondria often display altered morphology, decreased ATP-generating capacity, heightened production of reactive oxygen species (ROS), abnormal cardiolipin levels, and impaired supercomplexes.
Although ROS are typically associated with pathological states, ROS levels in the heart per se are best characterized by the term 'hormesis': small amounts can evoke adaptive signalling and create beneficial, compensatory responses. Modest production of ROS has been shown to mediate beneficial myocardial signalling involved in physiological responses such as (transient) sympathetic drive102, many preconditioning paradigms103, cardiac mitochondrial quality control104, and exercise105. Exercise training is known to augment endogenous ROS-scavenging mechanisms in the heart105,106,107, restore bioenergetic efficiency in porcine models of HFpEF108, and improve symptoms and quality of life in trials involving patients with HFrEF109,110 or HFpEF111. Consistent with the ROS hormesis concept, several studies have noted that administration of high doses of ROS scavengers can abolish the beneficial effects of exercise112,113, including humans taking oral vitamin C or E supplements114.
Mitochondrial production of ROS depends on the mitochondrial membrane potential. Increased expression of mitochondrial uncoupling proteins in HF115 might be a compensatory mechanism to reduce ROS by 'uncoupling to survive'116, whereby a reduction in mitochondrial membrane potential is postulated to lower ROS emission from mitochondria. This view is popular and almost dogmatic, but the decrease in ROS production by uncoupling is a prominent effect during mitochondrial state 4 respiration (no ADP). Heart mitochondria, however, are never respiring in state 4. Pathological ROS production in cardiomyocytes is likely to be more closely linked to decreased or collapsed membrane potential and/or depletion of the NADPH pool117,118,119, whereby ROS production overwhelms endogenous scavenging through mitochondrial membrane-dependent mechanisms89.
The repeated lack of benefits of ROS scavenging compounds in clinical trials of patients with HF11,120,121 continues to plague cardiovascular drug development, suggesting that oxygen radical scavenging per se is not a plausible mechanism of action for long-term improvements in HF. Lack of tissue permeability, poor intracellular targeting, and ineffective therapeutic doses might contribute to the poor translation of benefits of antioxidants to date. This approach to therapy, however, might ultimately succeed when novel scavenging compounds that overcome permeability and targeting problems, such as XJB-5-131 (Refs 122,123), mitoTEMPO124,125, and EUK8/EUK134 (Refs 126,127,128), are tested in humans.
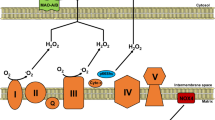
Abnormalities of mitochondrial ETC
Decrements in individual electron transport complexes, particularly complex I and/or IV activity, have been observed in animal models129 and humans35 with HF. Electron transport system proteins seem to aggregate into functional supercomplexes130,131,132, and a loss of mitochondrial supercomplexes, which is postulated to have a causal role in mitochondrial ROS generation133, has been noted in HF134.
Several approaches are being developed to improve the efficiency of the ETC in HF. The coenzyme Q (ubiquinol/ubiquinone CoQ) pool comprises a redox-cycling coenzyme found in the ETC. CoQ is typically synthesized de novo and undergoes a two-electron reduction from substrates fed into complexes I and II, and is then oxidized as it donates electrons into complex III. As a redox cycler, the ubiquinol/ubiquinone couple can both accept and donate electrons, depending on the redox potential135. Incomplete, one-electron reduction of CoQ produces semiquinone, itself a highly reactive radical. A reduced CoQ pool could potentially feed electrons 'backwards' towards complex I, which results in reverse electron transfer and ROS generation136. Decreased circulating CoQ has been observed in patients with HF137,138, with an inverse correlation observed between plasma CoQ and mortality139. In the Q-SYMBIO trial140, the efficacy of CoQ was tested in a small (n = 420), double-blind, placebo-controlled study in patients with HF and showed a reduction in mortality after 2 years of treatment. Although the Q-SYMBIO trial was fairly small, the promising findings triggered interest in the development of other CoQ analogues that more effectively target mitochondria. New quinone conjugates that are tethered to lipophilic, cationic triphenylphosphonium moieties, such as MitoQ, SkQ, and other plastoquinones, might improve the delivery of CoQ to mitochondria141,142,143, and have shown some promise in preclinical models of HF144. A potential problem with the use of these compounds is that they are self-limiting, in that they can depolarize mitochondria and inhibit mitochondrial respiration at high concentrations145. Several short-chain synthetic CoQ analogues are also in development, including EPI-743 (Ref. 146) and idebenone147. These compounds have shown promise in small trials of genetic mitochondrial disease148,149, but have not yet been tested in larger trials of human HF.
Aberrant mitochondrial membrane phospholipids in HF are integrally involved in ETC dysfunction. A membrane phospholipid integral to optimal function of the ETC and whose content and composition are altered in HF is cardiolipin. Cardiolipin resides in the inner mitochondrial membrane (Fig. 4) and, unlike most phospholipids that have two acyl tails, cardiolipin has four acyl chains. In mammalian hearts, these chains are enriched with linoleic acid (18:2)4. Cardiolipin decrements are observed in both paediatric150 and adult151,152 patients with HF. Cardiolipin is essential for the activity of ETC complexes, membrane transporters, mitochondrial ion homeostasis, and ROS production153. Given that most mitochondrial complexes associated with energy production are oligomers composed of many subunits, cardiolipin is proposed to act as molecular 'glue' holding these subunits together154,155,156. Approaches that target cardiolipin are likely to improve electron transport across the ETC and, in doing so, might be beneficial in treating HF.
A compound that targets cardiolipin in the mitochondria that is currently in clinical development is the cell-permeable peptide MTP-131 (also called elamipretide or Bendavia). An analogue of MTP-131 (SS-31) was serendipitously discovered by Szeto and Schiller in attempts to identify small peptides with opioid-receptor binding properties157. MTP-131 has no discernible opioid-receptor activity158, but was found to localize to the inner mitochondrial membrane159, reduce myocardial ischaemia–reperfusion injury112,160,161, improve renal function51,162, and restore skeletal muscle function163. MTP-131 is not a direct ROS scavenger164, and is postulated to act by interacting with cardiolipin165 to interrupt the vicious cycle of ROS-mediated cardiolipin oxidation and subsequent loss of energetics119,166. MTP-131-mediated improvements in mitochondrial energetics have been observed across a number of different tissues in animal models of disease, including the myocardium161,163,164. Of note, MTP-131 can improve mitochondrial bioenergetics by improving respiratory supercomplex formation (D. A. Brown, unpublished work).
MTP-131 is currently being investigated in several phase II clinical trials. Preclinical studies in mouse models of HF have demonstrated efficacy using MTP-131. In a mouse model of HF induced by aortic constriction, MTP-131 improved left ventricular function, reduced hypertrophic remodelling, and restored mitochondrial function167. In complementary studies, MTP-131 administration substantially reduced maladaptive remodelling, preserved cardiac function, lowered β-adrenergic-mediated calcium overload, and restored mitochondrial protein expression168,169,170. A substantial improvement in cardiac function with MTP-131 has been demonstrated in a porcine model of HFpEF171 and a canine model of HFrEF172. Beneficial improvements in ejection fraction were associated with improved activity or expression of mitochondrial complexes I, IV, and V, and a normalization of cardiolipin levels172. As the HF syndrome influences many different tissues (Fig. 3), the evidence that MTP-131 also improves skeletal muscle function, exercise capacity, and renal function adds to the promise of this emerging therapy51,163,173,174.
Blockers of the MPTP
The mitochondrial permeability transition pore (MPTP) is a nonspecific pore that opens in response to increased calcium levels and oxidative challenge, and is associated with ROS production, apoptotic cell death, and mitochondrial dysfunction. Increased proclivity of MPTP opening occurs in both acute and chronic heart disease, and numerous preclinical studies have demonstrated efficacy in cardiac pathology with MPTP blockers, such as cyclosporin, NIM811, and TRO40303 (reviewed previously175,176,177,178,179). Although the opening of the MPTP has historically been thought of as a pathological event leading to cell death, studies now suggest that transient MPTP opening might be a physiological 'reset' mechanism to prevent mitochondrial calcium overload. Rare, transient openings of the MPTP have been observed in individual mitochondria of primary cardiomyocytes180. Small, brief MPTP openings were found to be more frequent in HF cardiomyocytes, and were associated with transient mitochondrial depolarization and mitochondrial calcium release. If opening of these pores might be a normal compensatory mechanism akin to 'pressure release valves', the concept of treating HF by blocking them becomes increasingly difficult. Ongoing uncertainty regarding the molecular identity of the MPTP further complicates the development of novel therapies that act on the pore176,181,182,183,184,185. The MPTP seems to be comprised of ATP synthase (complex V) dimers and to be gated by mitochondrial matrix calcium content via cyclophillin D186,187.
Clinical studies have failed to demonstrate efficacy in most188,189, but not all190,191, studies; however, most of these studies focused on reducing acute cardiac ischaemia–reperfusion injury and not in limiting left ventricular dysfunction in HF. Chronic administration of cyclosporin has been linked with renal pathology and immunosuppression192,193, and cyclosporin was found to evoke systemic hypertension in porcine models of HFpEF194. Accordingly, cyclosporin is not an appropriate approach for the long-term management of HF. Further work with alternative MPTP blockers is needed to determine whether inhibiting or delaying MPTP opening is a clinically plausible approach to alter the progression of HF.
Cellular/mitochondrial ion homeostasis
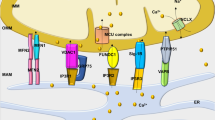
Aberrant handling of several different ions within the mitochondria has been observed, mostly in animal models of HF. Heightened levels of free iron can increase ROS through Fenton chemistry. Changes in cellular iron handling have been noted in HF7,195, and orally-available iron chelators such as deferiprone seem to redistribute iron from tissues, including the mitochondrial space, into the circulation196. Although a potential exists to treat HF by chelating cellular iron, no study to date has shown functional improvements of the failing heart, although several clinical trials are currently underway.
Impaired cellular calcium handling that leads to decrements in excitation–contraction coupling is noted across HF aetiologies, and contributes to poor cardiac mechanics and to arrhythmogenesis197,198,199,200. Mitochondria can directly influence cellular calcium dynamics, because many of the membrane-bound pumps required for cytosolic calcium release and removal are energy-dependent and ROS-dependent. Altered calcium handling has been implicated in HFpEF, in which abnormal calcium dynamics impair relaxation. Short-term administration of ivabradine to slow the heart rate led to modest benefits in patients with HFpEF, ostensibly by providing more time for calcium-dependent relaxation201. The vast majority of calcium resequestration into the sarcoplasmic reticulum, obligatory for diastolic relaxation, occurs through SERCA2a, which has been shown to be downregulated in HF202,203,204. Overexpressing SERCA2a has shown promise in animal models of HF205,206, although several barriers (such as the development of neutralizing antibodies) still exist before gene transfer realizes its full translational potential207. Furthermore, increased ROS can oxidize proteins associated with the ryanodine receptor calcium-release channel, which can lead to calcium leaking out of the sarcoplasmic reticulum during diastole208. Increased intracellular sodium levels in HF209,210,211,212 also contribute to poor calcium handling through mechanisms involving sodium–calcium exchange. Given that calcium is central to maintaining bioenergetic supply–demand matching21,213, sodium overload alters cellular and mitochondrial calcium fluxes and impairs bioenergetic supply–demand matching in HF214. Although very early in development, inhibitors of the mitochondrial sodium–calcium–(lithium) exchanger215, such as CGP-37157, have been shown to improve cardiac function in preclinical models of HF216,217. Inhibiting the sarcolemmal sodium–calcium exchanger might also be a promising approach, as demonstrated in a preclinical model of HFpEF218.
Another compound in clinical development to improve cardiac efficiency in HF is omecamtiv mecarbil (CK-1827452). This drug increases the calcium sensitivity of the myofilaments219, which prolongs the duration of systole in animal models and in human HF220,221,222. Two substantial phase IIb, double-blind, randomized studies comparing omecamtiv mecarbil and placebo have been conducted. In the ATOMIC-HF trial223, omecamtiv mecarbil was administered for 48 h intravenously to patients with acute HF. Overall, the study was neutral (with some evidence of a symptomatic benefit at higher doses), but suggested omecamtiv mecarbil was safe. In the COSMIC-HF trial224, an oral formulation of omecamtiv mecarbil was associated with improvements in cardiac function over 20 weeks, with an effect that persisted for 4 weeks after stopping the drug, suggesting that improved function had produced favourable structural remodelling. Despite the promise of omecamtiv mecarbil, concerns about elevated levels of serum troponin225, metabolic inefficiency226, and impaired cardiac relaxation227 must be assuaged by larger clinical trials to understand fully whether this approach can improve prognosis in HF.
Conclusions
The vast majority of HF trials over the past decade have been neutral, and event rates remain unacceptably high. Perhaps most alarming, no proven therapies exist for patients with worsening chronic HF or HFpEF — populations that collectively comprise the majority of the total HF population. Moreover, although systemic blockade of maladaptive neurohormonal responses has improved outcomes in HFrEF, these agents also lower blood pressure and/or heart rate, and development of new haemodynamically active drugs for stepwise addition to existing therapies raises safety and tolerability concerns. Therefore, an ideal novel therapy would be haemodynamically neutral and target the myocardium as the centrepiece of the therapeutic mechanism. In this context, overwhelming evidence from both preclinical and clinical studies indicates bioenergetic insufficiency in HF. Studies using preclinical models of the disease continue to advance our understanding of the cellular and molecular mechanisms that contribute to poor bioenergetics of the failing heart. Considerable potential exists to fill this unmet need, mitigate the economic burdens, and reduce symptoms in patients with HF by focusing on the development of new therapeutic modalities that target mitochondrial abnormalities in HF.
References
Jessup, M. et al. 2009 focused update: ACCF/AHA guidelines for the diagnosis and management of heart failure in adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 119, 1977–2016 (2009).
Hunt, S. A. et al. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation 112, e154–e235 (2005).
Wilcox, J. E. et al. “Targeting the Heart” in heart failure: myocardial recovery in heart failure with reduced ejection fraction. JACC Heart Fail. 3, 661–669 (2015).
Braunschweig, F., Cowie, M. R. & Auricchio, A. What are the costs of heart failure? Europace 13, ii13–ii17 (2011).
Yancy, C. W. et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 62, e147–e239 (2013).
Meta-analysis Global Group in Chronic Heart Failure. The survival of patients with heart failure with preserved or reduced left ventricular ejection fraction: an individual patient data meta-analysis. Eur. Heart J. 33, 1750–1757 (2012).
Bayeva, M., Gheorghiade, M. & Ardehali, H. Mitochondria as a therapeutic target in heart failure. J. Am. Coll. Cardiol. 61, 599–610 (2013).
Neely, J. R., Liebermeister, H., Battersby, E. J. & Morgan, H. E. Effect of pressure development on oxygen consumption by isolated rat heart. Am. J. Physiol. 212, 804–814 (1967).
Gheorghiade, M. et al. Developing new treatments for heart failure: focus on the heart. Circ. Heart Fail. 9, e002727 (2016).
Vaduganathan, M., Butler, J., Pitt, B. & Gheorghiade, M. Contemporary drug development in heart failure: call for hemodynamically neutral therapies. Circ. Heart Fail. 8, 826–831 (2015).
Downey, J. M. & Cohen, M. V. Why do we still not have cardioprotective drugs? Circ. J. 73, 1171–1177 (2009).
Senni, M., Gavazzi, A., Gheorghiade, M. & Butler, J. Heart failure at the crossroads: moving beyond blaming stakeholders to targeting the heart. Eur. J. Heart Fail. 17, 760–763 (2015).
Fordyce, C. B. et al. Cardiovascular drug development: is it dead or just hibernating? J. Am. Coll. Cardiol. 65, 1567–1582 (2015).
Amidon, S. & Amidon, T. The Sublime Engine: A Biography of the Human Heart. (Rodale Books, 2011).
Tornroth-Horsefield, S. & Neutze, R. Opening and closing the metabolite gate. Proc. Natl Acad. Sci. USA 105, 19565–19566 (2008).
Balaban, R. S. Cardiac energy metabolism homeostasis: role of cytosolic calcium. J. Mol. Cell. Cardiol. 34, 1259–1271 (2002).
Barth, E., Stammler, G., Speiser, B. & Schaper, J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell. Cardiol. 24, 669–681 (1992).
Schaper, J., Meiser, E. & Stammler, G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 56, 377–391 (1985).
Opie, L. The Heart: Physiology, from Cell to Circulation 3rd edn, (Lipincott−Raven, 1998).
Balaban, R. S. Domestication of the cardiac mitochondrion for energy conversion. J. Mol. Cell. Cardiol. 6, 832–841 (2009).
Balaban, R. S. The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim. Biophys. Acta 1787, 1334–1341 (2009).
Suliman, H. B. & Piantadosi, C. A. Mitochondrial quality control as a therapeutic target. Pharmacol. Rev. 68, 20–48 (2016).
Lesnefsky, E. J., Chen, Q. & Hoppel, C. L. Mitochondrial metabolism in aging heart. Circ. Res. 118, 1593–1611 (2016).
Gottlieb, R. A. & Bernstein, D. Mitochondrial remodeling: rearranging, recycling, and reprogramming. Cell Calcium 60, 88–101 (2016).
Shirihai, O. S., Song, M. & Dorn, G. W. II. How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 116, 1835–1849 (2015).
Dorn, G. W., II & Kitsis, R. N. The mitochondrial dynamism-mitophagy-cell death interactome: multiple roles performed by members of a mitochondrial molecular ensemble. Circ. Res. 116, 167–182 (2015).
Biala, A. K., Dhingra, R. & Kirshenbaum, L. A. Mitochondrial dynamics: orchestrating the journey to advanced age. J. Mol. Cell. Cardiol. 83, 37–43 (2015).
Dhingra, R. & Kirshenbaum, L. A. Regulation of mitochondrial dynamics and cell fate. Circ. J. 78, 803–810 (2014).
Thomas, R. L. & Gustafsson, A. B. Mitochondrial autophagy — an essential quality control mechanism for myocardial homeostasis. Circ. J. 77, 2449–2454 (2013).
Sebastiani, M. et al. Induction of mitochondrial biogenesis is a maladaptive mechanism in mitochondrial cardiomyopathies. J. Am. Coll. Cardiol. 50, 1362–1369 (2007).
Goh, K. Y. et al. Impaired mitochondrial network excitability in failing guinea-pig cardiomyocytes. Cardiovasc. Res. 109, 79–89 (2016).
Sabbah, H. N. et al. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. J. Mol. Cell. Cardiol. 24, 1333–1347 (1992).
Lehman, J. J. & Kelly, D. P. Gene regulatory mechanisms governing energy metabolism during cardiac hypertrophic growth. Heart Fail. Rev. 7, 175–185 (2002).
Lai, L. et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ. Heart Fail. 7, 1022–1031 (2014).
Lemieux, H., Semsroth, S., Antretter, H., Hofer, D. & Gnaiger, E. Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int. J. Biochem. Cell Biol. 43, 1729–1738 (2011).
Nicholls, D. G. & Ferguson, S. J. Bioenergetics 3rd edn (Academic, 2002).
Schon, E. A., DiMauro, S. & Hirano, M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat. Rev. Genet. 13, 878–890 (2012).
Ott, M., Amunts, A. & Brown, A. Organization and regulation of mitochondrial protein synthesis. Annu. Rev. Biochem. 85, 77–101 (2016).
Bates, M. G. et al. Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur. Heart J. 33, 3023–3033 (2012).
Margulis, L. Symbiotic theory of the origin of eukaryotic organelles; criteria for proof. Symp. Soc. Exp. Biol. 29 21–38 (1975).
Sato, M. & Sato, K. Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochim. Biophys. Acta 1833, 1979–1984 (2013).
Taylor, R. W. & Turnbull, D. M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 6, 389–402 (2005).
Bacman, S. R., Williams, S. L., Pinto, M., Peralta, S. & Moraes, C. T. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 19, 1111–1113 (2013).
Reddy, P. et al. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 161, 459–469 (2015).
Paull, D. et al. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature 493, 632–637 (2013).
Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191, 144–148 (1961).
Carrasco, A. J. et al. Adenylate kinase phosphotransfer communicates cellular energetic signals to ATP-sensitive potassium channels. Proc. Natl Acad. Sci. USA 98, 7623–7628 (2001).
Wu, F., Zhang, J. & Beard, D. A. Experimentally observed phenomena on cardiac energetics in heart failure emerge from simulations of cardiac metabolism. Proc. Natl Acad. Sci. USA 106, 7143–7148 (2009).
Neubauer, S. et al. Downregulation of the Na+-creatine cotransporter in failing human myocardium and in experimental heart failure. Circulation 100, 1847–1850 (1999).
Abozguia, K. et al. Reduced in vivo skeletal muscle oxygen consumption in patients with chronic heart failure — a study using Near Infrared Spectrophotometry (NIRS). Eur. J. Heart Fail. 10, 652–657 (2008).
Eirin, A. et al. Mitochondrial protection restores renal function in swine atherosclerotic renovascular disease. Cardiovasc. Res. 103, 461–472 (2014).
Anderson, E. J. et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 3, 573–581 (2009).
Zhang, Q. et al. Catechin ameliorates cardiac dysfunction in rats with chronic heart failure by regulating the balance between Th17 and Treg cells. Inflamm. Res. 63, 619–628 (2014).
Ramirez-Sanchez, I. et al. (−)-Epicatechin rich cocoa mediated modulation of oxidative stress regulators in skeletal muscle of heart failure and type 2 diabetes patients. Int. J. Cardiol. 168, 3982–3990 (2013).
Sung, M. M. & Dyck, J. R. Therapeutic potential of resveratrol in heart failure. Ann. NY Acad. Sci. 1348, 32–45 (2015).
Magyar, K. et al. Cardioprotection by resveratrol: a human clinical trial in patients with stable coronary artery disease. Clin. Hemorheol. Microcirc. 50, 179–187 (2012).
Stanley, W. C., Recchia, F. A. & Lopaschuk, G. D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 85, 1093–1129 (2005).
Doenst, T., Nguyen, T. D. & Abel, E. D. Cardiac metabolism in heart failure: implications beyond ATP production. Circ. Res. 113, 709–724 (2013).
Fukushima, A., Milner, K., Gupta, A. & Lopaschuk, G. D. Myocardial energy substrate metabolism in heart failure: from pathways to therapeutic targets. Curr. Pharm. Des. 21, 3654–3664 (2015).
Ventura-Clapier, R., Garnier, A. & Veksler, V. Energy metabolism in heart failure. J. Physiol. 555, 1–13 (2004).
Aubert, G., Vega, R. B. & Kelly, D. P. Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart. Biochim. Biophys. Acta 1833, 840–847 (2013).
de las Fuentes, L. et al. Myocardial fatty acid metabolism: independent predictor of left ventricular mass in hypertensive heart disease. Hypertension 41, 83–87 (2003).
Goikoetxea, M. J. et al. Altered cardiac expression of peroxisome proliferator-activated receptor-isoforms in patients with hypertensive heart disease. Cardiovasc. Res. 69, 899–907 (2006).
Sack, M. N. et al. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 94, 2837–2842 (1996).
Sihag, S., Cresci, S., Li, A. Y., Sucharov, C. C. & Lehman, J. J. PGC-1 α and ERR α target gene downregulation is a signature of the failing human heart. J. Mol. Cell. Cardiol. 46, 201–212 (2009).
Lehman, J. J. et al. Peroxisome proliferator-activated receptor γ coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 106, 847–856 (2000).
Sarma, S., Ardehali, H. & Gheorghiade, M. Enhancing the metabolic substrate: PPAR-alpha agonists in heart failure. Heart Fail. Rev. 17, 35–43 (2012).
Jaswal, J. S., Keung, W., Wang, W., Ussher, J. R. & Lopaschuk, G. D. Targeting fatty acid and carbohydrate oxidation — a novel therapeutic intervention in the ischemic and failing heart. Biochim. Biophys. Acta 1813, 1333–1350 (2011).
Igarashi, N. et al. Influence of β-adrenoceptor blockade on the myocardial accumulation of fatty acid tracer and its intracellular metabolism in the heart after ischemia−reperfusion injury. Circ. J. 70, 1509–1514 (2006).
Fillmore, N. & Lopaschuk, G. D. Malonyl CoA: a promising target for the treatment of cardiac disease. IUBMB Life http://dx.doi.org/10.1002/iub.1253 (2014).
Stanley, W. C. et al. Malonyl-CoA decarboxylase inhibition suppresses fatty acid oxidation and reduces lactate production during demand-induced ischemia. Am. J. Physiol. Heart Circ. Physiol. 289, H2304–H2309 (2005).
Dyck, J. R. et al. Malonyl coenzyme A decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ. Res. 94, e78–e84 (2004).
Kolwicz, S. C. Jr., Airhart, S. & Tian, R. Ketones step to the plate: a game changer for metabolic remodeling in heart failure? Circulation 133, 689–691 (2016).
Bedi, K. C. Jr. et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation 133, 706–716 (2016).
Pinti, M. V., Hathaway, Q. A. & Hollander, J. M. Role of microRNA in metabolic shift during heart failure. Am. J. Physiol. Heart Circ. Physiol. http://dx.doi.org/10.1152/ajpheart.00341.2016 (2016).
Opie, L. H., Thandroyen, F. T., Muller, C. & Bricknell, O. L. Adrenaline-induced “oxygen-wastage” and enzyme release from working rat heart. Effects of calcium antagonism, β-blockade, nicotinic acid and coronary artery ligation. J. Mol. Cell. Cardiol. 11, 1073–1094 (1979).
Francis, G. S. et al. Plasma norepinephrine, plasma renin activity, and congestive heart failure. Relations to survival and the effects of therapy in V-HeFT II. The V-HeFT VA Cooperative Studies Group. Circulation 87, VI40–V148 (1993).
Triposkiadis, F. et al. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J. Am. Coll. Cardiol. 54, 1747–1762 (2009).
Menon, B. et al. Expression of the cytoplasmic domain of β1 integrin induces apoptosis in adult rat ventricular myocytes (ARVM) via the involvement of caspase-8 and mitochondrial death pathway. Basic Res. Cardiol. 101, 485–493 (2006).
Rosca, M. G. & Hoppel, C. L. Mitochondrial dysfunction in heart failure. Heart Fail. Rev. 18, 607–622 (2013).
Leger, B. et al. Chronic formoterol administration reduces cardiac mitochondrial protein synthesis and oxidative capacity in mice. Int. J. Cardiol. 146, 270–272 (2011).
Izem-Meziane, M. et al. Catecholamine-induced cardiac mitochondrial dysfunction and mPTP opening: protective effect of curcumin. Am. J. Physiol. Heart Circ. Physiol. 302, H665–674 (2012).
Nagasaka, S. et al. Protein kinase A catalytic subunit alters cardiac mitochondrial redox state and membrane potential via the formation of reactive oxygen species. Circ. J. 71, 429–436 (2007).
Remondino, A. et al. β-adrenergic receptor-stimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of the mitochondrial pathway. Circ. Res. 92, 136–138 (2003).
Communal, C., Colucci, W. S. & Singh, K. p38 mitogen-activated protein kinase pathway protects adult rat ventricular myocytes against β -adrenergic receptor-stimulated apoptosis. Evidence for Gi-dependent activation. J. Biol. Chem. 275, 19395–19400 (2000).
Communal, C., Singh, K., Sawyer, D. B. & Colucci, W. S. Opposing effects of β1- and β2-adrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin-sensitive G protein. Circulation 100, 2210–2212 (1999).
Liaudet, L., Calderari, B. & Pacher, P. Pathophysiological mechanisms of catecholamine and cocaine-mediated cardiotoxicity. Heart Fail. Rev. 19, 815–824 (2014).
Feldman, D. S., Carnes, C. A., Abraham, W. T. & Bristow, M. R. Mechanisms of disease: β-adrenergic receptors — alterations in signal transduction and pharmacogenomics in heart failure. Nat. Clin. Pract. Cardiovasc. Med. 2, 475–483 (2005).
Aon, M. A., Cortassa, S. & O'Rourke, B. Redox-optimized ROS balance: a unifying hypothesis. Biochim. Biophys. Acta 6, 865–877 (2010).
Brown, D. A., Sabbah, H. N. & Shaikh, S. R. Mitochondrial inner membrane lipids and proteins as targets for decreasing cardiac ischemia/reperfusion injury. Pharmacol. Ther. 140, 258–266 (2013).
Murphy, E. & Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia−reperfusion injury. Physiol. Rev. 88, 581–609 (2008).
Murphy, E. & Steenbergen, C. Ion transport and energetics during cell death and protection. Physiology (Bethesda) 23, 115–123 (2008).
Murphy, M. P. How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 (2009).
Walters, A. M., Porter, G. A. Jr & Brookes, P. S. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ. Res. 111, 1222–1236 (2012).
Nabeebaccus, A., Zhang, M. & Shah, A. M. NADPH oxidases and cardiac remodelling. Heart Fail. Rev. 16, 5–12 (2011).
Orr, A. L. et al. Inhibitors of ROS production by the ubiquinone-binding site of mitochondrial complex I identified by chemical screening. Free Radic. Biol. Med. 65, 1047–1059 (2013).
Zorov, D. B., Juhaszova, M. & Sollott, S. J. Mitochondrial ROS-induced ROS release: an update and review. Biochim. Biophys. Acta 1757, 509–517 (2006).
Aon, M. A., Cortassa, S., Akar, F. G. & O'Rourke, B. Mitochondrial criticality: a new concept at the turning point of life or death. Biochim. Biophys. Acta 1762, 232–240 (2006).
Ide, T. et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 88, 529–535 (2001).
Ide, T. et al. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ. Res. 86, 152–157 (2000).
Ide, T. et al. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ. Res. 85, 357–363 (1999).
Rosca, M. G., Tandler, B. & Hoppel, C. L. Mitochondria in cardiac hypertrophy and heart failure. J. Mol. Cell. Cardiol. 55, 31–41 (2013).
Alleman, R. J., Katunga, L. A., Nelson, M. A., Brown, D. A. & Anderson, E. J. The “Goldilocks Zone” from a redox perspective — adaptive versus deleterious responses to oxidative stress in striated muscle. Front. Physiol. 5, 358 (2014).
Song, M. et al. Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ. Res. 115, 348–353 (2014).
Frasier, C. R., Moore, R. L. & Brown, D. A. Exercise-induced cardiac preconditioning: how exercise protects your achy-breaky heart. J. Appl. Physiol. 111, 905–915 (2011).
Brown, D. A., Jew, K. N., Sparagna, G. C., Musch, T. I. & Moore, R. L. Exercise training preserves coronary flow and reduces infarct size following ischemia−reperfusion in rat heart. J. Appl. Physiol. 95, 2510–2518 (2003).
Brown, D. A. & Moore, R. L. Perspectives in innate and acquired cardioprotection: cardioprotection acquired through exercise. J. Appl. Physiol. 103, 1894–1899 (2007).
Marshall, K. D. et al. Heart failure with preserved ejection fraction: chronic low-intensity interval exercise training preserves myocardial O2 balance and diastolic function. J. Appl. Physiol. 114, 131–147 (2013).
Flynn, K. E. et al. Effects of exercise training on health status in patients with chronic heart failure: HF-ACTION randomized controlled trial. JAMA 301, 1451–1459 (2009).
O'Connor, C. M. et al. Efficacy and safety of exercise training in patients with chronic heart failure: HF-ACTION randomized controlled trial. JAMA 301, 1439–1450 (2009).
Edelmann, F. et al. Exercise training improves exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction: results of the Ex-DHF (Exercise training in Diastolic Heart Failure) pilot study. J. Am. Coll. Cardiol. 58, 1780–1791 (2011).
Frasier, C. R. et al. Redox-dependent increases in glutathione reductase and exercise preconditioning: role of NADPH oxidase and mitochondria. Cardiovasc. Res. 98, 47–55 (2013).
Nelson, M. J., Harris, M. B., Boluyt, M. O., Hwang, H. S. & Starnes, J. W. Effect of N-2-mercaptopropionyl glycine on exercise-induced cardiac adaptations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R993–R1000 (2011).
Ristow, M. et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl Acad. Sci. USA 106, 8665–8670 (2009).
Akhmedov, A. T., Rybin, V. & Marin-Garcia, J. Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart. Heart Fail. Rev. 20, 227–249 (2015).
Brand, M. D. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp. Gerontol. 35, 811–820 (2000).
Slodzinski, M. K., Aon, M. A. & O'Rourke, B. Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J. Mol. Cell. Cardiol. 45, 650–660 (2008).
Brown, D. A. et al. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J. Mol. Cell. Cardiol. 48, 673–679 (2010).
Nickel, A. G. et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 22, 472–484 (2015).
Yusuf, S., Dagenais, G., Pogue, J., Bosch, J. & Sleight, P. Vitamin E supplementation and cardiovascular events in high-risk patients. Heart Outcomes Prevention Evaluation Study Investigators. N. Engl. J. Med. 342, 154–160 (2000).
Tsujita, K. et al. Effects of edaravone on reperfusion injury in patients with acute myocardial infarction. Am. J. Cardiol. 94, 481–484 (2004).
Escobales, N. et al. Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J. Mol. Cell. Cardiol. 77, 136–146 (2014).
Javadov, S. et al. Mitochondria-targeted antioxidant preserves contractile properties and mitochondrial function of skeletal muscle in aged rats. Oncotarget 6, 39469–39481 (2015).
Dikalova, A. E. et al. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 107, 106–116 (2010).
Liang, H. L., Sedlic, F., Bosnjak, Z. & Nilakantan, V. SOD1 and MitoTEMPO partially prevent mitochondrial permeability transition pore opening, necrosis, and mitochondrial apoptosis after ATP depletion recovery. Free Radic. Biol. Med. 49, 1550–1560 (2010).
Koyama, H. et al. Antioxidants improve the phenotypes of dilated cardiomyopathy and muscle fatigue in mitochondrial superoxide dismutase-deficient mice. Molecules 18, 1383–1393 (2013).
Kawakami, S. et al. Antioxidant, EUK-8, prevents murine dilated cardiomyopathy. Circ. J. 73, 2125–2134 (2009).
van Empel, V. P. et al. EUK-8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload-induced heart failure in the harlequin mouse mutant. J. Am. Coll. Cardiol. 48, 824–832 (2006).
Rosca, M., Minkler, P. & Hoppel, C. L. Cardiac mitochondria in heart failure: normal cardiolipin profile and increased threonine phosphorylation of complex IV. Biochim. Biophys. Acta 1807, 1373–1382 (2011).
Acin-Perez, R. et al. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell 13, 805–815 (2004).
Acin-Perez, R., Fernandez-Silva, P., Peleato, M. L., Perez-Martos, A. & Enriquez, J. A. Respiratory active mitochondrial supercomplexes. Mol. Cell 32, 529–539 (2008).
Lapuente-Brun, E. et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340, 1567–1570 (2013).
Maranzana, E., Barbero, G., Falasca, A. I., Lenaz, G. & Genova, M. L. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid. Redox Signal. 19, 1469–1480 (2013).
Rosca, M. G. et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. 80, 30–39 (2008).
Nicholls, D. G. & Ferguson, S. J. Bioenergetics 4th edn (Academic, 2013).
Chouchani, E. T. et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 (2014).
Okonko, D. O. & Shah, A. M. Heart failure: mitochondrial dysfunction and oxidative stress in CHF. Nat. Rev. Cardiol. 12, 6–8 (2015).
Molyneux, S. L. et al. Coenzyme Q10: an independent predictor of mortality in chronic heart failure. J. Am. Coll. Cardiol. 52, 1435–1441 (2008).
McMurray, J. J. et al. Coenzyme Q10, rosuvastatin, and clinical outcomes in heart failure: a pre-specified substudy of CORONA (controlled rosuvastatin multinational study in heart failure). J. Am. Coll. Cardiol. 56, 1196–1204 (2010).
Mortensen, S. A. et al. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q-SYMBIO: a randomized double-blind trial. JACC Heart Fail. 2, 641–649 (2014).
Murphy, M. P. Targeting lipophilic cations to mitochondria. Biochim. Biophys. Acta 1777, 1028–1031 (2008).
Smith, R. A. et al. Mitochondria-targeted antioxidants in the treatment of disease. Ann. NY Acad. Sci. 1147, 105–111 (2008).
Skulachev, V. P. et al. An attempt to prevent senescence: a mitochondrial approach. Biochim. Biophys. Acta 1787, 437–461 (2009).
Graham, D. et al. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 54, 322–328 (2009).
Lyamzaev, K. G. et al. Novel mitochondria-targeted antioxidants: plastoquinone conjugated with cationic plant alkaloids berberine and palmatine. Pharm. Res. 28, 2883–2895 (2011).
Enns, G. M. Treatment of mitochondrial disorders: antioxidants and beyond. J. Child Neurol. 29, 1235–1240 (2014).
Jaber, S. & Polster, B. M. Idebenone and neuroprotection: antioxidant, pro-oxidant, or electron carrier? J. Bioenerg. Biomembr. 47, 111–118 (2015).
Sadun, A. A. et al. Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch. Neurol. 69, 331–338 (2012).
Lerman-Sagie, T. et al. Dramatic improvement in mitochondrial cardiomyopathy following treatment with idebenone. J. Inherit Metab. Dis. 24, 28–34 (2001).
Chatfield, K. C. et al. Dysregulation of cardiolipin biosynthesis in pediatric heart failure. J. Mol. Cell. Cardiol. 74, 251–259 (2014).
Saini-Chohan, H. K. et al. Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J. Lipid Res. 50, 1600–1608 (2009).
Sparagna, G. C. & Lesnefsky, E. J. Cardiolipin remodeling in the heart. J. Cardiovasc. Pharmacol. 53, 290–301 (2009).
Chicco, A. J. & Sparagna, G. C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 292, C33–C44 (2007).
Pfeiffer, K. et al. Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880 (2003).
Schagger, H. Respiratory chain supercomplexes of mitochondria and bacteria. Biochim. Biophys. Acta 1555, 154–159 (2002).
Zhang, M., Mileykovskaya, E. & Dowhan, W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 277, 43553–43556 (2002).
Szeto, H. H. & Birk, A. V. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin. Pharmacol. Ther. 96, 672–683 (2014).
Szeto, H. H. & Schiller, P. W. Novel therapies targeting inner mitochondrial membrane — from discovery to clinical development. Pharm. Res. 28, 2669–2671 (2011).
Szeto, H. H. Mitochondria-targeted cytoprotective peptides for ischemia−reperfusion injury. Antioxid. Redox Signal. 10, 601–619 (2008).
Sloan, R. C. et al. Mitochondrial permeability transition in the diabetic heart: contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J. Mol. Cell. Cardiol. 52, 1009–1018 (2012).
Kloner, R. A. et al. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective peptide. J. Am. Heart Assoc. 1, e001644 (2012).
Szeto, H. H. et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 22, 1041–1052 (2011).
Siegel, M. P. et al. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 12, 763–771 (2013).
Brown, D. A. et al. Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia. J. Cardiovasc. Pharmacol. Ther. 19, 121–132 (2013).
Birk, A. V., Chao, W. M., Bracken, C., Warren, J. D. & Szeto, H. H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 171, 2017–2028 (2013).
Birk, A. V. et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 24, 1250–1261 (2013).
Dai, D. F. et al. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J. Am. Coll. Cardiol. 58, 73–82 (2011).
Dai, D. F. et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart Fail. 6, 1067–1076 (2013).
Shi, J. et al. Bendavia restores mitochondrial energy metabolism gene expression and suppresses cardiac fibrosis in the border zone of the infarcted heart. Life Sci. 141, 170–178 (2015).
Andersson, D. C. et al. Mitochondrial production of reactive oxygen species contributes to the β-adrenergic stimulation of mouse cardiomycytes. J. Physiol. 589, 1791–1801 (2011).
Eirin, A. et al. Mitochondrial targeted peptides attenuate residual myocardial damage after reversal of experimental renovascular hypertension. J. Hypertens. 32, 154–165 (2014).
Sabbah, H. N. et al. Chronic therapy with elamipretide (MTP-131), a novel mitochondria-targeting peptide, improves left ventricular and mitochondrial function in dogs with advanced heart failure. Circ. Heart Fail. 9, e002206 (2016).
Min, K. et al. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. 111, 1459–1466 (2011).
Powers, S. K. et al. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit. Care Med. 39, 1749–1759 (2011).
Halestrap, A. P., Clarke, S. J. & Javadov, S. A. Mitochondrial permeability transition pore opening during myocardial reperfusion — a target for cardioprotection. Cardiovasc. Res. 61, 372–385 (2004).
Halestrap, A. P. & Pasdois, P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta 1787, 1402–1415 (2009).
Kowaltowski, A. J., Castilho, R. F. & Vercesi, A. E. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 495, 12–15 (2001).
Sharov, V. G., Todor, A., Khanal, S., Imai, M. & Sabbah, H. N. Cyclosporine A attenuates mitochondrial permeability transition and improves mitochondrial respiratory function in cardiomyocytes isolated from dogs with heart failure. J. Mol. Cell. Cardiol. 42, 150–158 (2007).
Sharov, V. G., Todor, A. V., Imai, M. & Sabbah, H. N. Inhibition of mitochondrial permeability transition pores by cyclosporine A improves cytochrome c oxidase function and increases rate of ATP synthesis in failing cardiomyocytes. Heart Fail. Rev. 10, 305–310 (2005).
Lu, X., Kwong, J. Q., Molkentin, J. D. & Bers, D. M. Individual cardiac mitochondria undergo rare transient permeability transition pore openings. Circ. Res. 118, 834–841 (2016).
Baines, C. P. The molecular composition of the mitochondrial permeability transition pore. J. Mol. Cell. Cardiol. 46, 850–857 (2009).
Halestrap, A. P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 46, 821–831 (2009).
Baines, C. P. et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662 (2005).
Baines, C. P., Kaiser, R. A., Sheiko, T., Craigen, W. J. & Molkentin, J. D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9, 550–555 (2007).
Kokoszka, J. E. et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465 (2004).
Giorgio, V. et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl Acad. Sci. USA 110, 5887–5892 (2013).
Carraro, M. et al. Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition. J. Biol. Chem. 289, 15980–15985 (2014).
Ghaffari, S., Kazemi, B., Toluey, M. & Sepehrvand, N. The effect of prethrombolytic cyclosporine-A injection on clinical outcome of acute anterior ST-elevation myocardial infarction. Cardiovasc. Ther. 31, e34–e39 (2013).
MITOCARE Study Group. Rationale and design of the 'MITOCARE' Study: a phase II, multicenter, randomized, double-blind, placebo-controlled study to assess the safety and efficacy of TRO40303 for the reduction of reperfusion injury in patients undergoing percutaneous coronary intervention for acute myocardial infarction. Cardiology 123, 201–207 (2012).
Mewton, N. et al. Effect of cyclosporine on left ventricular remodeling after reperfused myocardial infarction. J. Am. Coll. Cardiol. 55, 1200–1205 (2010).
Piot, C. et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 359, 473–481 (2008).
Naesens, M., Kuypers, D. R. & Sarwal, M. Calcineurin inhibitor nephrotoxicity. Clin. J. Am. Soc. Nephrol. 4, 481–508 (2009).
Tabara, L. C. et al. Mitochondria-targeted therapies for acute kidney injury. Expert Rev. Mol. Med. 16, e13 (2014).
Hiemstra, J. A. et al. A new twist on an old idea part 2: cyclosporine preserves normal mitochondrial but not cardiomyocyte function in mini-swine with compensated heart failure. Physiol. Rep. 2, e12050 (2014).
Bayeva, M., Sawicki, K. T., Butler, J., Gheorghiade, M. & Ardehali, H. Molecular and cellular basis of viable dysfunctional myocardium. Circ. Heart Fail. 7, 680–691 (2014).
Sohn, Y. S., Breuer, W., Munnich, A. & Cabantchik, Z. I. Redistribution of accumulated cell iron: a modality of chelation with therapeutic implications. Blood 111, 1690–1699 (2008).
O'Rourke, B. et al. Mechanisms of altered excitation−contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ. Res. 84, 562–570 (1999).
Wilson, L. D. et al. Heart failure enhances susceptibility to arrhythmogenic cardiac alternans. Heart Rhythm 6, 251–259 (2009).
Brown, D. A. & Cascio, W. E. 'Leaky' ryanodine receptors and sudden cardiac death. Cardiovasc. Res. 84, 343–344 (2009).
Brown, D. A. & O'Rourke, B. Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 88, 241–249 (2010).
Kosmala, W. et al. Effect of If-channel inhibition on hemodynamic status and exercise tolerance in heart failure with preserved ejection fraction: a randomized trial. J. Am. Coll. Cardiol. 62, 1330–1338 (2013).
Gorski, P. A., Ceholski, D. K. & Hajjar, R. J. Altered myocardial calcium cycling and energetics in heart failure — a rational approach for disease treatment. Cell. Metab. 21, 183–194 (2015).
Kawase, Y. & Hajjar, R. J. The cardiac sarcoplasmic/endoplasmic reticulum calcium ATPase: a potent target for cardiovascular diseases. Nat. Clin. Pract. Cardiovasc. Med. 5, 554–565 (2008).
Kho, C., Lee, A. & Hajjar, R. J. Altered sarcoplasmic reticulum calcium cycling — targets for heart failure therapy. Nat. Rev. Cardiol. 9, 717–733 (2012).
Gong, H. B., Wang, L., Lv, Q. & Wang, J. Improved systolic function of rat cardiocytes during heart failure by overexpression of SERCA2a. Eur. Rev. Med. Pharmacol. Sci. 20, 1590–1596 (2016).
Mattila, M., Koskenvuo, J., Soderstrom, M., Eerola, K. & Savontaus, M. Intramyocardial injection of SERCA2a-expressing lentivirus improves myocardial function in doxorubicin-induced heart failure. J. Gene Med. 18, 124–133 (2016).
Greenberg, B. et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 387, 1178–1186 (2016).
Belevych, A. E. et al. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc. Res. 84, 387–395 (2009).
Despa, S., Islam, M. A., Weber, C. R., Pogwizd, S. M. & Bers, D. M. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 105, 2543–2548 (2002).
Pieske, B. & Houser, S. R. [Na+]i handling in the failing human heart. Cardiovasc. Res. 57, 874–886 (2003).
Pieske, B., Houser, S. R., Hasenfuss, G. & Bers, D. M. Sodium and the heart: a hidden key factor in cardiac regulation. Cardiovasc. Res. 57, 871–872 (2003).
Pieske, B. et al. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation 106, 447–453 (2002).
Griffiths, E. J. Mitochondrial calcium transport in the heart: physiological and pathological roles. J. Mol. Cell. Cardiol. 46, 789–803 (2009).
Maack, C. et al. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 99, 172–182 (2006).
Palty, R. et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl Acad. Sci. USA 107, 436–441 (2010).
Nicolau, S. M. et al. Mitochondrial Na+/Ca2+-exchanger blocker CGP37157 protects against chromaffin cell death elicited by veratridine. J. Pharmacol. Exp. Ther. 330, 844–854 (2009).
Liu, T., Brown, D. A. & O'Rourke, B. Role of mitochondrial dysfunction in cardiac glycoside toxicity. J. Mol. Cell. Cardiol. 49, 728–736 (2010).
Primessnig, U. et al. Novel pathomechanisms of cardiomyocyte dysfunction in a model of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 18, 987–997 (2016).
Mamidi, R., Gresham, K. S., Li, A., dos Remedios, C. G. & Stelzer, J. E. Molecular effects of the myosin activator omecamtiv mecarbil on contractile properties of skinned myocardium lacking cardiac myosin binding protein-C. J. Mol. Cell. Cardiol. 85, 262–272 (2015).
Malik, F. I. et al. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science 331, 1439–1443 (2011).
Cleland, J. G. et al. The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: a double-blind, placebo-controlled, crossover, dose-ranging phase 2 trial. Lancet 378, 676–683 (2011).
Greenberg, B. H. et al. Safety and tolerability of omecamtiv mecarbil during exercise in patients with ischemic cardiomyopathy and angina. JACC Heart Fail. 3, 22–29 (2015).
Teerlink, J. R. et al. Acute treatment with omecamtiv mecarbil to increase contractility in acute heart failure: the ATOMIC-AHF study. J. Am. Coll. Cardiol. 67, 1444–1455 (2016).
Teerlink, J. R. et al. Chronic oral study of myosin activation to increase contractility in heart failure (COSMIC-HF): final results from a double-blind, randomized, placebo-controlled, multicenter study [abstract]. Presented at American Heart Association Scientific Sessions 2015.
Moin, D. S., Sackheim, J., Hamo, C. E. & Butler, J. Cardiac myosin activators in systolic heart failure: more friend than foe? Curr. Cardiol. Rep. 18, 100 (2016).
Bakkehaug, J. P. et al. Myosin activator omecamtiv mecarbil increases myocardial oxygen consumption and impairs cardiac efficiency mediated by resting myosin ATPase activity. Circ. Heart Fail. 8, 766–775 (2015).
Nagy, L. et al. The novel cardiac myosin activator omecamtiv mecarbil increases the calcium sensitivity of force production in isolated cardiomyocytes and skeletal muscle fibres of the rat. Br. J. Pharmacol. http://dx.doi.org/10.1111/bph.13235 (2015).
Phan, T. T. et al. Heart failure with preserved ejection fraction is characterized by dynamic impairment of active relaxation and contraction of the left ventricle on exercise and associated with myocardial energy deficiency. J. Am. Coll. Cardiol. 54, 402–409 (2009).
Esposito, A. et al. Impaired left ventricular energy metabolism in patients with hypertrophic cardiomyopathy is related to the extension of fibrosis at delayed gadolinium-enhanced magnetic resonance imaging. Heart 95, 228–233 (2009).
Smith, C. S., Bottomley, P. A., Schulman, S. P., Gerstenblith, G. & Weiss, R. G. Altered creatine kinase adenosine triphosphate kinetics in failing hypertrophied human myocardium. Circulation 114, 1151–1158 (2006).
Weiss, R. G., Gerstenblith, G. & Bottomley, P. A. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc. Natl Acad. Sci. USA 102, 808–813 (2005).
Crilley, J. G. et al. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J. Am. Coll. Cardiol. 41, 1776–1782 (2003).
Beer, M. et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with 31P-SLOOP magnetic resonance spectroscopy. J. Am. Coll. Cardiol. 40, 1267–1274 (2002).
Beer, M. et al. [Cardiac energy metabolism in heart valve diseases with 31P MR spectroscopy [German]. Radiologe 40, 162–167 (2000).
Lamb, H. J. et al. Diastolic dysfunction in hypertensive heart disease is associated with altered myocardial metabolism. Circulation 99, 2261–2267 (1999).
Conway, M. A., Bottomley, P. A., Ouwerkerk, R., Radda, G. K. & Rajagopalan, B. Mitral regurgitation: impaired systolic function, eccentric hypertrophy, and increased severity are linked to lower phosphocreatine/ATP ratios in humans. Circulation 97, 1716–1723 (1998).
Jung, W. I. et al. 31P NMR spectroscopy detects metabolic abnormalities in asymptomatic patients with hypertrophic cardiomyopathy. Circulation 97, 2536–2542 (1998).
Starling, R. C., Hammer, D. F. & Altschuld, R. A. Human myocardial ATP content and in vivo contractile function. Mol. Cell. Biochem. 180, 171–177 (1998).
Neubauer, S. et al. Cardiac high-energy phosphate metabolism in patients with aortic valve disease assessed by 31P-magnetic resonance spectroscopy. J. Investig. Med. 45, 453–462 (1997).
Neubauer, S. et al. Contributions of 31P-magnetic resonance spectroscopy to the understanding of dilated heart muscle disease. Eur. Heart J. 16 (Suppl. O), 115–118 (1995).
Yabe, T., Mitsunami, K., Inubushi, T. & Kinoshita, M. Quantitative measurements of cardiac phosphorus metabolites in coronary artery disease by 31P magnetic resonance spectroscopy. Circulation 92, 15–23 (1995).
Yabe, T. et al. Detection of myocardial ischemia by 31P magnetic resonance spectroscopy during handgrip exercise. Circulation 89, 1709–1716 (1994).
Sakuma, H. et al. 31P MR spectroscopy in hypertrophic cardiomyopathy: comparison with Tl-201 myocardial perfusion imaging. Am. Heart J. 125, 1323–1328 (1993).
Schaefer, S. et al. Metabolic response of the human heart to inotropic stimulation: in vivo phosphorus-31 studies of normal and cardiomyopathic myocardium. Magn. Reson. Med. 25, 260–272 (1992).
de Roos, A. et al. Cardiac metabolism in patients with dilated and hypertrophic cardiomyopathy: assessment with proton-decoupled P-31 MR spectroscopy. J. Magn. Reson. Imaging 2, 711–719 (1992).
Neubauer, S. et al. 31P magnetic resonance spectroscopy in dilated cardiomyopathy and coronary artery disease. Altered cardiac high-energy phosphate metabolism in heart failure. Circulation 86, 1810–1818 (1992).
Regitz, V. & Fleck, E. Myocardial adenine nucleotide concentrations and myocardial norepinephrine content in patients with heart failure secondary to idiopathic dilated or ischemic cardiomyopathy. Am. J. Cardiol. 69, 1574–1580 (1992).
Conway, M. A. et al. Detection of low phosphocreatine to ATP ratio in failing hypertrophied human myocardium by 31P magnetic resonance spectroscopy. Lancet 338, 973–976 (1991).
Hardy, C. J., Weiss, R. G., Bottomley, P. A. & Gerstenblith, G. Altered myocardial high-energy phosphate metabolites in patients with dilated cardiomyopathy. Am. Heart J. 122, 795–801 (1991).
Schaefer, S. et al. In vivo phosphorus-31 spectroscopic imaging in patients with global myocardial disease. Am. J. Cardiol. 65, 1154–1161 (1990).
Acknowledgements
The roundtable discussion in Stresa, Italy, was organized by Logica Med LLC and funded by an unrestricted grant from Stealth BioTherapeutics. We thank Fumiko Inoue (Logica Med) for her help in organizing the roundtable meeting. The authors also acknowledge BioCentric, Inc. for their assistance with developing previous versions of the manuscript Figures. D.A.B. has received research grants from the NIH (NHLBI R01 HL123647 and R15 HL122922) and Stealth BioTherapeutics. B.L.S. is supported by research grants from the NIH (NIA R01 AG049762, NHLBI R01 HL131458, R01HL126928, and R01HL107715) and Stealth BioTherapeutics. S.R.S. is supported by grants from the NIH (R01 HL123647, R15 HL122922, and R01 AT008375). J.B.has received research support from the NIH and the European Union. A.A.V. is supported by a grant from the European Commission (FP7-242209-BIOSTAT-CHF).
Author information
Authors and Affiliations
Contributions
D.A.B. wrote the manuscript, and all authors reviewed and edited it before submission.
Corresponding author
Ethics declarations
Competing interests
D.A.B. has received consulting income from Stealth BioTherapeutics. J.G.F.C. reports consultation with Amgen, Biotronik, GSK, Medtronic, Novartis, Servier, Sorin, and Stealth BioTherapeutics. W.S.C. is a consultant to BMS, Cardioxyl, Johnson & Johnson, Mast, Merck, and Novartis. J.B. is a consultant to Amgen, Bayer, Boehringer Ingelheim, Cardiocell, Celladon, Novartis, Relypsa, Trevena, Z Pharma, and Zensun. A.A.V. has received consultancy fees and/or research grants from Alere, Amgen, Bayer, Boehringer, Cardio3Biosciences, Celladon, GSK, Merck/MSD, Novartis, Servier, Singulex, Sphingotec, Trevena, Vifor, and ZS Pharma. B.Pieske reports speaker's bureau and/or advisory/steering committee honoraria from Abbott Vascular, AstraZeneca, Bayer Healthcare, Novartis Pharma, Servier, and Stealth BioTherapeutics. M.G. has been a consultant for Abbott Laboratories, Astellas, AstraZeneca, Bayer HealthCare, CorThera, Cytokinetics, DebioPharm, Errekappa Terapeutici, GlaxoSmithKline, Ikaria, Johnson & Johnson, Medtronic, Merck, Novartis Pharma, Otsuka Pharmaceuticals, Palatin Technologies, Pericor Therapeutics, Protein Design Laboratories, Sanofi-Aventis, Sigma Tau, Solvay Pharmaceuticals, Takeda Pharmaceutical, and Trevena Therapeutics. The other authors declare no competing interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brown, D., Perry, J., Allen, M. et al. Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol 14, 238–250 (2017). https://doi.org/10.1038/nrcardio.2016.203
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrcardio.2016.203
This article is cited by
-
Serum metabolite signatures of cardiac function and morphology in individuals from a population-based cohort
Biomarker Research (2024)
-
Upregulation of the AMPK-FOXO1-PDK4 pathway is a primary mechanism of pyruvate dehydrogenase activity reduction in tafazzin-deficient cells
Scientific Reports (2024)
-
2-APQC, a small-molecule activator of Sirtuin-3 (SIRT3), alleviates myocardial hypertrophy and fibrosis by regulating mitochondrial homeostasis
Signal Transduction and Targeted Therapy (2024)
-
A self-reinforcing cycle hypothesis in heart failure pathogenesis
Nature Cardiovascular Research (2024)
-
Metabolomic profiling identifies novel metabolites associated with cardiac dysfunction
Scientific Reports (2024)
