
Key Points
-
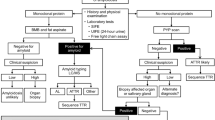
Cardiac amyloidosis should be suspected in any patient with heart failure and preserved ejection fraction or infiltrative cardiomyopathy
-
Histological diagnosis of amyloid requires further investigation to determine the protein subunit type, because the therapies vary widely
-
Preferred therapies for immunoglobulin light-chain amyloidosis involve standard-dose or high-dose chemotherapy with stem-cell rescue
-
Investigational therapies for transthyretin-related cardiomyopathy are diflunisal or tafamidis, and multiple new therapies for transthyretin-related amyloidosis and antibody therapy for immunoglobulin light-chain amyloidosis are being developed
Abstract
Amyloid cardiomyopathy should be suspected in any patient who presents with heart failure and preserved ejection fraction. In patients with echocardiographic evidence of ventricular thickening and without a clear history of hypertension, infiltrative cardiomyopathy should be considered. If imaging suggests the presence of amyloid deposits, confirmation by biopsy is required, although endomyocardial biopsy is generally not necessary. Assessment of aspirated subcutaneous fat and bone-marrow biopsy samples verifies the diagnosis in 40–80% of patients, dependent on the type of amyloidosis. Mass spectroscopy can be used to determine the protein subunit and classify the disease as immunoglobulin light-chain amyloidosis or transthyretin-related amyloidosis associated with mutant or wild-type TTR (formerly known as familial amyloid cardiomyopathy and senile cardiac amyloidosis, respectively). In this Review, we discuss the characteristics of cardiac amyloidosis, and present a structured approach to both the assessment of patients and treatment with emerging therapies and organ transplantation.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

References
Gertz, M. A. et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18–22 April 2004. Am. J. Hematol. 79, 319–328 (2005).
Monge, M. et al. Localized amyloidosis of the genitourinary tract: report of 5 new cases and review of the literature. Medicine (Baltimore) 90, 212–222 (2011).
Sommer, P., Kumar, G., Lipchik, R. J. & Patel, J. J. Tracheobronchial amyloidosis managed with multimodality therapies. Ther. Adv. Respir. Dis. 8, 48–52 (2014).
Gertz, M. A. Immunoglobulin light chain amyloidosis: update on diagnosis, prognosis, and treatment. Am. J. Hematol. 88, 416–425 (2013).
Sikkink, L. A. & Ramirez-Alvarado, M. Cytotoxicity of amyloidogenic immunoglobulin light chains in cell culture. Cell Death Dis. 1, e98 (2010).
Levinson, R. T. et al. Role of mutations in the cellular internalization of amyloidogenic light chains into cardiomyocytes. Sci. Rep. 3, 1278 (2013).
Ramirez-Alvarado, M. Amyloid formation in light chain amyloidosis. Curr. Top. Med. Chem. 12, 2523–2533 (2012).
Pinney, J. H. et al. Systemic amyloidosis in England: an epidemiological study. Br. J. Haematol. 161, 525–532 (2013).
Bhole, M. V., Sadler, R. & Ramasamy, K. Serum-free light-chain assay: clinical utility and limitations. Ann. Clin. Biochem. 51, 528–542 (2014).
Kourelis, T. V. et al. Coexistent multiple myeloma or increased bone marrow plasma cells define equally high-risk populations in patients with immunoglobulin light chain amyloidosis. J. Clin. Oncol. 31, 4319–4324 (2013).
Kyle, R. A. & Gertz, M. A. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin. Hematol. 32, 45–59 (1995).
Anrade, C. A peculiar form of peripheral neuropathy: familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 75, 408–427 (1952).
Azevedo, E. M., Scaff, M., Canelas, H. M. & Spina-Franca, A. Type I primary neuropathic amyloidosis [Portuguese]. Arq. Neuropsiquiatr. 33, 105–118 (1975).
Rowczenio, D. & Wechalekar, A. Mutations in hereditary amyloidosis [online], (2010).
Saraiva, M. J. Transthyretin mutations in health and disease. Hum. Mutat. 5, 191–196 (1995).
Rapezzi, C. et al. Gender-related risk of myocardial involvement in systemic amyloidosis. Amyloid 15, 40–48 (2008).
Zeldenrust, S. R. Genotype–phenotype correlation in FAP. Amyloid 19 (Suppl. 1), 22–24 (2012).
Sattianayagam, P. T. et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur. Heart J. 33, 1120–1127 (2012).
Arruda-Olson, A. M. et al. Genotype, echocardiography, and survival in familial transthyretin amyloidosis. Amyloid 20, 263–268 (2013).
Ruberg, F. L. et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). Am. Heart J. 164, 222–228.e1 (2012).
Reddi, H. V. et al. Homozygosity for the V122I mutation in transthyretin is associated with earlier onset of cardiac amyloidosis in the African American population in the seventh decade of life. J. Mol. Diagn. 16, 68–74 (2014).
Pukitis, A. et al. Effect of infliximab induction therapy on secondary systemic amyloidosis associated with Crohn's disease: case report and review of the literature. J. Gastrointestin. Liver Dis. 22, 333–336 (2013).
Kristen, A. V. et al. Transthyretin valine-94-alanine, a novel variant associated with late-onset systemic amyloidosis with cardiac involvement. Amyloid 14, 283–287 (2007).
Pinney, J. H. et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J. Am. Heart Assoc. 2, e000098 (2013).
Swiecicki, P. L. et al. Hereditary amyloidosis: a single-institution experience with 284 patients [abstract OP-53]. Presented at the XIVth International Symposium on Amyloidosis.
Ng, B., Connors, L. H., Davidoff, R., Skinner, M. & Falk, R. H. Senile systemic amyloidosis presenting with heart failure: a comparison with light chain-associated amyloidosis. Arch. Intern. Med. 165, 1425–1429 (2005).
Takeda, M. et al. MRI differentiation of cardiomyopathy showing left ventricular hypertrophy and heart failure: differentiation between cardiac amyloidosis, hypertrophic cardiomyopathy, and hypertensive heart disease. Jpn. J. Radiol. 31, 693–700 (2013).
Mookadam, F., Haley, J. H., Olson, L. J., Cikes, M. & Mookadam, M. Dynamic left ventricular outflow tract obstruction in senile cardiac amyloidosis. Eur. J. Echocardiogr. 7, 465–468 (2006).
Potysova, Z. et al. Renal AA amyloidosis: survey of epidemiologic and laboratory data from one nephrology centre. Int. Urol. Nephrol. 41, 941–945 (2009).
Girnius, S., Dember, L., Doros, G. & Skinner, M. The changing face of AA amyloidosis: a single center experience. Amyloid 18 (Suppl. 1), 226–228 (2011).
Browning, M. J. et al. Ten years' experience of an amyloid clinic: a clinicopathological survey. Q. J. Med. 54, 213–227 (1985).
Louros, N. N. et al. An N-terminal pro-atrial natriuretic peptide (NT-proANP) 'aggregation-prone' segment involved in isolated atrial amyloidosis. FEBS Lett. 588, 52–57 (2014).
Podduturi, V., Armstrong, D. R., Hitchcock, M. A., Roberts, W. C. & Guileyardo, J. M. Isolated atrial amyloidosis and the importance of molecular classification. Proc. (Bayl. Univ. Med. Cent.) 26, 387–389 (2013).
Millucci, L. et al. Prevalence of isolated atrial amyloidosis in young patients affected by congestive heart failure. ScientificWorldJournal 2012, 293863 (2012).
Ariyarajah, V. et al. The association of atrial tachyarrhythmias with isolated atrial amyloid disease: preliminary observations in autopsied heart specimens. Cardiology 113, 132–137 (2009).
Steensma, D. P. “Congo” red: out of Africa? Arch. Pathol. Lab. Med. 125, 250–252 (2001).
Benson, M. D., Breall, J., Cummings, O. W. & Liepnieks, J. J. Biochemical characterisation of amyloid by endomyocardial biopsy. Amyloid 16, 9–14 (2009).
Arbustini, E. et al. Cardiac immunocyte-derived (AL) amyloidosis: an endomyocardial biopsy study in 11 patients. Am. Heart J. 130, 528–536 (1995).
Sloan, K. P., Bruce, C. J., Oh, J. K. & Rihal, C. S. Complications of echocardiography-guided endomyocardial biopsy. J. Am. Soc. Echocardiogr. 22, 324.e1–324.e4 (2009).
Gertz, M. A. Immunoglobulin light chain amyloidosis: update on diagnosis, risk-stratification, and management. Am. J. Hematol. 86, 180–186 (2011).
Fine, N. M. et al. Yield of noncardiac biopsy for the diagnosis of transthyretin cardiac amyloidosis. Am. J. Cardiol. 113, 1723–1727 (2014).
Brambilla, F., Lavatelli, F., Merlini, G. & Mauri, P. Clinical proteomics for diagnosis and typing of systemic amyloidoses. Proteomics Clin. Appl. 7, 136–143 (2013).
Chee, C. E., Lacy, M. Q., Dogan, A., Zeldenrust, S. R. & Gertz, M. A. Pitfalls in the diagnosis of primary amyloidosis. Clin. Lymphoma Myeloma Leuk. 10, 177–180 (2010).
Maleszewski, J. J. et al. Relationship between monoclonal gammopathy and cardiac amyloid type. Cardiovasc. Pathol. 22, 189–194 (2013).
Paueksakon, P., Fogo, A. B. & Sethi, S. Leukocyte chemotactic factor 2 amyloidosis cannot be reliably diagnosed by immunohistochemical staining. Hum. Pathol. 45, 1445–1450 (2014).
Mollee, P., Renaut, P., Gottlieb, D. & Goodman, H. How to diagnose amyloidosis. Intern. Med. J. 44, 7–17 (2014).
Hoshii, Y., Nanbara, H., Cui, D., Takahashi, M. & Ikeda, E. Immunohistochemical examination of Aκ amyloidosis with antibody against adjacent portion of the carboxy terminus of immunoglobulin kappa light chain. Med. Mol. Morphol. 45, 124–128 (2012).
Satoskar, A. A. et al. Strong transthyretin immunostaining: potential pitfall in cardiac amyloid typing. Am. J. Surg. Pathol. 35, 1685–1690 (2011).
Vrana, J. A. et al. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood 114, 4957–4959 (2009).
Nasr, S. H. et al. The diagnosis and characteristics of renal heavy-chain and heavy/light-chain amyloidosis and their comparison with renal light-chain amyloidosis. Kidney Int. 83, 463–470 (2013).
Laffer, U. Intra-portal chemoprevention and therapy of liver metastases [German]. Z. Gastroenterol. Verh. 24, 189–191 (1989).
Theis, J. D. et al. Proteome of amyloidosis: Mayo Clinic experience in 4139 cases [abstract OP-19] [online], (2014).
Guan, J. et al. Stanniocalcin1 is a key mediator of amyloidogenic light chain induced cardiotoxicity. Basic Res. Cardiol. 108, 378 (2013).
Shi, J. et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38α MAPK pathway. Proc. Natl Acad. Sci. USA 107, 4188–4193 (2010).
Mohammed, S. F. et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2, 113–122 (2014).
Murtagh, B. et al. Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Am. J. Cardiol. 95, 535–537 (2005).
Mohty, D. et al. Cardiac amyloidosis: updates in diagnosis and management. Arch. Cardiovasc. Dis. 106, 528–540 (2013).
Russo, C., Green, P. & Maurer, M. The prognostic significance of central hemodynamics in patients with cardiac amyloidosis. Amyloid 20, 199–203 (2013).
Wittich, C. M., Neben-Wittich, M. A., Mueller, P. S., Gertz, M. A. & Edwards, W. D. Deposition of amyloid proteins in the epicardial coronary arteries of 58 patients with primary systemic amyloidosis. Cardiovasc. Pathol. 16, 75–78 (2007).
Seward, J. B. & Casaclang-Verzosa, G. Infiltrative cardiovascular diseases: cardiomyopathies that look alike. J. Am. Coll. Cardiol. 55, 1769–1779 (2010).
Bellavia, D. et al. Detection of left ventricular systolic dysfunction in cardiac amyloidosis with strain rate echocardiography. J. Am. Soc. Echocardiogr. 20, 1194–1202 (2007).
Buss, S. J. et al. Longitudinal left ventricular function for prediction of survival in systemic light-chain amyloidosis: incremental value compared with clinical and biochemical markers. J. Am. Coll. Cardiol. 60, 1067–1076 (2012).
Bellavia, D. et al. Evidence of impaired left ventricular systolic function by Doppler myocardial imaging in patients with systemic amyloidosis and no evidence of cardiac involvement by standard two-dimensional and Doppler echocardiography. Am. J. Cardiol. 101, 1039–1045 (2008).
Al-Zahrani, G. B. et al. Doppler myocardial imaging compared to standard two-dimensional and Doppler echocardiography for assessment of diastolic function in patients with systemic amyloidosis. J. Am. Soc. Echocardiogr. 22, 290–298 (2009).
Nesbitt, G. C. & Mankad, S. Strain and strain rate imaging in cardiomyopathy. Echocardiography 26, 337–344 (2009).
Bellavia, D. et al. Independent predictors of survival in primary systemic (AL) amyloidosis, including cardiac biomarkers and left ventricular strain imaging: an observational cohort study. J. Am. Soc. Echocardiogr. 23, 643–652 (2010).
Bellavia, D. et al. Comparison of right ventricular longitudinal strain imaging, tricuspid annular plane systolic excursion, and cardiac biomarkers for early diagnosis of cardiac involvement and risk stratification in primary systematic (AL) amyloidosis: a 5-year cohort study. Eur. Heart J. Cardiovasc. Imaging 13, 680–689 (2012).
Lee, G. Y. et al. Cardiac amyloidosis without increased left ventricular wall thickness. Mayo Clin. Proc. 89, 781–789 (2014).
Suresh, R. et al. Advanced cardiac amyloidosis associated with normal interventricular spetal thickness: an uncommon presentation of infiltrative cardiomyopathy. J. Am. Soc. Echocardiogr. 27, 440–447 (2014).
Hazenberg, B. P. et al. Diagnostic performance and prognostic value of extravascular retention of 123I-labeled serum amyloid P component in systemic amyloidosis. J. Nucl. Med. 48, 865–872 (2007).
Hawkins, P. N. et al. Scintigraphic imaging and turnover studies with iodine-131 labelled serum amyloid P component in systemic amyloidosis. Eur. J. Nucl. Med. 25, 701–708 (1998).
Sachchithanantham, S. & Wechalekar, A. D. Imaging in systemic amyloidosis. Br. Med. Bull. 107, 41–56 (2013).
Bokhari, S. et al. 99mTc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ. Cardiovasc. Imaging 6, 195–201 (2013).
Gertz, M. A., Brown, M. L., Hauser, M. F. & Kyle, R. A. Utility of technetium Tc 99m pyrophosphate bone scanning in cardiac amyloidosis. Arch. Intern. Med. 147, 1039–1044 (1987).
Aljaroudi, W. A. et al. Role of imaging in the diagnosis and management of patients with cardiac amyloidosis: state of the art review and focus on emerging nuclear techniques. J. Nucl. Cardiol. 21, 271–283 (2014).
Storandt, M., Mintun, M. A., Head, D. & Morris, J. C. Cognitive decline and brain volume loss as signatures of cerebral amyloid-β peptide deposition identified with Pittsburgh compound B: cognitive decline associated with Abeta deposition. Arch. Neurol. 66, 1476–1481 (2009).
Antoni, G. et al. In vivo visualization of amyloid deposits in the heart with 11C-PIB and PET. J. Nucl. Med. 54, 213–220 (2013).
Wang, J. et al. Noninvasive diagnosis of cardiac amyloidosis by MRI and echochardiography. J. Huazhong Univ. Sci. Technolog. Med. Sci. 30, 536–540 (2010).
Cheng, A. S., Banning, A. P., Mitchell, A. R., Neubauer, S. & Selvanayagam, J. B. Cardiac changes in systemic amyloidosis: visualisation by magnetic resonance imaging. Int. J. Cardiol. 113, E21–E23 (2006).
Aquaro, G. D. et al. Myocardial signal intensity decay after gadolinium injection: a fast and effective method for the diagnosis of cardiac amyloidosis. Int. J. Cardiovasc. Imaging 30, 1105–1115 (2014).
Pouchot, J. & Arlet, J. B. Biological treatment in adult-onset Still's disease. Best Pract. Res. Clin. Rheumatol. 26, 477–487 (2012).
Rubinshtein, R. et al. Comparison of magnetic resonance imaging versus Doppler echocardiography for the evaluation of left ventricular diastolic function in patients with cardiac amyloidosis. Am. J. Cardiol. 103, 718–723 (2009).
Syed, I. S. et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc. Imaging 3, 155–164 (2010).
Giesbrandt, K. J., Bolan, C. W., Shapiro, B. P., Edwards, W. D. & Mergo, P. J. Diffuse diseases of the myocardium: MRI-pathologic review of cardiomyopathies with dilatation. Am. J. Roentgenol. 200, W274–W282 (2013).
Harvey-Taylor, J., Zhang, Y., Kuderer, V. & Cooke, D. T. Diagnosis of systemic amyloidosis and amyloidosis mediated cardiomyopathy by VATS pleural biopsy for chronic pleural effusion. J. Thorac. Dis. 5, E112–E114 (2013).
Finocchiaro, G. et al. Long term survival in patients with cardiac amyloidosis: prevalence and characterisation during follow-up. Heart Lung Circ. 22, 647–654 (2013).
Chaulagain, C. P. & Comenzo, R. L. New insights and modern treatment of AL amyloidosis. Curr. Hematol. Malig. Rep. 8, 291–298 (2013).
Dispenzieri, A. et al. Prognostication of survival using cardiac troponins and N-terminal pro-brain natriuretic peptide in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood 104, 1881–1887 (2004).
Gertz, M. A. et al. Clinical outcome of immunoglobulin light chain amyloidosis affecting the kidney. Nephrol. Dial. Transplant. 24, 3132–3137 (2009).
Park, M. A. et al. Primary (AL) hepatic amyloidosis: clinical features and natural history in 98 patients. Medicine (Baltimore) 82, 291–298 (2003).
Rajkumar, S. V., Gertz, M. A. & Kyle, R. A. Prognosis of patients with primary systemic amyloidosis who present with dominant neuropathy. Am. J. Med. 104, 232–237 (1998).
Dispenzieri, A. et al. High sensitivity cardiac troponin T in patients with immunoglobulin light chain amyloidosis. Heart 100, 383–388 (2014).
Kumar, S. et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J. Clin. Oncol. 30, 989–995 (2012).
Halwani, O. & Delgado, D. H. Cardiac amyloidosis: an approach to diagnosis and management. Expert Rev. Cardiovasc. Ther. 8, 1007–1013 (2010).
Nash, K. L., Brij, S. O. & Clesham, G. J. Cardiac amyloidosis and the use of diuretic and ACE inhibitor therapy in severe heart failure. Int. J. Clin. Pract. 51, 384–385 (1997).
Bouhour, J. B., Haddak, M. & Lefevre, M. Risks of beta-blockers and calcium inhibitors in amyloid cardiopathy [French]. Presse Med. 15, 981 (1986).
Desport, E. et al. AL amyloidosis. Orphanet J. Rare Dis. 7, 54 (2012).
Lin, G., Dispenzieri, A., Kyle, R., Grogan, M. & Brady, P. A. Implantable cardioverter defibrillators in patients with cardiac amyloidosis. J. Cardiovasc. Electrophysiol. 24, 793–798 (2013).
Hess, E. P. & White, R. D. Out-of-hospital cardiac arrest in patients with cardiac amyloidosis: presenting rhythms, management and outcomes in four patients. Resuscitation 60, 105–111 (2004).
Swiecicki, P. L. et al. Left ventricular device implantation for advanced cardiac amyloidosis. J. Heart Lung Transplant. 32, 563–568 (2013).
Feng, D. et al. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation 116, 2420–2426 (2007).
Zubkov, A. Y., Rabinstein, A. A., Dispenzieri, A. & Wijdicks, E. F. Primary systemic amyloidosis with ischemic stroke as a presenting complication. Neurology 69, 1136–1141 (2007).
Feng, D. et al. Intracardiac thrombosis and anticoagulation therapy in cardiac amyloidosis. Circulation 119, 2490–2497 (2009).
Kumar, S. K. et al. Recent improvements in survival in primary systemic amyloidosis and the importance of an early mortality risk score. Mayo Clin. Proc. 86, 12–18 (2011).
Gertz, M. A. How to manage primary amyloidosis. Leukemia 26, 191–198 (2012).
Nelson, M. R. et al. Histologic remission of cardiac amyloidosis: a case report. Amyloid 19, 106–109 (2012).
Palladini, G. et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J. Clin. Oncol. 30, 4541–4549 (2012).
Merlini, G., Seldin, D. C. & Gertz, M. A. Amyloidosis: pathogenesis and new therapeutic options. J. Clin. Oncol. 29, 1924–1933 (2011).
Kumar, S. K. et al. Lenalidomide, cyclophosphamide, and dexamethasone (CRd) for light-chain amyloidosis: long-term results from a phase 2 trial. Blood 119, 4860–4867 (2012).
Tapan, U. et al. Increases in B-type natriuretic peptide (BNP) during treatment with lenalidomide in AL amyloidosis. Blood 116, 5071–5072 (2010).
Mikhael, J. R. et al. Cyclophosphamide-bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyloidosis. Blood 119, 4391–4394 (2012).
Landau, H. et al. Bortezomib and dexamethasone consolidation following risk-adapted melphalan and stem cell transplantation for patients with newly diagnosed light-chain amyloidosis. Leukemia 27, 823–828 (2013).
Gertz, M. et al. Troponin T level as an exclusion criterion for stem cell transplantation in light-chain amyloidosis. Leuk. Lymphoma 49, 36–41 (2008).
Gertz, M. A. et al. Trends in day 100 and 2-year survival after auto-SCT for AL amyloidosis: outcomes before and after 2006. Bone Marrow Transplant. 46, 970–975 (2011).
Gertz, M. A. et al. Trend toward improved day 100 and two-year survival following stem cell transplantation for AL: a comparison before and after 2006. Amyloid 18 (Suppl. 1), 137–138 (2011).
Bellavia, D. et al. Utility of Doppler myocardial imaging, cardiac biomarkers, and clonal immunoglobulin genes to assess left ventricular performance and stratify risk following peripheral blood stem cell transplantation in patients with systemic light chain amyloidosis (AL). J. Am. Soc. Echocardiogr. 24, 444–454 (2011).
Singla, A. et al. Incidence of supraventricular arrhythmias during autologous peripheral blood stem cell transplantation. Biol. Blood Marrow Transplant. 19, 1233–1237 (2013).
Bleeker, J. S. et al. Evaluation of pretransplant factors predicting cardiac dysfunction following high-dose melphalan conditioning and autologous peripheral blood stem cell transplantation. Eur. J. Haematol. 89, 228–235 (2012).
Madan, S. et al. High-dose melphalan and peripheral blood stem cell transplantation for light-chain amyloidosis with cardiac involvement. Blood 119, 1117–1122 (2012).
Gertz, M. A. et al. Refinement in patient selection to reduce treatment-related mortality from autologous stem cell transplantation in amyloidosis. Bone Marrow Transplant. 48, 557–561 (2013).
Jimenez-Zepeda, V. H. et al. Autologous stem cell transplant is an effective therapy for carefully selected patients with AL amyloidosis: experience of a single institution. Br. J. Haematol. 164, 722–728 (2014).
Dispenzieri, A. et al. The activity of lenalidomide with or without dexamethasone in patients with primary systemic amyloidosis. Blood 109, 465–470 (2007).
Dispenzieri, A. et al. Discordance between serum cardiac biomarker and immunoglobulin-free light-chain response in patients with immunoglobulin light-chain amyloidosis treated with immune modulatory drugs. Am. J. Hematol. 85, 757–759 (2010).
Dispenzieri, A. et al. Activity of pomalidomide in patients with immunoglobulin light-chain amyloidosis. Blood 119, 5397–5404 (2012).
Liedtke, M. et al. Preliminary cardiac biomarker responses demonstrated in an ongoing phase I study of NEOD001 in patients with AL amyloidosis and persistent organ dysfunction [abstract PB-48] [online], (2014).
Gertz, M. A. & Dispenzieri, A. Immunoglobulin light-chain amyloidosis: growing recognition, new approaches to therapy, active clinical trials. Oncology (Williston Park) 26, 152–161 (2012).
Lacy, M. Q. et al. Autologous stem cell transplant after heart transplant for light chain (AL) amyloid cardiomyopathy. J. Heart Lung Transplant. 27, 823–829 (2008).
Gray Gilstrap, L. et al. Predictors of survival to orthotopic heart transplant in patients with light chain amyloidosis. J. Heart Lung Transplant. 33, 149–156 (2014).
Varr, B. C. et al. Heart transplantation and cardiac amyloidosis: approach to screening and novel management strategies. J. Heart Lung Transplant. 31, 325–331 (2012).
Raichlin, E. et al. Combined heart and liver transplantation: a single-center experience. Transplantation 88, 219–225 (2009).
Merlini, G. et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J. Cardiovasc. Transl. Res. 6, 1011–1020 (2013).
Coelho, T. et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J. Neurol. 260, 2802–2814 (2013).
Merlini, G. et al. Survival in patients with transthyretin familial amyloid polyneuropathy receiving tafamidis treatment [abstract OP-65]. Presented at the XIVth International Symposium on Amyloidosis.
Maurer, M. S., Judge, D. P., Rosas, G. R., Mandel, F. S. & Aarts, J. Interim analysis of long-term, open-label tafamidis treatment in transthyretin amyloid cardiomyopathy after up to 5 years of treatment [abstract OP-66]. Presented at the XIVth International Symposium on Amyloidosis.
Coelho, T. et al. Familial amyloid polyneuropathy treatment with tafamidis: evaluation of one year treatment at Porto, Portugal [abstract OP-67]. Presented at the XIVth International Symposium on Amyloidosis.
Obici, L. et al. A phase II study of doxycycline plus tauroursodeoxycholic acid in transthyretin amyloidosis [abstract OP-68]. Presented at the XIVth International Symposium on Amyloidosis.
Quarta, C. C. et al. The prevalence of cardiac amyloidosis in familial amyloidotic polyneuropathy with predominant neuropathy: the Diflunisal Trial [abstract OP-69]. Presented at the XIVth International Symposium on Amyloidosis.
Berk, J. L. et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 310, 2658–2667 (2013).
Ackermann, E. J., Hughes, S., Yamashita, M. & Monia, B. P. Clinical development of ISIS-TTRRx: a second generation antisense therapy for the treatment of transthyretin-associated diseases [abstract OP-71]. Presented at the XIVth International Symposium on Amyloidosis.
Suhr, O. et al. Further analysis of phase II trial of patisiran, a novel RNAi therapeutic for the treatment of familial amyloidotic polyneuropathy [abstract OP-72]. Presented at the XIVth International Symposium on Amyloidosis.
Author information
Authors and Affiliations
Contributions
M.A.G. researched data for the article, discussed its content, and wrote, reviewed, and edited the manuscript before submission. A.D. and T.S. also wrote the manuscript, and reviewed and edited it before submission.
Corresponding author
Ethics declarations
Competing interests
M.A.G. declares that he has received honoraria from Celgene, ISIS, Millennium, Neotope, Novartis, and Onyx. A.D. declares that she has received research funding from Celgene, Janssen, Millennium, and Pfizer. T.S. declares no competing interests.
Rights and permissions
About this article

Cite this article
Gertz, M., Dispenzieri, A. & Sher, T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol 12, 91–102 (2015). https://doi.org/10.1038/nrcardio.2014.165
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrcardio.2014.165
This article is cited by
-
Targeted sequencing of selected functional genes in patients with wild-type transthyretin amyloidosis
BMC Research Notes (2023)
-
Coronary microvascular disease in hypertrophic and infiltrative cardiomyopathies
Journal of Nuclear Cardiology (2023)
-
Late gadolinium enhanced cardiac MR derived radiomics approach for predicting all-cause mortality in cardiac amyloidosis: a multicenter study
European Radiology (2023)
-
Pulmonary 99mTc-HMDP uptake correlates with restrictive ventilatory defects and abnormal lung reactance in transthyretin cardiac amyloidosis patients
Respiratory Research (2022)
-
The roles of global longitudinal strain imaging in contemporary clinical cardiology
Journal of Medical Ultrasonics (2022)
