
Abstract
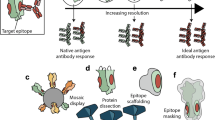
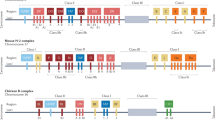
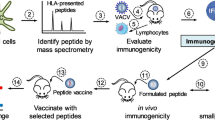
The Global HIV Vaccine Enterprise convened a meeting of a Working Group in July 2009 to discuss recent progress in rational design of the components of an HIV vaccine, such as inserts, vectors and adjuvants,and in understanding antigen processing and presentation to T and B cells. This Report summarizes the key points of that discussion, and subsequent discussions with the Chairs of the other Enterprise Working Groups, the Enterprise Science Committee, the Enterprise Council and the broader scientific community during open sessions at scientific conferences.
Similar content being viewed by others
Article PDF
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mascola, J., King, R., Steinman, R. et al. Immunogens and Antigen Processing: Report from a Global HIV Vaccine Enterprise Working Group. Nat Prec (2010). https://doi.org/10.1038/npre.2010.4796.1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/npre.2010.4796.1
