
Abstract
Petunia hybrida is a popular bedding plant that has a long history as a genetic model system. We report the whole-genome sequencing and assembly of inbred derivatives of its two wild parents, P. axillaris N and P. inflata S6. The assemblies include 91.3% and 90.2% coverage of their diploid genomes (1.4 Gb; 2n = 14) containing 32,928 and 36,697 protein-coding genes, respectively. The genomes reveal that the Petunia lineage has experienced at least two rounds of hexaploidization: the older gamma event, which is shared with most Eudicots, and a more recent Solanaceae event that is shared with tomato and other solanaceous species. Transcription factors involved in the shift from bee to moth pollination reside in particularly dynamic regions of the genome, which may have been key to the remarkable diversity of floral colour patterns and pollination systems. The high-quality genome sequences will enhance the value of Petunia as a model system for research on unique biological phenomena such as small RNAs, symbiosis, self-incompatibility and circadian rhythms.
Similar content being viewed by others


The garden petunia, Petunia hybrida, with its diversity of colour and morphology is the world's most popular bedding plant with an annual wholesale value exceeding US$130 million in the USA alone1. Petunia has a long history as a model species for scientific research. To the scientific community, Petunia is best known for the discovery of RNAi2,3. This breakthrough was the culmination of decades-long research on the synthesis and regulation of the floral pigments and as a consequence anthocyanin biosynthesis remains one of the best-known pathways of secondary metabolism in any plant species4. Development, transposon activity, genetic self-incompatibility, and interactions with microbes, herbivores and pollinators have also been active research topics utilizing Petunia as model system.
The genus Petunia is a member of the Solanaceae family native to South America. It forms a separate and early branching clade within the family with a base chromosome number of x = 7 rather than the typical x = 12 found for most Solanaceae crown-group species, including important crops such as tomato, potato, tobacco, pepper and eggplant5. The commercial P. hybrida is derived from crosses between a white-flowered, moth-pollinated P. axillaris, and species of the P. integrifolia clade, a group of closely related bee-pollinated species and subspecies (Fig. 1)6,7. The first hybrids were produced by European horticulturalists in the early nineteenth century, probably multiple times from different accessions of the two parent clades7,8. The remarkable phenotypic diversity in today's commercial garden petunias is the result of almost two centuries of intense commercial breeding. Here, we present the genome sequences of P. axillaris N and P. inflata S6, two inbred laboratory accessions representing the parents of P. hybrida (Fig. 1).

a, P. inflata S6, P. axillaris N and their F1. b, Selected individuals from P. inflata S6 × P. axillaris N F2 population. c, Commercial P. hybrida accessions. d, P. hybrida accessions and mutants. Row 1, from left to right: Mitchell (W115); R27; transposon line W138; R143; vacuolar ph3 mutant with pale colour compared with the isogenic R143. Mitchell, R27 and R143 were used for transcriptomics analysis. Row 2, from left to right: V26; V26 with CHS RNAi transgene (images provided by J. Kooter, VU Amsterdam); homeotic mutant pMADS3RNAi/fbp6; an2 mutant; homeotic mutant blind.
Results and discussion
Sequencing, assembly and annotation
For P. axillaris N, we performed a hybrid de novo assembly using a combination of short read (Illumina; coverage 137X) and long read technologies (PacBio; coverage 21X), whereas for P. inflata S6 we produced exclusively short reads (Illumina; coverage 135X) and performed a short read de novo assembly (for details see Supplementary Note 1). The resulting high-quality assemblies have a size of 1.26 Gb for P. axillaris and 1.29 Gb for P. inflata (Table 1). The estimated size of both genomes is 1.4 Gb, using a k-mer size of 31, which is consistent with previous microdensity measurements9. We have remapped Illumina reads to the assemblies and called single nucleotide polymorphism (SNPs) to estimate the level of heterozygosity, which is estimated as 0.03% for both accessions. Moreover, we mapped the 248 Core Eukaryotic Genes (CEGs) to assess the completeness of both assemblies and found 239 (94%) and 243 (98%) in the assembly of P. axillaris and P. inflata, respectively. The estimated unassembled fraction of the genome comprises ∼140 Mb for P. axillaris (181 Mb if sequence gaps of 41 Mb are included) and ∼110 Mb for P. inflata (197 Mb with sequence gaps of 87 Mb), which is likely to be due to the large numbers of repetitive sequences (see below). Genome annotation identified 32,928 protein-coding genes for P. axillaris and 36,697 protein-coding genes for P. inflata with an average of 5.2 and 5.1 exons per protein coding gene and an average predicted protein size of 393 and 386 amino acids, respectively.
Repeat landscape of Petunia genomes
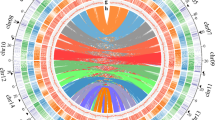
Petunia genomes are rich in repetitive DNA (as are most other plant genomes), but its presence at 60–65% of the assembled genome is relatively low considering its genome size (Fig. 2a; Supplementary Note 2), indicating a larger gene, regulatory and low copy sequence space. Long terminal repeats (LTR)-retroelement-related sequences are abundant near centromeres (Fig. 2b), and within the assemblies, equal numbers of fragments and full-length Ty3/Gypsy-like and Ty1/Copia-like elements were detected. Repeat cluster analysis of unassembled reads supported the amount and complexity of the diverse and rearranged repeat landscape of Petunia. Petunia chromosomes average 200 Mb in length (three times that of Solanum lycopersicum or S. tuberosum), as a larger genome is distributed over 7 rather than 12 chromosomes (Fig. 2b). Chromosomal organization in Petunia is thus different compared to other Solanaceae and this together with high DNA transposon frequency and mobility has an effect on genome evolution, meiotic recombination and homogenization events10.

a, Comparative genome organization of Solanum lycopersicum, P. axillaris and Nicotiana tomentosiformis. The circles are proportional to genome size; regulatory sequences and repeat classes are shown in the segments19,29. b, Fluorescent in situ hybridization (FISH) to P. axillaris chromosomes (grey). Red: four pericentromeric Petunia vein clearing virus (PVCV) sites; green: dispersed Gypsy-like retroelement junction probe at all centromeres (overlapping yellow signals); blue: 5S rDNA. Scale bar, 10 µm. c, Distribution of dTph1-like transposons in P. axillaris and P. inflata. d, Duplicated gene families in functional categories showing Petunia-specific and balanced families. e, Venn diagram based on the gene family cluster analysis from five Solanaceae species. The numbers below the species name indicate the number of protein-coding genes (top) and number of gene family clusters (bottom).
DNA transposons
DNA transposons are five times more abundant in the Petunia genome than in Nicotiana tomentosiformis and S. lycopersicum (Fig. 2a). The identification and cloning of the small endogenous non-autonomous hAT-like defective transposon of petunia hybrida1 (dTph1), which is highly mobile in the P. hybrida line W138 (Fig. 1d), has allowed the development of efficient tools for forward and reverse genetics11. The P. axillaris and P. inflata genomes contain 16 and 21 dTph1 copies, respectively (Fig. 2c and Supplementary Note 3). This is similar to the numbers in most P. hybrida accessions, but far fewer than in the old P. hybrida accession R27 or the hyperactive accession W138 with over 200 copies. Comparison of dTph1 insertion loci in P. axillaris and P. inflata with W138 provides evidence that both species indeed contributed to W138. dTph1 distribution patterns in wild P. axillaris accessions from Uruguay showed comparable low dTph1 copy numbers and a very low overall locus diversity, suggesting that dTph1 transposition activity is largely suppressed in natural populations, but was reactivated after the interspecific crosses leading to the domesticated P. hybrida. Seven previously identified dTph1-like elements and one newly discovered element, dTPh12, are present in both genomes, demonstrating their ancient origin (Fig. 2c.) The expansion of different transposable elements—dTph1 in W138 and dTph7 in the two wild species—suggests that, despite extensive homology in their terminal inverted repeat regions, they may require different transacting factors for their mobility.
Endogenous pararetroviruses
Integrated copies of Caulimoviridae are widespread in plant nuclear genomes including the Solanaceae12. These DNA viruses are characterized by a gag region with RNA binding domains and a pol region that codes for reverse transcriptase and RNase H (ref. 13). The P. axillaris and P. inflata genomes show near-complete but also degenerated and rearranged copies of Petunia vein clearing virus (PVCV, a Petuvirus14; Supplementary Note 2). Their structures suggest that the behaviour and mode of integration are similar for both species, and parallel the types of complex rearrangements seen in the banana genome15. Fluorescent in situ hybridization of these sequences (Fig. 2b) showed signals near the centromeres of two chromosome pairs in P. axillaris adjacent to LTR retroelements. Phylogenetic analysis of single insertions showed repeated incidents of homogenization. Such homologous sequences contributed to the tandem array structures found in P. hybrida that are prerequisites of inducible and disease generating viruses14.
Gene families and tandem duplications
Polypeptide sequences from P. axillaris, P. inflata, S. lycopersicum, S. tuberosum, Nicotiana benthamiana and Arabidopsis thaliana were clustered into gene families. This analysis (Supplementary Note 4) grouped 39.2% of the genes into 27,600 gene families, ranging in size from 2 to 1,026 members. Most gene families followed the accepted evolutionary lineage (Fig. 3a), with the Petunia, Solanum and Solanaceae clades sharing gene families far more often than other species groupings (Fig. 2e). Two contrasting sets of gene families that are almost mutually exclusive were found: Petunia-specific families and balanced shared families (Fig. 2d). The size distributions of tandem gene arrays in P. axillaris, P. inflata and S. lycopersicum were quite similar, with each species containing about 8,000 genes in 3,000 tandem arrays.

a, Paleohexapolyploid history of the Solanaceae family, showing the gamma hexaploidy event shared with most eudicots and the family-specific Solanaceae-α hexaploidy event. We place Solanaceae-α before the divergence of Petunia and the x = 12 crown-group (∼30 and 49 Myr ago (Ma))5. b, Differential gene fractionation of Petunia (P. axillaris, shortened to P. axi.) and tomato (S. lycopersicum, S. lyco.) in comparison with grape (V. vitifera). One grape genomic region is syntenic to three regions of Petunia and tomato. Genes in red represent shared-retained genes of Petunia and tomato whereas green (retained in Petunia/lost in tomato) and purple (retained in tomato/lost in Petunia) represent independently fractionated genes. For details see Supplementary Note 5.
Paleopolyploidy history of Petunia
Analysis of the Petunia data allowed us to infer the history of polyploidy not only for Petunia but for the entire Solanaceae. Polyploidy is ubiquitous among angiosperms, with many independent lineage-specific paleopolyploidy events associated with changes in genome structure and gene retention and loss16,17. Most paleopolyploidy events are the result of ancient genome duplications (paleotetraploidies), but ancient triplications (paleohexaploidies) have also been identified, for example the gamma event near the origin of Eudicots (Fig. 3a) first detected by analysis of the Vitis vinifera (grape) genome18. Similarly, genome analysis of S. lycopersicum suggested that there was a triplication at some point during the evolution of the Solanaceae family19. Petunia as a sister to the x = 12 crown-group clade of the Solanaceae5 is an ideal species to investigate the timing and nature of this event (Fig. 3a).
Using whole-genome synteny analyses of our de novo assemblies, we identified genomic regions of collinearity between S. lycopersicum and P. axillaris, using V. vinifera as an outgroup (Supplementary Note 5). Inferring their relative timing by analysing synonymous changes (Ks), we show that Petunia shares the older gamma paleopolyploidy event with other higher eudicots, and the more recent paleohexaploidy event with S. lycopersicum. We then can infer that the Solanaceae event occurred at least 30 Myr ago (Fig. 3a). Microsynteny analysis shows the process of gene fractionation following the polyploidization event, and reveals that the S. lycopersicum genome has retained fewer genes than the Petunia genome, thus contributing to the relatively large genic fraction found in Petunia (Fig. 2a). From the fractionation patterns observed, (Fig. 3b), we predict a first and common incomplete gene fractionation step in both Petunia and S. lycopersicum and a second step after their divergence in S. lycopersicum only. This may have contributed to the separation of the lineages, similar to that observed in Saccharomyces yeasts20 but until now not yet described in flowering plants.
Origin of the P. hybrida genomes
Comparisons of the two genome sequences with transcriptomics data from three unrelated P. hybrida lines, namely Mitchell, R27 and R143 (Fig. 1d, see Supplementary Note 6) revealed a complex history of the garden petunia. The majority of the ∼20,000 analysed genes could be assigned to P. axillaris (∼15,000), with only ∼600 genes assigned to P. inflata. This indicates that the P. inflata parent makes only a minor contribution to the P. hybrida gene space. One possible explanation for this preponderance of the white parent genome could be that breeding for different colours and colour patterns required a background with recessive mutations in the pigmentation pathway. About 2,000 P. hybrida genes contain a high percentage of non-specific SNPs potentially derived from an unknown ancestor.
Approximately 1,500 genes of mixed parentage were identified, with blocks of SNPs similar to P. axillaris and other blocks similar to P. inflata (Fig. 4). These unusual constellations are conserved between the three P. hybrida accessions and may involve gene conversion, random repair of heteroduplexes, contributions of unknown parents or unknown mechanisms. Gene conversion events have been previously reported in plastids21 and polyploids22 but they have not been reported before in hybrids (or species of hybrid origin). Definitive answers, especially to the question whether this phenomenon is restricted to transcribed regions will require transcriptome and whole-genome sequencing of multiple P. hybrida accessions.

a,b, Two examples of genes with mixed parentage in P. hybrida accessions Mitchell, R143 and R27. a, PME inhibitor; Peaxi162Scf00002g00042 and Peinf101Scf01857g01001 for P. axillaris N and P. inflata S6, respectively. b, Stress-induced phosphoprotein; Peaxi162Scf00002g00511 and Peinf101Scf01857g08047 for P. axillaris N and P. inflata S6, respectively. Green and blue circles represent SNPs specific to P. axillaris N and P. inflata S6, respectively. Small black arrows represent SNPs present only in the P. hybrida lines.
Genes encoding pollinator attraction traits
Bee-pollinated P. inflata has purple flowers that produce only a limited amount of scent, whereas the flowers of the hawkmoth-pollinated P. axillaris are strongly scented and white (Fig. 1a). Colour and scent influence the attraction of pollinators and thereby cause reproductive isolation and ultimately speciation. Speciation of P. axillaris from a P. inflata-like ancestor involved the loss of anthocyanin pigments and the gain of volatiles4. Thus the genes that caused the changes in these two traits are potential speciation genes. The anthocyanin backbone is synthesized from phenylalanine by nine enzymatic steps followed by specific decorations of the backbone that modify the absorption spectrum. To address how the change in anthocyanin pigmentation of Petunia flowers evolved, we compared all known regulatory and structural genes (Supplementary Note 7).
Both Petunia genomes contain a complete set of functional genes for the core pathway (CHS, CHI, DFR, ANS, 3GT, 5GT and AAT); however, some of the decorating enzymes are compromised in P. axillaris. The steps in the pathway, from DFR on, are regulated by a ternary complex consisting of MYB, bHLH and WD40 transcription factors. The bHLH and WD40 components are functional, but in all P. axillaris accessions, the MYB factor AN2 has been inactivated because of independent mutations in the coding region23,24 (Fig. 1d). The only known function of AN2 is to regulate anthocyanin synthesis in petal lobes and this lack of pleiotropic effects makes AN2 a preferred target of selection in the natural habitat.
In P. hybrida, four related MYB factors activate the anthocyanin biosynthetic pathway in different tissues: AN2 controls anthocyanin deposition in the petal limb, AN4 in the anthers and DPL and PHZ in green tissues. Unlike AN2, the AN4, DPL and PHZ coding sequences have remained intact in P. axillaris. Based on P. hybrida data, differential expression of AN4 might be responsible for the shift in anther colour from purple in P. inflata to yellow in P. axillaris.
The genomic regions containing these four MYB genes have undergone massive rearrangements since the separation of the two species estimated at 0.9 Myr ago, possibly influenced by transposon or retroelement activities found in the vicinity (Fig. 5a). As a consequence, the synteny between the corresponding regions of P. axillaris and P. inflata has been largely destroyed and gene spacing altered. P. axillaris AN4 is duplicated and inactivated subsequently in anthers because of large insertions of transposon-like sequences in the promoter. Similar insertion events are visible around the other anthocyanin MYB genes. Instead, the genomic regions containing other anthocyanin regulators (AN1, JAF13, AN11) and other MYBs involved in vacuolar pH regulation and scent production show strong conservation of the synteny between the two Petunia species. Thus, the AN2-like MYBs reside in an exceptionally dynamic region of the genome. Although lack of pleiotropy makes AN2-like MYBs preferential targets of selection, genomic rearrangements may have provided the mechanism responsible for the remarkable spatial and temporal diversity of anthocyanin pigmentation patterns.

a, Genome dynamics at different MYB gene regions. Genomic regions around AN2-like genes are highly rearranged with few conserved genes, whereas synteny is conserved around the MYB ODO1 involved in scent production. Black arrows, MYB genes. Different coloured arrows, other syntenic genes. Purple blocks, various repeat sequences. b, Biosynthesis of 2-phenylacetaldehyde is different in Petunia and S. lycopersicum. Red and blue arrows depict enzymatic steps characterized in S. lycopersicum and Petunia, respectively. The black arrow represents a predicted activity in S. lycopersicum. c, Biosynthesis of eugenol in Petunia. Although tomato also makes eugenol, homologues of the two genes involved seem to be absent. AADC, aromatic l-amino acid decarboxylase; PAAS, phenylacetaldehyde synthase; CFAT, CoA:coniferyl alcohol acetyltransferase; EGS, eugenol synthase.
Exceptional dynamics of the regions containing the MYB regulators of the anthocyanin pathway is not restricted to Petunia. The regions in S. lycopersicum share little synteny with either of the two Petunia species indicating that large rearrangements occurred after the separation of the genera. In the more distantly related Mimulus guttatus, we also find duplications and rearrangements to have taken place after the separation of the ancestors of Solanaceae and Phrymaceae. Thus, genome dynamics of AN2-type MYB factors may be a general mechanism that caused the diversity of floral pigmentation patterns across angiosperms.
P. axillaris emits an abundant blend of floral benzenoid and phenylpropanoid volatiles whereas P. inflata only emits benzaldehyde. A comparison of all structural and regulatory genes known to be involved in floral scent synthesis indicates that all the known biosynthetic and regulatory genes encode functional proteins (Supplementary Note 8). Thus, the increase in complexity and concentration of volatiles accompanying the shift to moth pollination in P. axillaris involved mutations in cis-acting regulatory elements or the mutation of as yet unknown transcriptional regulators.
Petunia uses a single enzyme for the biosynthesis of 2-phenylactealdehyde25 whereas S. lycopersicum utilizes an amino acid decarboxylase plus a yet unidentified amine oxidase (Fig. 5b)26. Interestingly, the S. lycopersicum genome does harbour a homologue of the Petunia gene, but this is predicted to be 124 amino acids shorter than its Petunia homologue and presumably inactive. Furthermore, although S. lycopersicum is also known to produce eugenol27, homologues of the two involved enzymes appear to be absent (Fig. 5c). Thus, the Solanaceae have evolved multiple strategies for the synthesis of C6–C2 and C6–C3 compounds.
Petunia as a model for comparative research of gene function
High throughput DNA sequencing makes it possible to compare DNA sequences and RNA expression patterns across a wide variety of taxa. However, functional analysis is necessary to determine if sequence conservation can be equated with conservation of gene function. Good examples are the AP2 and BL/FIS (MIR169) genes, which, although very well conserved at the sequence level, can perform divergent developmental functions in different species28.
In general, a larger diversity of genetic model systems will be essential to link sequence information with function. Ease of cultivation and propagation, highly efficient genetics and transformation make Petunia an attractive model system for comparative analysis of gene function (see Boxes 1boxed-textboxed-text–4). The availability of high-quality genome sequences further increases the utility of the asterid Petunia not only for testing the generality of conclusions based on the rosid Arabidopsis or the monocot rice, but also for studying biological phenomena in a species with different genome organization, biochemistry, development, ecology and evolution.
Methods
Genome sequencing, assembly and annotation
Plants were grown and DNA was extracted following the methods described at Supplementary Note 1.
Illumina libraries with 0.17-, 0.35-, 0.5-, 0.8-, 1-, 2-, 5-, 8- and 15-kb inserts were sequenced at BGI-Shenzghen and University of Illinois, Roy J. Carver Biotechnology. PacBio P. axillaris DNA library was sequenced with P4/C2 chemistry.
Illumina reads were processed using Fastq-mcf (quality filtering; https://code.google.com/p/ea-utils/wiki/FastqMcf), PRINSEQ (duplication filtering; http://prinseq.sourceforge.net/) and Musket (error correction; http://musket.sourceforge.net/). Pacbio reads were processed using the SMRT Analysis pipeline (v.2.0.1; https://github.com/PacificBiosciences/SMRT-Analysis).
Both genomes were assembled with SOAPdenovo30 with different k-mer sizes. For both genomes, k-mer = 79 showed the best statistics. Gaps between contigs were completed using GapCloser30. Additionally for P. axillaris, PacBio reads were integrated in four different steps: (1) Rescaffolding of the Illumina contigs using the PacBio reads and the AHA assembler31; (2) Gap filling using PBJelly32; (3) Rescaffolding using the Illumina pair data and SSPACE33; (4) Last round of gap filling using PBJelly32.
Genome size estimation was performed through the k-mers abundance distribution34 (k-mer = 31). Heterozygosity was estimated mapping the Illumina reads to the assemblies using Bowtie235, calling SNPs using FreeBayes36 and annotating the SNPs using SnpEff37.
The genome structural annotation was performed using Maker-P38: (1) SNAP and Augustus as ab initio gene predictors; (2) Exonerate as experimental based predictor with 454 and Illumina RNASeq reads and protein sequences from different protein datasets. RNAseq Illumina data was mapped using Tophat239. tRNAs were annotated using tRNAscan (http://lowelab.ucsc.edu/tRNAscan-SE/).
The gene functional annotation was performed by sequence homology search with different protein datasets using BlastP40 and protein domains search using InterProScan41. Functional annotations were integrated using AHRD (https://github.com/groupschoof/AHRD). See Supplementary Note 1.
Repetitive elements analysis
Repeat annotation was performed using RepeatModeler (v1.0.8; http://www.repeatmasker.org/RepeatModeler.html), RepeatMasker (v4.0.5; http://www.repeatmasker.org) with the repeat database Repbase (release 20140131; http://www.girinst.org/repbase/) and Geneious (v7.1.4; http://www.geneious.com). Identification of PVCV-like and EPRV elements was performed using BlastN and TBlastN40. The identified sequences were aligned with ClustalW (MEGA5 package; http://www.megasoftware.net/) and then manually curated. RepeatExplorer (http://www.repeatexplorer.org/) and other methods were used to extend the analysis to unassembled repeats. Fluorescent in situ hybridization was performed in root tips from young P. axillaris and P. inflata plants for 5S rDNA and three PVCV viral probes following the procedure described in Supplementary Note 2.
The detection of dTph1 loci in P. hybrida W138 was performed through a BLAST40 search of the P. axillaris and P. inflata dTph1 elements including the 500 bp of flanking sequence against the TFS W138 collection43. Polymorphisms found in the genomic flanking regions were used to identify the species of origin. dTph1 elements were identified in a P. axillaris population using a modification of the methodology described in Supplementary Note 3.
Whole-genome duplication, tandem duplications and gene family analysis
Whole-genome collinear analysis was performed using SynMap and microsynteny analysis were performed using GEvo in the comparative genomics platform, CoGe42. See Supplementary Note 5.
The gene family analysis included Solanum lycopersicum, S. tuberosum, Nicotiana benthamiana and Arabidopsis thaliana protein sets using BlastP (v2.2.27)40 on an all-versus-all comparison and grouping the genes into families with OrthoMCL, v2.0.8. See Supplementary Note 4.
Small RNA sequencing and analysis
Total RNA was purified and small RNA libraries were prepared and sequenced and analysed following the methods described in Supplementary Note 9. Annotation and identification was performed using Perl scripts, mirDeep-P (v1.3; http://sourceforge.net/projects/mirdp/), Bowtie (v1.0.1) and CLCbio, based on identity to miRNAs in Arabidopsis and Solanaceae spp. Secondary structures of pre-miRNAs were predicted with RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). MiRNA target genes were predicted using TargetFinder (v1.6). See Supplementary Note 9.
Petunia hybrida transcripts comparison
Petunia hybrida (accessions Mitchell, R27 and R143) reads were mapped to the P. axillaris genome (v1.6.2) using Bowtie235. SNPs were called using FreeBayes36 and annotated using Snpeff37. Exons and genes were assigned to P. axillaris or P. inflata based in the SNP data using a Perl script. Five categories were used: Homozygous P. axillaris; Homozygous P. inflata; Heterozygous P. axillaris/P. inflata; Homozygous P. axillaris/P. inflata and unclear assignment. Homozygous SNPs for the genes with exons from both species were confirmed aligning P. hybrida EST using Exonerate (v2.2 l; http://www.ebi.ac.uk/about/vertebrate-genomics/software/exonerate). Gene Set Enrichment Analysis (GSEA) was performed using the Bioconductor package TopGO (v2.22.0; http://bioconductor.org/packages/topGO/). See Supplementary Note 6.
Gene data mining
The specific identification of genes for P. axillaris and P. inflata genomes for colour and scent, root-specific pathways, self-incompatibility and circadian clock was performed through a BlastN/BlastP sequence homology search. Blast GUI, JBrowser (http://jbrowse.org/) and WebApollo (http://genomearchitect.org/) were installed in a server to search and manually curate the gene structures of the identified genes. See Supplementary Notes 7, 8 and 10–12.
The P. axillaris and P. inflata genome sequences are available on the Sol Genomics Network (SGN) at https://solgenomics.net/organism/Petunia_axillaris/genome and https://solgenomics.net/organism/Petunia_inflata/genome, respectively.
References
National Agricultural Statistics Service. Floriculture Crops 2014 Summary (US Department of Agriculture, 2015); http://usda.mannlib.cornell.edu/usda/nass/FlorCrop//2010s/2015/FlorCrop-06-04-2015.pdf
Napoli, C., Lemieux, C. & Jorgensen, R. Introduction of chimeric chalcone synthase gene into Petunia results in reversible co-suppression of homologous genes in trans. Plant Cell 2, 279–289 (1990).
van der Krol, A. R., Mur, L. A., Beld, M., Mol, J. N. M. & Stuitje, A. R. Flavonoid genes in Petunia: addition of a limited number of gene copies may lead to a suppression of gene expression. Plant Cell 2, 291–299 (1990).
Sheehan, H., Hermann, K. & Kuhlemeier, C. Color and scent: how single genes influence pollinator attraction. Cold Spring Harb. Symp. Quant. Biol. 77, 117–133 (2012).
Saerkinen, T., Bohs, L., Olmstead, R. G. & Knapp, S. A phylogenetic framework for evolutionary study of the nightshades (Solanaceae) a dated 1000-tip tree. BMC Evol Biol 13, 214 (2013).
Segatto, A. L. A., Ramos-Fregonezi, A. M. C., Bonatto, S. L. & Freitas, L. B. Molecular insights into the purple-flowered ancestor of garden petunias. Am. J. Bot. 101, 119–127 (2014).
Stehmann, J. R., Lorenz-Lemke, A. P., Freitas, L. B. & Semir, J. in Petunia: Evolutionary, Developmental and Physiological Genetics 2nd edn (eds Gerats, T. & Strommer, J. ) 1–28 (Springer, 2009).
Sink, K. C. in Petunia: Monographs on Theoretical and Applied Genetics Vol. 9 (ed. Sink, K. C. ) 3–9 (Springer, 1984).
White, J. & Rees, H. Chromosome weights and measures in Petunia. Heredity 58, 139–143 (1987).
Heslop-Harrison, J. P. Genome evolution: extinction, continuation or explosion? Curr. Opin. Plant Biol. 15, 115–121 (2012).
Vandenbussche, M. et al. Generation of a 3D indexed Petunia insertion database for reverse genetics. Plant J. 54, 1105–1114 (2008).
Geering, A. D. W. et al. Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nature Commun. 5, 5269 (2014).
Hansen, C. & Heslop-Harrison, J. Sequences and phylogenies of plant pararetroviruses, viruses, and transposable elements. Adv. Bot. Res. 41, 165–193 (2004).
Richert-Pöggeler, K. R., Noreen, F., Schwarzacher, T., Harper, G. & Hohn, T. Induction of infectious petunia vein clearing (pararetro) virus from endogenous provirus in petunia. EMBO J. 22, 4836–4845 (2003).
D'Hont, A. et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213–217 (2012).
Jiao, Y. et al. Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100 (2011).
Soltis, D. E., Bell, C. D., Kim, S. & Soltis, P. S. Origin and early evolution of angiosperms. Ann. NY Acad. Sci. 1133, 3–25 (2008).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Scannell, D. R., Byrne, K. P., Gordon, J. L., Wong, S. & Wolfe, K. H. Multiple rounds of speciation associated with reciprocal gene loss in polyploid yeasts. Nature 440, 341–345 (2006).
Khakhlova, O. & Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94 (2006).
Salmon, A., Udall, J. A., Jeddeloh, J. A. & Wendel, J. Targeted capture of homoeologous coding and noncoding sequence in polyploid cotton. G3 2, 921–930 (2012).
Quattrocchio, F. et al. Molecular analysis of the anthocyanin2 gene of Petunia and its role in the evolution of flower color. Plant Cell 11, 1433–1444 (1999).
Hoballah, M. E. et al. Single gene-mediated shift in pollinator attraction in Petunia. Plant Cell 19, 779–790 (2007).
Kaminaga, Y. et al. Plant phenylacetaldehyde synthase is a bifunctional homotetrameric enzyme that catalyzes phenylalanine decarboxylation and oxidation. J. Biol. Chem. 281, 23357–23366 (2006).
Tieman, D. et al. Tomato aromatic amino acid decarboxylases participate in synthesis of the flavor volatiles 2-phenylethanol and 2-phenylacetaldehyde. Proc. Natl Acad. Sci. USA 103, 8287–8292 (2006).
Tikunov, Y. M. et al. Non-smoky glycosyltransferase1 prevents the release of smoky aroma from tomato fruit. Plant Cell 25, 3067–3078 (2013).
Cartolano, M. et al. A conserved microRNA module exerts homeotic control over Petunia hybrida and Antirrhinum majus floral organ identity. Nature Genet. 39, 901–905 (2007).
Sierro, N. et al. Reference genomes and transcriptomes of Nicotiana sylvestris and Nicotiana tomentosiformis. Genome Biol. 14, R60 (2013).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
Bashir, A. et al. A hybrid approach for the automated finishing of bacterial genomes. Nature Biotechnol. 30, 701–707 (2012).
English, A. C. et al. Mind the gap: upgrading genomes with pacific biosciences RS long-Read sequencing technology. PloS One 7, e47768 (2012).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Li, R. et al. The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317 (2010).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359 (2012).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. Preprint at http://arxiv.org/abs/1207.3907 (2012).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6, 80–92 (2014).
Campbell, M. S. et al. MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 164, 513–524 (2014).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Mulder, N. & Apweiler, R. InterPro and InterProScan Vol. 396, 59–70 (Humana, 2007).
Lyons, E. & Freeling, M. How to usefully compare homologous plant genes and chromosomes as DNA sequences. Plant J. 53, 661–673 (2008).
Van den Broeck, D. et al. Transposon display identifies individual transposable elements in high copy number lines. Plant J. 13, 121–129 (1998).
Antolin-Llovera, M. et al. Knowing your friends and foes – plant receptor-like kinases as initiators of symbiosis or defence. New Phytol. 204, 791–802 (2014).
Limpens, E., van Zeijl, A. & Geurts, R. Lipochitooligosaccharides modulate plant host immunity to enable endosymbioses. Ann. Rev. Phytopatol. 53, 15.1–15.24 (2015).
Sims, T. L. & Robbins, T. P in Petunia: Evolutionary, Developmental and Physiological Genetics (eds Gerats, T. & Strommer, J. ) 85–106 (Springer, 2009).
Clark, K. R., Okuley, J. J., Collins, P. D. & Sims, T. L. Sequence variability and developmental expression of S-alleles in self-incompatible and pseudo-self-compatible Petunia. Plant Cell 2, 815–826 (1990).
Kessler, D., Diezel, C., Clark, D. G., Colquhoun, T. A. & Baldwin, I. T. Petunia flowers solve the defence/apparency dilemma of pollinator attraction by deploying complex floral blends. Ecol. Lett. 16, 299–306 (2013).
Sponberg, S., Dyhr, J. P., Hall, R. W. & Daniel, T. L. Luminance-dependent visual processing enables moth flight in low light. Science 348, 1245–1248 (2015).
Lou, P. et al. Genetic architecture of the circadian clock and flowering time in Brassica rapa. Theor. Appl. Genet. 123, 397–409 (2011).
Acknowledgements
We dedicate this work to Tom Gerats in honour of his lifelong contributions to petunia research, and we thank him for generously sharing his vast knowledge with all of us.
We thank H. Puchta for advice on Fig. 4, R. Köpfli for IT and graphics support, and K. Esfeld for carefully reading the manuscript. This work was carried out without dedicated funding. We acknowledge following agencies who support our work on Petunia: NWO-ALW grant 022.001.018 (M.B); Marie Curie Independent Fellowship (L.G.); Swiss NSF grant 31003A_159493 and NCCR Plant Survival (C.K.); NWO-TOP grant 854.11.006 (R.K.); Borsa di Studio Lanzi per Genetica Agraria, Accademia dei Lincei (V.P.); NWO-ALW grant 820.02.015 (K.V.); CNRS ATIP-AVENIR award (M.V.); Deutsche Forschungsgemeinschaft grant DR411/2-1 (U.D. and P.F.); Swiss NSF grant 31003A_135778 (D.R.).
Author information
Authors and Affiliations
Contributions
The authors are listed in alphabetical order except for the first two and the last three. A.B., F.Q., T.Si. and C.K. conceived and planned the work. All authors wrote or commented on the main text and supplementary notes; A.B., M.M., A.A., L.B., C.B., M.Bl., M.Bo., D.B., N.D., N.F-P., L.G., J.H., J.H.-H., M.J., R.K., X.L., E.L., D.M., E.M., N.M., P.M., J.M., E.N., V.P., Q.Q., D.R., M.R., K.R-P., T.R., E.S., R.S., T.Sc., C.S., H.T., S.U., M.V., K.V., G.V., R.W., J.W., Z.Y., J.Z. and F.Q. performed the experiments and analysed the data; R.B., M.D., X.L., M.P., M.S., Z.Y., T.Si. and C.K. contributed sequencing data and analysis tools.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Note 1
Assembly and annotation of the Petunia genomes (PDF 362 kb)
Supplementary Note 2
Analysis of Petunia vein clearing virus (PVCV) sequences, retroelements and tandem repeats in Petunia axillaris N and P. inflata S6 (PDF 2373 kb)
Supplementary Note 3
Genome wide analysis of Petunia dTPH transposable elements in Petunia axillaris and Petunia inflata (PDF 1341 kb)
Supplementary Note 4
Analysis of tandem duplications and gene families in Petunia species as compared to other Solanaceae and Arabidopsis (PDF 329 kb)
Supplementary Note 5
Incomplete gene fractionation after paleopolyploidy: the first study case in flowering plants revealed by comparison of the Petunia axillaris N. and Solanum lycopersicum genomes (PDF 1915 kb)
Supplementary Note 6
Analysis of the genomic origin of Petunia hybrida (PDF 1569 kb)
Supplementary Note 7
The genes behind the different colors of P. axillaris and P. inflata flowers (PDF 2831 kb)
Supplementary Note 8
Extreme variation in volatile production in wild petunias: what can we learn from their genomes? (PDF 704 kb)
Supplementary Note 9
Identification of conserved miRNAs in Petunia axillaris and P. inflata young flower buds and their verification in the Petunia genome sequence (PDF 3854 kb)
Supplementary Note 10
Genomic insight into the pathways that control adventitious root formation and arbuscular mycorrhiza in petunia (PDF 1982 kb)
Supplementary Note 11
Characterization of S-loci in P. inflata and P. axillaris identifies multiple linked S-locus F-box genes (PDF 818 kb)
Supplementary Note 12
Genetic structure of the circadian clock in Petunia (PDF 1116 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bombarely, A., Moser, M., Amrad, A. et al. Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nature Plants 2, 16074 (2016). https://doi.org/10.1038/nplants.2016.74
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/nplants.2016.74
This article is cited by
-
Chromosome level genome assembly of endangered medicinal plant Anisodus tanguticus
Scientific Data (2024)
-
Genetics and evolutionary insights from Solanaceae genome sequences
Plant Systematics and Evolution (2024)
-
Transcriptomic analysis implicates ABA signaling and carbon supply in the differential outgrowth of petunia axillary buds
BMC Plant Biology (2023)
-
Genetic architecture of a pollinator shift and its fate in secondary hybrid zones of two Petunia species
BMC Biology (2023)
-
Comparative and expression analyses of AP2/ERF genes reveal copy number expansion and potential functions of ERF genes in Solanaceae
BMC Plant Biology (2023)
