
Abstract
The Para rubber tree (Hevea brasiliensis) is an economically important tropical tree species that produces natural rubber, an essential industrial raw material. Here we present a high-quality genome assembly of this species (1.37 Gb, scaffold N50 = 1.28 Mb) that covers 93.8% of the genome (1.47 Gb) and harbours 43,792 predicted protein-coding genes. A striking expansion of the REF/SRPP (rubber elongation factor/small rubber particle protein) gene family and its divergence into several laticifer-specific isoforms seem crucial for rubber biosynthesis. The REF/SRPP family has isoforms with sizes similar to or larger than SRPP1 (204 amino acids) in 17 other plants examined, but no isoforms with similar sizes to REF1 (138 amino acids), the predominant molecular variant. A pivotal point in Hevea evolution was the emergence of REF1, which is located on the surface of large rubber particles that account for 93% of rubber in the latex (despite constituting only 6% of total rubber particles, large and small). The stringent control of ethylene synthesis under active ethylene signalling and response in laticifers resolves a longstanding mystery of ethylene stimulation in rubber production. Our study, which includes the re-sequencing of five other Hevea cultivars and extensive RNA-seq data, provides a valuable resource for functional genomics and tools for breeding elite Hevea cultivars.
Similar content being viewed by others


The rubber tree (Hevea brasiliensis, hereafter referred to as Hevea) is a member of the spurge family (Euphorbiaceae), along with several other economically important species such as cassava (Manihot esculenta) and the castor oil plant (Ricinus communis). Natural rubber (cis-1, 4-polyisoprene) makes up about one-third of the volume of latex that is essentially cytoplasm of the articulated laticifers in Hevea. The latex is extracted by tapping the bark, a non-destructive method of harvesting that facilitates continual production. As an industrial commodity, natural rubber is an elastomer with physical and chemical properties that cannot be fully matched by synthetic rubber1. In contrast to synthetics, the production of natural rubber is sustainable and environment friendly2. The commercial cultivation of Hevea, a native to the Amazon Basin, began in 1896 on a plantation scale in Malaya (now Malaysia) and expanded to other Southeast Asian countries that lead in world natural rubber production today3. Decades of selective breeding have resulted in a gradual improvement in rubber productivity, from 650 kg ha–1 derived from unselected seedlings during the 1920s to 2,500 kg ha–1 yielded by elite cultivars by the 1990s4. Nevertheless, the field production achieved so far is still well below the theoretical yield of 7,000–12,000 kg ha–1, as has been suggested for the rubber tree5. Meanwhile, conventional rubber breeding has been stagnating in the introduction of high-yield cultivars. The reasons include a narrow genetic basis for exploiting breeding potential and difficulty in introducing wild germplasms because of the genetic burden in removing unfavourable alleles6. The incorporation of marker-assisted selection and transgenic techniques offers promise to improve breeding efficiency for latex yield, and sequencing of the Hevea genome would uncover even more avenues leading to this end.
The first draft Hevea genome was released by a Malaysian team7 that was participant to the recent boom in transcriptomic and proteomic studies of the species8–11. However, its low sequence coverage (∼13×) and a lack of large insert libraries (such as fosmid- or BAC-based clone libraries) have limited the success of genome assembly (a scaffold N50 size of 2,972 bp), precluding its application for furthering quality research in the field.
Here, we report a high-quality genome assembly of Hevea Reyan7-33-97, an elite cultivar widely planted in China12,13 based on sequence data from both whole-genome shotgun (WGS) and pooled BAC clones. This assembly contains 7,453 scaffolds (N50 = 1.28 Mb), has a length of 1.37 Gb and covers ∼94% of the predicted genome size (1.46 Gb). Together with analysis of data from re-sequencing five other cultivars and comprehensive transcriptomic surveys, we aim to obtain new insights into the physiology of laticifers and molecular details of rubber biosynthesis, especially in relation to ethylene-stimulated rubber production.
Results
Genome assembly and annotation
We assembled the Hevea genome (cultivar Reyan7-33-97) based on 138 Gb (94× genome coverage) processed shotgun sequences (Illumina GA2 and Hiseq 2000; Supplementary Table 1). We also generated paired-end reads for five other Hevea cultivars (PR107, Reyan8-79, RRIM600, Wenchang11 and Yunyan77-4) with high-coverage of 38× to 59× (Supplementary Fig. 1 and Supplementary Table 2). Based on a 17-mer sequence library, the estimated genome size of Reyan7-33-97 is 1.46 Gb, whereas that of the other five cultivars ranges from 1.41 Gb to 1.55 Gb (Supplementary Fig. 2). To assist with scaffold construction, we generated an additional 55-Gb high-quality mate-pair data (insert sizes from 800 to 10,000 bp; 682× physical coverage; Supplementary Table 1 and Supplementary Fig. 3), and obtained a preliminary assembly with a scaffold N50 value of 55 kb. We further improved the assembly by adding pooled BAC sequences (Supplementary Fig. 4 and Supplementary Note 1) from 47,616 BAC clones (mean insert size = 135 kb) (Supplementary Table 3 and Supplementary Fig. 5) that generated pseudo-mate-pair reads of 10 to 200 kb insert lengths to assist scaffolding (Supplementary Fig. 6). This yielded a 1.37-Gb genome assembly that covers 93.8% of the estimated genome size, and contains 7,453 scaffolds (N50 = 1.28 Mb) and 84,285 contigs (N50 = 30.6 kb) (Table 1).
To validate quality of the assembly, we first aligned all Hevea DNA, expressed sequence tag (EST) and protein sequences available in the public domain to show mapping rates of 91.5, 97.9 and 100%, respectively (Supplementary Figs 7 and 8). Of the 1.11-Gb Hevea genome assembled by the Malaysian group7, only 25.2 Mb failed to be aligned to our assembly. The unalignable portions may reflect real sequence differences between cultivars, RRIM600 used by the Malaysian team and Reyan7-33-97 in the present study, as also reflected in the genome size difference (Supplementary Fig. 2). Next, we aligned 18 BAC scaffolds with low repetitive sequences to our genome assembly, and yielded alignments from 91.69% to 99.74% (Supplementary Fig. 9). Finally, the transcripts we assembled also showed excellent alignment to the genome; of 84,241 transcripts, 98.32% were mapped (transcript coverage > 80% and identity > 99%; Supplementary Table 4 and Supplementary Note 4).
To assist genome annotation and gene expression analysis, we combined three methods: ab initio, 84,241 unique transcripts (Supplementary Table 5 and Supplementary Fig. 10) and protein homologues (Supplementary Fig. 11) to define 43,792 protein-coding genes (Table 1), of which 39,919 were found in the NCBI non-redundant protein database (NRPD, Mar. 2015, Supplementary Table 6). A gene family survey clustered 16,315 gene families, among which 1,077 are Hevea specific and 10,675 are shared with Manihot esculenta, Ricinus communis, Populus trichocarpa and Linum usitatissimum (Fig. 1a and Supplementary Note 9). In addition, a homologue search for non-coding RNAs predicted 167 ribosomal RNAs, 591 miRNAs, 10 SPPRNAs, 3,445 SnoRNAs, 4 tmRNAs, 697 tRNAs, 219 snRNAs and 217 other types of RNAs (Supplementary Table 7).

a, A Venn diagram of shared orthologues among five species. Each number represents a gene family number. b, Density distributions of Ks and 4DTv for paralogous genes. The peak values are shown in insets. Hbr, H. brasiliensis; Mes, M. esculenta; Rco, R. communis; Ptr, P. trichocarpa; Lus, L. usitatissimum. c, Collinear region representation of Hbr scaffold0195 to the corresponding Mes and Ptr scaffolds. The outer circle with different colours denotes scaffolds of different species. Each scaffold is scaled, and the scale label is 1 × 10–6 of actual size. The inner circle shows the genes in the corresponding scaffolds, and genes in positive and negative strands are represented by red and black colours, respectively. The collinear blocks between two species are linked by curved ribbons. d, Phylogenetic tree of 13 species based on orthologues of single-gene families. The number in each node indicates the number of gene families. The number at the root (10,758) represents the number of gene families in the common ancestor. The values above each branch denote gene family gain/loss number at each round of genome duplication after diversifying from the common ancestor. The green numbers below each branch denote speculated divergent time of each node. The clades are marked by four different block colours in the tree: the last one (red) is a monocot species, O. sativa, used as an outgroup; the first three are Rosids clades in dicots, including Fabidae (green), COM (yellow) and Malvidae (brown). Bootstrap values for each node are above 50%.
Repeat-driven genome expansion
We identified 71% of the genome length as repeats (Table 1 and Supplementary Table 8). Among the five species of Malpighiales (Hevea, Manihot, Ricinus, Populus and Linum), Hevea not only has the largest genome but also the greatest repeat content (Supplementary Table 9). Of the repeat sequences, two types of long-terminal repeat retrotransposons (LTR-RTs), Gypsy (691,209 in number) and Copia (114,165), are most abundant, and they total more than half (∼0.7 Gb; 50.9%) of the genome assembly, substantially higher than that of the other four Malpighiales species. Among the five Malpighiales species, Hevea harbours the largest set (35,079) of LTR-RTs (Supplementary Table 10), and both Gypsy and Copia show a peak substitution rate around 0.05, suggesting a burst of LTR-RT insertion at that point (of time) (Supplementary Fig. 12), which occurred five million years (Myr) ago as calculated (see Methods). Similar to Hevea, three other Malpighiales species (Manihot, Ricinus and Populus, with Linum being the exception) have more Gypsy than Copia, and share nearly identical substitution rates for the two elements (Supplementary Fig. 12).
Genome duplication, collinearity and phylogeny
We explored gene-based collinearity and calculated synonymous substitution rate (Ks) as well as fourfold synonymous third-codon transversion (4DTv) rate among paralogues and orthologues. With the exception of Ricinus, all the other four Malpighiales species display an obvious two-peak pattern for the distributions of Ks/4DTv, with the left sharp peak representing the recent species-specific duplication and the right smooth one representing the eurosid duplication (Fig. 1b). The most ancient peak, representing γ duplication, is rather obscure and mingled with the right peak. A recent phylogenomic analyses inferred the clades Euphorbiaceae (Hevea, Manihot), Salicaceae (Populus) and Linaceae (Linum) to have emerged 89.9, 58.0 and 39.5 Myr ago, respectively14. Hence, Linum is the youngest of the five species, as also reflected by its lower Ks and 4DTv peak values (Fig. 1b). If the diversification time is assumed to a maximum of 89.9 million years, according to the Ks value of the second peak, the Hevea synonymous substitution constant is estimated as 7.5 × 10–9 per site per year. Thus, the recent Hevea-specific duplication might have occurred in ∼15.3 Myr ago, nearly 10 million years earlier than the burst of Hevea LTR insertion. However, we are still not sure whether the two duplication events in Hevea are whole genome duplication because only 30.0% and 5.8% genes are covered by the first and second peaks, respectively. In comparison, the relative rates in Manihot, Populus and Linum are much higher; for example, 92.8% and 56.8% genes are attributable to the two peaks of Populus (Supplementary Table 12).
Plotting collinear regions of Hevea with itself and five other species reveals obvious conserved gene clusters (Supplementary Fig. 13). The absence of collinearity with Ricinus is unexpected, which might indicate a lack of species-specific duplication in Ricinus as revealed by its Ks/4DTv distribution pattern (Fig. 1b). Among Hevea, Manihot and Populus (Supplementary Fig. 14), the collinearity size of Hevea is significantly larger than that of Manihot (P = 1.3 × 10–8) or Populus (P = 1.79× 10–7). The largest collinearity region exists between Hevea scaffold0195 (1,174,012 bp; 108 genes) and Manihot scaffold06656 (752,726 bp; 81 genes) (Fig. 1c), where 71 genes are overall matched. In Populus, four scaffolds are collinear to the Hevea scaffold0195, indicating structural variations between Hevea and Populus.
To analyse the gene gain–loss event, we used 72 single-copy gene families to construct a maximum likelihood phylogeny between Hevea and 12 other species (Fig. 1d), representing a typical Rosid clade sequenced so far in Eudicots. Taxonomically, Hevea, Manihot, Ricinus, Populus and Linum belong to the Celastrales–Oxalidales–Malpighiales (COM). The phylogenetic placement of the COM clade remains controversial in angiosperms. Here, the COM clade was placed in Malvidae, consistent with a recent study using single- and multicopy nuclear loci15. Among the 13 species examined, Hevea and rice are representative of the most and fewest gene families, respectively. In most species, the gene family gain is less than gene loss; the only exceptions are in rice and soybean. The divergence time for the Euphorbiaceae family is estimated to be 57.7 Myr ago (Fig. 1d), more recent than a previously reported dating of 89.9 Myr ago14, possibly because of the limited number of Euphorbiaceae species used here.
Genetic diversity among Hevea cultivars
The high-quality genome assembly allows us to characterize genetic diversity among the Hevea cultivars Reyan7-33-97 (RY73397), PR107, RRIM600, Wenchang11 (WC11), Reyan8-79 (RY879) and Yunyan77-4 (YY774) (Supplementary Table 2). Among them, PR107 and RRIM600 are cultivated globally, and the other four are elite cultivars bred and widely planted in China, with PR107 and/or RRIM600 as direct or indirect parents (Supplementary Fig. 1).
The density of single nucleotide polymorphisms (SNPs) contributed by all cultivars averages ∼2 SNPs per kilobase (Supplementary Table 12), and 95.1–95.6% SNPs occur in non-coding regions (Supplementary Table 13). YY774 has the most variety-specific SNPs (442,278), whereas WC11 has the least (132,006) (Fig. 2a). Phylogeny based on SNPs shows the genetic relationship among the cultivars (Fig. 2b) is consistent with their breeding history (Supplementary Fig. 1).

a, SNP number distribution in six cultivars. b, Phylogenetic analysis of cultivars based on SNPs and frequency distribution. Using homology SNP sites of the cultivars, the tree is constructed by MEGA5.1 with the Neighbour-Joining model and the bootstrap test (1,000 replicates). The series of plots on the right show SNP density (SNP per kb) and frequencies. SNP frequency is calculated based on a 50-kb sliding window in 1-kb steps. c, Gene Ontology (GO) enrichment in biological process for genes located in core SNP desert using GOEAST. Note that two biological processes, defence response (DR) and respiratory electron transport chain (RETC), are most enriched. LBP, leucine biosynthetic process; ASCPT, ATP synthesis coupled proton transport; CMO, cellulose microfibril organization; CG, cell growth.
Low genetic diversity regions or ‘SNP deserts’ often indicate selective sweeps16 and relate to domestication in many major crop species17,18. All re-sequenced Hevea cultivars exhibit a unimodal pattern in the distribution of SNPs with a single low-SNP peak at <1 SNP per kilobase (Fig. 2b), contrasting with bimodal curves observed in rice and date palm, where the low-SNP peak reflects trait selection in cultivation17,19. The SNP deserts account for 42% of the Hevea genome (Supplementary Table 14), a value significantly higher than that reported in rice (8%)17 or date palm (18%)19. Compared with the genome average, the SNP desert has a higher Ka/Ks (nonsynonymous/synonymous substitution) ratio (1.72 versus 1.27) (Supplementary Table 14). The low-SNP peak in various cultivars might be attributed to natural selection; artificial selection remains a possibility, although to a lesser extent due to a short domestication history of Hevea (since the late nineteenth century).
About half of SNP deserts (13,127 blocks, 261,150 kb) are conserved in all six cultivars (Supplementary Table 15), defined as shared or core SNP deserts representing most sequence signatures left mainly by natural selection after Hevea speciation. There are 3,820 genes (8.7% of the total genes) located in the core SNP desert (Supplementary Table 16) that are most enriched in respiratory electron transport chain and defence response functions (Fig. 2c). Of particular interest is the fact that the single most highly expressed gene in latex—by a wide margin—REF1 resides in the core SNP desert (Supplementary Tables 16 and 20). In this regard, the sister gene to REF1, SRPP1, is noteworthy by its absence in the SNP desert. Meanwhile, both genes showed the least sequence diversity among the six Hevea cultivars examined, suggesting the importance of their conservation in this species (Supplementary Table 17).
Rubber biosynthesis and expansion of the REF/SRPP family
In the laticifer network of Hevea, natural rubber is synthesized by a sequential condensation of isopentenyl diphosphates (IPPs) in cis-configuration to the priming allylic molecules (initiators)20. By convention, genes involved in the synthesis of IPP to the final rubber polymer are termed as rubber biosynthesis genes21. From our genome assembly, 84 rubber biosynthesis genes from 20 gene families were identified (Supplementary Table 18), including 18 in the cytosolic mevalonate (MVA) pathway and 22 in the plastidic 2-C-methyl-D-erythritol-4-phosphate (MEP) pathway for IPP synthesis, 15 for initiator synthesis in the cytosol and 39 for ‘rubber elongation’ on rubber particles (Fig. 3a). Of the 24 MEP genes, only two DXS (1-deoxy-D-xylulose 5-phosphate synthase) genes (DXS7 and DXS10) show substantial and preferential expression in latex (Fig. 3a and Supplementary Table 18). In contrast, at least one gene for each MVA pathway enzyme shows latex-biased abundant expression, supporting the proposition that it is the MVA rather than the MEP pathway that is involved in rubber biosynthesis22,23. Three gene families, REF/SRPP, CPT (cis-prenyltransferase) and DXS, have more than ten genes. Recently, a homologue of the human Nogo-B receptor was demonstrated to be essential for rubber biosynthesis in dandelion24. However, blast searching yielded no significant hits in our genome assembly or the public Hevea sequences, indicating a discrepancy in the mechanisms of rubber biosynthesis between these two species.

a, The rubber biosynthesis pathway and expression profiles (reads per kilobase per million reads mapped; RPKM) of the genes involved in rubber biosynthesis. Lx, latex; Bk, bark; Lf, leaf; Rt, root; FF, female flower; MF, male flower. b, Phylogeny of the REF/SRPP gene family. Full-length coding sequences of the 18 REF/SRPPs are aligned with MAFFT v7.205. The tree is constructed by using MEGA5.1, with the Neighbour-Joining model and bootstrap test with 1,000 replicates. c, Genomic location of the Hevea REF/SRPP genes. Scaffolds are represented as solid bars with their names on the left and length scale on the bottom. Note that most of the REF/SRPP genes, including the four laticifer-specific ones (REF1, SRPP1, REF3 and REF7), are located in a single scaffold (Scaffold1222).
The 18-member REF/SRPP family of Hevea is the largest compared with the other 17 sequenced plants examined this study (10 at most) (Supplementary Table 19), indicating an obvious expansion of this family in Hevea. The gene number of the REF/SRPP family does not seem to correlate with either genome size or polyploidy of a given species. For example, only four REF/SRPP genes are identified in the hexaploid Triticum aestivum genome (17 Gb) but six in the small diploid genome of Populus trichocarpa (480 Mb). When considering other rubber-producing plants, eight REF/SRPP genes are identified in Lactuca sativa and six in limited EST/mRNA sequences of Parthenium argentatum (Supplementary Table 19). These results suggest a possible link between expansion of the REF/SRPP family and the ability to produce rubber.
A close involvement of REF and SRPP in rubber synthesis has been proposed based on in vitro rubber biosynthesis assays25,26, and a positive correlation of REF expression with rubber yield27. Recent transgenic studies in Taraxacum brevicorniculatum28,29 also reveal their essential role in rubber biosynthesis. REF and SRPP share a conserved REF motif that is also retained in several stress-related or lipid droplet-associated proteins in plant cells25,30,31. Based on the presence of a carboxy (C) terminus found in SRPP and its absence in REF25,30, and the similarities among protein sequences, the 18 Hevea REF/SRPP genes are named SRPP1 to 10 and REF1 to 8 (Supplementary Fig. 15). Of these, REF1 (138 aa) and SRPP1 (204 aa) are the two most abundant isoforms (Supplementary Table 18), and correspond, respectively, to the well characterized REF and SRPP25,26. In the other 17 plants examined, the REF/SRPP family has isoforms with sizes similar to or larger than SRPP1, but no isoforms with similar size to REF1. The uniqueness of REF1 is highlighted by an alignment of REF/SRPP proteins from different plants (Supplementary Fig. 16).
The 18 REF/SRPP genes exhibit distinct expression patterns in seven Hevea tissues (Fig. 3a). Four isoforms, REF1, SRPP1, REF3 and REF7, have striking RPKM values of >7,000 in latex, this being especially true of REF1 (38,999). In total they account for 96.8% of expression of the REF/SRPP gene family in latex (Supplementary Table 18). In addition, their expression in latex is over tenfold higher than in any other tissue. Considering the fact that all other tissues also contain laticifers and small amounts of latex, we think that the expression of these genes in the tissues may have arisen from latex itself, and REF1, SRPP1, REF3 and REF7 are thus essentially laticifer-specific genes. When compared with all the genes expressed in latex, these four REF/SRPP genes rank among the top, with REF1 ranked first, SRPP1 sixth, REF3 ninth and REF7 the twelfth (Supplementary Table 20).
Phylogenetically, the Hevea REF/SRPP genes are classified into two major groups, with 14 in group I and four in group II (Fig. 3b). Of the group II genes, three (SRPP4, 6 and 10) are not expressed or expressed at very low levels in latex, and the remaining SRPP7 shows somewhat constitutive low expression across different tissues (Fig. 3a), therefore precluding the active participation of group II genes in rubber biosynthesis. group I genes are further divided into two distinct clades, 13 in clade 1 and only SRPP2 in clade 2. SRPP2 exhibited moderate expression across diverse tissues, excluding its special function in laticifers. Interestingly, all latex-specific isoforms (REF1, SRPP1, REF3 and REF7) are clustered in clade 1. When the REF/SRPPs from Hevea and 11 other plants were investigated for their phylogenetic relationship, all the 13 clade 1 isoforms were clustered into an independent clade whereas the remainder were scattered together with the homologues from other plants into different clades (Supplementary Fig. 17). These results suggest that the clade 1 REF/SRPP genes might have evolved independently towards functioning in rubber biosynthesis. The Hevea REF/SRPP family was further surveyed for their genomic location. Of particular note, 12 of the 13 clade 1 REF/SRPPs including the four laticifer-specific genes, are located in a single 205-kb Scaffold1222 whereas the remaining six are scattered into six different scaffolds (Fig. 3c and Supplementary Fig. 18). SRPP1 and the parallel precursor for REF1 appear to have evolved from the same ancestral SRPP gene, but the emergence of REF1 was a more recent event (Fig. 3b, upper part of clade 1).
Active ethylene signalling and response in laticifers
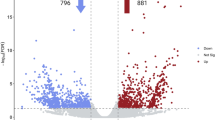
Since latex production is greatly increased by the ethylene-releasing agent ethephon, we identified 509 differentially expressed genes (DEGs) in latex that respond to ethylene treatment (Fig. 4a and Supplementary Table 21). About one-quarter of the 342 annotated DEGs (>2-fold expressional change) are transcription factors (53) and protein kinases (31) (Supplementary Tables 22 and 23). Both gene groups are essential in ethylene signal transduction and response in higher plants32,33.

a, Expression dynamics of differentially expressed genes (DEGs) within 24 h of ethylene treatment. The expression levels (RPKM) of the 509 DEGs are normalized based on the Z-score method (scale bar). The DEGs are divided into six clades; two (clades I and II) of them are downregulated genes whereas the remainder (clades III to VI) are upregulated. b, A model summarizing the mechanisms of ethylene stimulation of rubber production. The red and green text used for genes and enzymes denotes that their expression/activity is activated and inhibited by ethylene, respectively. The red background of molecules or events indicates that their quantity or strength is boosted by ethylene. Suc, sucrose; Met, methionine; ROS, reactive oxygen species; NIN, neutral invertase; GS, glutamine synthetase.
To understand ethylene-dependent mechanisms, we first identified the genes responsible for ethylene biosynthesis in Hevea and examined their expression patterns. The three enzymes that act sequentially to synthesize ethylene from methionine, that is S-adenosyl-l-methionine synthase (SAMS), 1-aminocyclopropane-1-carboxylic acid (ACC) synthase (ACS) and ACC oxidase (ACO)34 have 8, 14 and 16 genes respectively in Hevea (Supplementary Table 24). However, the expression of these gene families, especially that of SAMS (>fivefold) and ACO (>10-fold) is much lower in latex than other tissues. Such low expression or a lack of expression has been noticed recently for nine ACO genes in Hevea latex35. These results, together with the low oxygen condition in latex36 and the requirement of oxygen by ACO to produce ethylene34, point out a weak ethylene-synthesizing ability in laticifers. In addition, ethylene treatment had little effect on the expression of ACS, the rate-limiting enzyme of ethylene biosynthesis in latex (Supplementary Table 24). The primary ethylene signalling components, namely ETR, CTR1, EIN2 and EIN3/EIL1, were investigated for their expression in latex in response to ethylene treatment (Supplementary Table 25). Four out of the eight receptor genes (ETR) showed enhanced expression after ethylene treatment, indicating an ethylene response triggered in laticifers since increased expression has been observed for one or more ETRs in every known ethylene response33. Of particular interest, expressions of two EIN2s and one EIL1 were apparently boosted in latex after ethylene treatment. As the central transducer and master transcription factors in ethylene signalling, EIN2 and EIN3/EIL1 genes are mainly controlled by ethylene at a post-translational level32,33. Hitherto, none of these genes identified in other plants has been found to be transcriptionally regulated by ethylene32. The transcriptional enhancement of EIN2 and EIL1 in latex suggests an active ethylene signalling in laticifers.
Downstream of the primary ethylene signalling pathway, ethylene-responsive element binding factors (ERFs) are implicated. A total of 225 Hevea AP2/ERF members, including 181 ERFs, 35 AP2s, 7 RAVs and 2 Soloists (Supplementary Table 26) were identified, representing the largest assemblage in this grouping as compared to other plants (Supplementary Table 27)37. Nearly one-tenth of the AP2/ERF members showed a more than twofold change in expression after ethylene treatment; 10 of them were upregulated and 11 downregulated (Supplementary Table 28). Interestingly, of the seven downregulated ERF genes, four belong to repressor subgroups (ERF-IIa and ERF-VIIIa) of ethylene response38. The depression of their expression is expected to be beneficial for activating downstream ethylene response.
Ethylene treatment triggers a number of stimulated downstream biochemical cascades in relation to latex production: sugar loading and its catabolism9,36,39,40, water uptake41, energy availability42, cytosolic alkalinization42, nitrogen assimilation43 and defence responses44. Many genes implicated in these events were among the DEGs identified in this study (Supplementary Tables 22 and 23), but not the rubber biosynthesis genes, which is consistent with the previous proposition that ethylene has little direct effect on genes in rubber biosynthesis45. The defence response genes involved in producing and scavenging reactive oxygen species (ROS) were further analysed for their importance in the functioning of laticifers20. Three DEGs in ROS production were identified, among which an l-ascorbate oxidase (scaffold2147_8831) and an NADPH oxidase (scaffold0143_441741) appeared upregulated, whereas a lipoxygenase (scaffold4250_5149) was downregulated (Supplementary Tables 22 and 23). Of the six ROS-scavenging DEGs, five that include two thioredoxins (scaffolds0444_166127 and 0520_1896151), a glutaredoxin (scaffold4986_4172), a caleosin-related peroxygenase (scaffold1473_35058) and polyamine oxidase (scaffold0036_2884533) were upregulated, and one, a thioredoxin (scaffold0794_475278), was downregulated. Both the number of DEGs involved in ROS scavenging and their expression levels after ethylene treatment were much higher than those involved in ROS production. We illustrate in Fig. 4b the mechanisms underlying ethylene stimulation of latex production in Hevea.
Discussion
First, an effort to improve genome assembly is especially meaningful in plant genomes with high repeat content46. The Hevea genome contains 71% repetitive content and our BAC-pool sequencing allowed an over 20-fold increase in scaffold N50 length, which can be exploited in sequencing other repeat-rich large plant genomes. Second, expansion and divergence of the REF/SRPP family and its correlation with rubber biosynthesis leads to new insights into the physiology of rubber-producing laticifers. Third, an in-depth RNA-seq analysis assisted by the high-quality genome assembly has allowed us to gain new understanding of the mechanisms underlying ethylene stimulation of rubber production.
What sets the rubber tree apart from the numerous other rubber-bearing plants47 is its ability to produce prodigious amounts of rubber. In this regard, the Hevea genome provides a good resource to understand how the tree has managed to accomplish this uncommon feat. An appraisal of data from the genome points to the REF/SRPP gene family, which encompasses the most highly expressed genes in the latex. The divergence of this gene family into several laticifer-specific abundant isoforms suggests extraordinary selection pressure in play. Rubber contained in latex provides Hevea with a protective function against boring pests (such as beetles) with its coagulating ability to entrap them in the exuding latex which then self-seals the wound48. From the viewpoint of evolutionary advantage to the species, this is the clear benefit that the rubber tree receives. From the anthropological perspective, this adaptation has fortuitous consequences in that it provides mankind with the building blocks of a global industry.
A search for REF and SRPP in the Hevea SNP desert is instructive. REF heads the list in gene expression in the SNP desert, implying that it is an active gene that has descended from a recent, single selection of an ancestral sequence. On the other hand, the significance of SRPP in relation to the SNP desert lies in its absence. The phylogenetic dendrogram in Fig. 3b offers an explanation. An SRPP isoform is an ancestral protein in the REF/SRPP gene family. A mutation event occurred relatively recently whereby SRPP was truncated to give rise to REF. Deletion of the SRPP C terminus that resulted in REF isoforms appears to have occurred more than once in the rubber tree's evolutionary history, but the isoforms that prevail in modern Hevea are REF1 and SRPP1. The late appearance of REF also explains why, among the other 17 plants examined, the REF/SRPP family has isoform sizes similar to or larger than SRPP1, but none with similar size to REF1. The breakout event that produced REF is important to the modern Hevea's capacity for high rubber production. Hevea is unique in its high rubber production arguably because REF is unique to Hevea.
There are two classes of rubber particles in Hevea latex: the large particles (LRPs) with REF located on their surfaces and the small particles (SRPs) that are coated with SRPP25,49. SRPs are far superior in number, accounting for 94% of all rubber particles in the latex, whereas LRPs constitute only the remaining 6% of the rubber particles. However, it is precisely this 6% of rubber particles by number that makes up 93% of the rubber by volume in the latex50. REF1 might contribute to rubber biosynthesis by facilitating the biogenesis of LRPs or the ‘growing’ of SRPs to LRPs. Two pieces of evidence support this presumption: (1) the amount of REF1 protein in the whole latex has been found to be proportional to the rubber content26; (2) REF1 gene expression was reported to correlate with yield levels of Hevea cultivars27. Taking a hypothetical stance, if REF had not evolved to facilitate the formation of LRPs, the typical tapped latex that has a dry rubber content (drc) of 33% would have less than 3% drc for the same number of SRPs per unit volume. In other words, the entire natural rubber industry is essentially founded on a very small proportion of rubber particles that are LRPs.
In Hevea planting, the ethylene generating compound, ethephon, is commonly applied to the bark of Hevea to lengthen the flow duration and to aid latex regeneration after tapping. Although bearing a weak capability for ethylene synthesis that is little affected by ethylene treatment too, the laticifers reveal an active ethylene signalling capability as evidenced by transcriptional accentuation of the ethylene signal receptor and transduction genes on ethylene stimulation. The low ethylene-synthesizing ability in laticifers, but an active ethylene signalling mechanism that is responsive to exogenous ethylene could shed new light on how ethylene triggers a significant increase in latex flow and rubber production.
The data from this study, together with other public resources, pave a way for whole genome association studies, germplasm improvement and genetic modification of Hevea to meet increasing global demand for natural rubber.
Methods
Plant materials
Genomic DNA was prepared from young leaves of six Hevea cultivars (Reyan7-33-97, PR107, RRIM600, Reyan8-79, Wenchang11 and Yunyan77-4) (Supplementary Fig. 1) using the CTAB method51. RNA samples were prepared from mature leaf, male flower, female flower, mature seed, trunk bark, latex and feeder root of ten-year-old mature Reyan7-33-97 trees that had been tapped for two years, or from the latex collected from Reyan7-33-97 trees treated with 1.5% ethephon (2-chloroethylphosphonic acid, an ethylene generator) as described previously39.
Shotgun library construction and sequencing
For genome sequencing, paired-end libraries (<600 bp) were constructed by the standard A-tailing Illumina protocol. Mate-paired libraries (800 bp to 10 kb) were constructed by a modified SOLiD mate-pair library preparation protocol52. The Reyan7-33-97 genome was sequenced by the IlluminaGA2 and Hiseq2000 systems, whereas the genomes of re-sequenced cultivars were sequenced solely on the Hiseq2000 platform. Raw reads were preprocessed by an in-house perlscript, and then proofread by the ErrorCorrection module in the SOAPdenovo package53. For RNA-seq, RNA samples were used to prepare libraries and sequenced using the Illumina Hiseq2000 system. Raw RNA-seq reads were processed to trim terminal low quality bases and adapter sequences via an in-house custom pipeline. The clean reads were then mapped to the Hevea genome using GSNAP54. Cufflinks55 was used to compute the transcripts expression levels in reads per kilobase per million reads mapped (RPKM) and to identify the differentially expressed genes (DEGs) upon ethylene stimulation. Hierarchical clustering of DEGs was conducted using custom R scripts.
Genome assembly and validation
The pre-processed paired-end reads were assembled using SOAPdenovo (69-mer size), and mate-pair reads were used to construct scaffolds using SSPACE56. Scaffold gaps were closed with Gapfiller57 and the cd-hit software was employed to filter chimeric scaffolds58. A BAC-pooling sequencing strategy was employed to improve the assembly further (Supplementary Fig. 4 and Supplementary Note 1). Public databases, BAC and transcript sequences were used to validate the genome assembly (Supplementary Note 4).
Genome annotation
Genome repeat sequences were annotated de novo by using RepeatMasker (http://www.repeatmasker.org). The protein-coding genes were predicted based on the repeat-masked genome through a combination of ab initio, conserved protein homologues and assembled transcripts. The non-coding RNAs were annotated by employing the INFERNAL software59 to search against the Rfam database (Supplementary Note 5).
Estimation of whole-genome duplication
The all-to-all BLASTP program was used to identify the homolog pairs in Hevea proteins and the MCscan program60 was used to search for collinearity blocks which were then filtered using the criterion of fewer than ten non-collinear genes between any two collinear genes. The same method was used to identify the syntenic blocks in L. usitatissimum, P. trichocarpa, R. communis and M. esculenta, or between Hevea and any of the four species. The collinearity region sizes for Hevea, M. esculenta and P. trichocarpa were estimated statistically by the Wilcox test. To estimate the duplication event, we calculated the synonymous Ks and fourfold synonymous third-codon transversion position (4DTv) using KaKs_calculator61 with the NG model and an in-house perl script, respectively.
Phylogenetic analysis
Twelve species, Medicago truncatula, Glycine max, Fragaria vesca, Vitis vinifera, Oryza sativa, L. usitatissimum, P. trichocarpa, R. communis, M. esculenta, Citrus sinensis, Carica papaya and Arabidopsis thaliana, were selected to construct a phylogenetic tree with Hevea using 72 single-copy gene families (Supplementary Note 7). These species represent the typical Rosids sequenced to date in the Eudicots. O. sativa was designated as an outgroup.
Genetic heterogeneity of Hevea cultivars
Paired-end sequences (35×) from each of the six Hevea cultivars were mapped to the Reyan7-33-97 reference genome using BWA (ref. 62). The mapping results were transformed into bam format and sorted with SAMtools63. SNP calling results were filtered against the following criteria: (1) each SNP supported by at least five non-redundant reads; (2) average mapping quality more than 40; and (3) two SNPs within 10 bp to be excluded. Based on the SNP calling results, SNP deserts (<1 SNP per kb) were identified in each sequenced cultivar using a perl script. Genes located in SNP deserts were subjected to functional annotation and GO enrichment analysis using GOEAST (http://omicslab.genetics.ac.cn/GOEAST/).
Genes associated with rubber biosynthesis, ethylene synthesis and signalling
The 20 gene families that have been reported as involved in rubber biosynthesis were identified from the genome (Supplementary Note 10). Ethylene synthesis and signalling genes that had been characterized in A. thaliana64 were retrieved for their corresponding protein sequences from the Arabidopsis Information Resource (TAIR) (https://www.arabidopsis.org/ index.jsp). The retrieved A. thaliana proteins were processed with Interproscan65 and Blastp searched against Hevea proteins. Hits sharing >30% amino acid identity and >50% amino acid alignment length with the A. thaliana homologues were further checked for pfam domain architecture. Those having the same pfam domains as their A. thaliana homologues were regarded as proteins involved in ethylene synthesis and signalling in Hevea (Supplementary Table 25).
The H. brasiliensis genome and RNA-seq sequences have been deposited in GenBank/DDBJ/EMBL under the accession codes of LVXX01000000 and SRP069104, respectively.
References
van Beilen, J. B. & Poirier, Y. Establishment of new crops for the production of natural rubber. Trends Biotechnol. 25, 522–529 (2007).
Jones, K. Natural rubber as a green commodity—Part II. Rubber Dev. 47, 37–41 (1994).
Chan, H. in Milestones in Rubber Research (ed. Ong, E. L. ) 2–15 (Malaysian Rubber Board, 2000).
Priyadarshan, P. & Goncalves, P. d.S. Hevea gene pool for breeding. Genet. Resour. Crop Ev. 50, 101–114 (2003).
Paardekooper, E. in Rubber (eds Webster, C. & Baulkwill, W. ) 349–414 (Longman Scientific and Technical, 1989).
Clément-Demange, A., Priyadarshan, P., Hoa, T. T. T. & Venkatachalam, P. Hevea rubber breeding and genetics. Plant Breeding Rev. 29, 177 (2007).
Rahman, A. Y. et al. Draft genome sequence of the rubber tree Hevea brasiliensis. BMC Genomics 14, 75 (2013).
Wang, X. et al. Comprehensive proteomics analysis of laticifer latex reveals new insights into ethylene stimulation of natural rubber production. Sci. Rep. 5, 13778 (2015).
Liu, J. P., Xia, Z. Q., Tian, X. Y. & Li, Y. J. Transcriptome sequencing and analysis of rubber tree (Hevea brasiliensis Muell.) to discover putative genes associated with tapping panel dryness (TPD). BMC Genomics 16, 398 (2015).
Hurtado Paez, U. A., Garcia Romero, I. A., Restrepo Restrepo, S., Aristizabal Gutierrez, F. A. & Montoya Castano, D. Assembly and analysis of differential transcriptome responses of Hevea brasiliensis on interaction with Microcyclus ulei. PloS ONE 10, e0134837 (2015).
Chao, J., Chen, Y., Wu, S. & Tian, W. M. Comparative transcriptome analysis of latex from rubber tree clone CATAS8-79 and PR107 reveals new cues for the regulation of latex regeneration and duration of latex flow. BMC Plant Biol. 15, 104 (2015).
Wang, S., Xu, Y., Luo, S. & Gao, X. Adaptability of new desirable rubber clone Reyan7-33-97 in central mountainous areas of Hainan province. Chin. J. Trop. Agr. 29, 1–4 (2009).
Huang, H., Liang, M., Wu, Y., Li, D. & He, J. Selection and breeding of a moderate scale clone SCATC7-33-97. Chin. J. Trop. Crops 15, 1–6 (1994).
Xi, Z. et al. Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proc. Natl Acad. Sci. USA 109, 17519–17524 (2012).
Sun, M. et al. Deep phylogenetic incongruence in the angiosperm clade Rosidae. Mol. Phylogenet. Evol. 83, 156–166 (2015).
Kaplan, N. L., Hudson, R. & Langley, C. The “hitchhiking effect” revisited. Genetics 123, 887–899 (1989).
Wang, L. et al. SNP deserts of Asian cultivated rice: genomic regions under domestication. J. Evol. Biol. 22, 751–761 (2009).
Tenaillon, M. I., U'Ren, J., Tenaillon, O. & Gaut, B. S. Selection versus demography: a multilocus investigation of the domestication process in maize. Mol. Biol. Evol. 21, 1214–1225 (2004).
Al-Mssallem, I. S. et al. Genome sequence of the date palm Phoenix dactylifera L. Nature Commun. 4, 2274 (2013).
d'Auzac, J. et al. The regulation of cis-polyisoprene production (natural rubber) from Hevea brasiliensis. Recent Res. Dev. Plant Physiol. 1, 273–332 (1997).
Chow, K. S. et al. Insights into rubber biosynthesis from transcriptome analysis of Hevea brasiliensis latex. J. Exp. Bot. 58, 2429–2440 (2007).
Sando, T. et al. Cloning and characterization of the 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway genes of a natural-rubber producing plant, Hevea brasiliensis. Biosci. Biotech. Bioch. 72, 2903–2917 (2008).
Sando, T. et al. Cloning and characterization of mevalonate pathway genes in a natural rubber producing plant, Hevea brasiliensis. Biosci. Biotech. Bioch. 72, 2049–2060 (2008).
Epping, J. et al. A rubber transferase activator is necessary for natural rubber biosynthesis in dandelion. Nature Plants 1,15048 (2015).
Oh, S. K. et al. Isolation, characterization, and functional analysis of a novel cDNA clone encoding a small rubber particle protein from Hevea brasiliensis. J. Biol. Chem. 274, 17132–17138 (1999).
Dennis, M. S. & Light, D. R. Rubber elongation factor from Hevea brasiliensis. Identification, characterization, and role in rubber biosynthesis. J. Biol. Chem. 264, 18608–18617 (1989).
Priya, P., Venkatachalam, P. & Thulaseedharan, A. Differential expression pattern of rubber elongation factor (REF) mRNA transcripts from high and low yielding clones of rubber tree (Hevea brasiliensis Muell. Arg.). Plant Cell Rep. 26, 1833–1838 (2007).
Hillebrand, A. et al. Down-regulation of small rubber particle protein expression affects integrity of rubber particles and rubber content in Taraxacum brevicorniculatum. PloS ONE 7, e41874 (2012).
Laibach, N., Hillebrand, A., Twyman, R. M., Prufer, D. & Schulze Gronover, C. Identification of a Taraxacum brevicorniculatum rubber elongation factor protein that is localized on rubber particles and promotes rubber biosynthesis. Plant J. 82, 609–620 (2015).
Berthelot, K. et al. Rubber elongation factor (REF), a major allergen component in Hevea brasiliensis latex has amyloid properties. PloS ONE 7, e48065 (2012).
Gidda, S. K. et al. Lipid droplet-associated proteins (LDAPs) are involved in the compartmentalization of lipophilic compounds in plant cells. Plant Signal. Behav. 8, e27141 (2013).
Yang, C., Lu, X., Ma, B., Chen, S. Y. & Zhang, J. S. Ethylene signaling in rice and Arabidopsis: conserved and diverged aspects. Mol. Plant 8, 495–505 (2015).
Klee, H. J. Ethylene signal transduction. Moving beyond Arabidopsis. Plant Physiol. 135, 660–667 (2004).
Yang, S. F. & Hoffman, N. E. Ethylene biosynthesis and its regulation in higher plants. Ann. Rev. Plant Physiol. 35, 155–189 (1984).
Zhu, J. H., Xu, J., Chang, W. J. & Zhang, Z. L. Isolation and molecular characterization of 1-aminocyclopropane-1-carboxylic acid synthase genes in Hevea brasiliensis. Int. J. Mol. Sci. 16, 4136–4149 (2015).
Tupy, J. in Physiology of Rubber Tree Latex (eds d'Auzac, J., Jacob, J-L & Chrestin, H. ) 179–218 (CRC, 1989).
Piyatrakul, P. et al. Sequence and expression analyses of ethylene response factors highly expressed in latex cells from Hevea brasiliensis. PloS ONE 9, e99367 (2014).
Nakano, T., Suzuki, K., Fujimura, T. & Shinshi, H. Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol. 140, 411–432 (2006).
Tang, C. et al. The sucrose transporter HbSUT3 plays an active role in sucrose loading to laticifer and rubber productivity in exploited trees of Hevea brasiliensis (para rubber tree). Plant Cell Environ. 33, 1708–1720 (2010).
Dusotoit-Coucaud, A. et al. Ethylene stimulation of latex yield depends on the expression of a sucrose transporter (HbSUT1B) in rubber tree (Hevea brasiliensis). Tree Physiol. 30, 1586–1598 (2010).
Tungngoen, K. et al. Involvement of HbPIP2;1 and HbTIP1;1 aquaporins in ethylene stimulation of latex yield through regulation of water exchanges between inner liber and latex cells in Hevea brasiliensis. Plant Physiol. 151, 843–856 (2009).
Amalou, Z., Bangratz, J. & Chrestin, H. Ethrel (ethylene releaser)-induced increases in the adenylate pool and transtonoplast delta pH within Hevea latex cells. Plant Physiol. 98, 1270–1276 (1992).
Pujade-Renaud, V. et al. Ethylene-Induced increase in glutamine synthetase activity and mRNA levels in Hevea brasiliensis latex cells. Plant Physiol. 105, 127–132 (1994).
Putranto, R. A. et al. Ethylene response factors are controlled by multiple harvesting stresses in Hevea brasiliensis. PloS ONE 10, e0123618 (2015).
Zhu, J. & Zhang, Z. Ethylene stimulation of latex production in Hevea brasiliensis. Plant Signal. Behav. 4, 1072–1074 (2009).
Feuillet, C., Leach, J. E., Rogers, J., Schnable, P. S. & Eversole, K. Crop genome sequencing: lessons and rationales. Trends Plant Sci. 16, 77–88 (2011).
Polhamus, L. G. Rubber. (Leonard Hill, London, 1962).
Sharples, A. The laticiferous system of Hevea brasiliensis and its protective function. Ann. Bot. 32, 247–251 (1918).
Berthelot, K. et al. Rubber particle proteins, HbREF and HbSRPP, show different interactions with model membranes. BBA-Biomembranes 1838, 287–299 (2014).
Yeang, H., Yip, E. & Hamzah, S. Characterisation of Zone 1 and Zone 2 rubber particles in Hevea brasiliensis latex. J. Nat. Rubb. Res. 10, 108–123 (1995).
Allen, G. C., Flores-Vergara, M. A., Krasynanski, S., Kumar, S. & Thompson, W. F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nature Protoc. 1, 2320–2325 (2006).
van Heesch, S. et al. Improving mammalian genome scaffolding using large insert mate-pair next-generation sequencing. BMC Genomics 14, 257 (2013).
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010).
Wu, T. D. & Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881 (2010).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protoc. 7, 562–578 (2012).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Nadalin, F., Vezzi, F. & Policriti, A. GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC bioinformatics 13 (Suppl. 14), S8 (2012).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488 (2008).
Zhang, Z. et al. KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics 4, 259–263 (2006).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Zhao, Q. & Guo, H. W. Paradigms and paradox in the ethylene signaling pathway and interaction network. Mol. Plant 4 626–634 (2011).
Li, W. et al. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 43, W580–W584 (2015).
Acknowledgements
We thank E. (Liansheng) Zheng for critically reading the manuscript. This research was financially supported by grants from the Fundamental Research Funds for Rubber Research Institute, CATAS, and the 863 program (2013AA102605).
Author information
Authors and Affiliations
Contributions
H.H. and C.T. conceptualized the research programme. C.T., S.H., H.H. and J.Y. designed experiments and coordinated the project. C.T. and S.H. supervised the data generation and analysis. M.Y., Y.F., C.T., Y.L., S.H.G. and H.M. performed most of the data analysis. B.Z. and X.T. organized the experiments for genome- and BAC- sequencing, RNA-seq and library construction. X.X., B.Z., Z.A., Y.Q., J.H.Y., X.L., J.Q. and Y.H. performed the field experiments, DNA and RNA extraction, and PCR amplification for target genes. Q.L., P.M., Z.H., M.S., W.J.L., X.Z., H.C., Y.L., J.Y., W.T., N.Z., R.Z.Z., D.L., P.H., Z.L., Z.Z., S.L., C.L., J.W., D.W., C.Q.L. and W.L. were partially involved in either experiments or data analysis. C.T., M.Y., Y.F, Y.H.Y., J.Y. and S.H. wrote the manuscript. All authors discussed results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figs 1–22 and Supplementary Notes. (PDF 6754 kb)
Supplementary Tables
Supplementary Tables 1–30. (XLSX 1002 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Tang, C., Yang, M., Fang, Y. et al. The rubber tree genome reveals new insights into rubber production and species adaptation. Nature Plants 2, 16073 (2016). https://doi.org/10.1038/nplants.2016.73
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/nplants.2016.73
This article is cited by
-
Genome-wide identification of rubber tree pathogenesis-related 10 (PR-10) proteins with biological relevance to plant defense
Scientific Reports (2024)
-
Response of the clone RRIM 3001 (Hevea brasiliensis) to three ethephon stimulation treatments and the identification of differentially expressed transcription factors for a water-based stimulant
Journal of Rubber Research (2024)
-
Effect of Michelia macclurei intercropping on soil properties and microbial population in young rubber plantation
Journal of Rubber Research (2024)
-
Preparation and properties of flame-retardant rubber composites based on steel slag filler
Journal of Iron and Steel Research International (2023)
-
From forest to plantation: a brief history of the rubber tree
Indian Journal of History of Science (2023)
