
Abstract
Protein structures are key to understanding biomolecular mechanisms and diseases, yet their interpretation is hampered by limited knowledge of their biologically relevant quaternary structure (QS). A critical challenge in inferring QS information from crystallographic data is distinguishing biological interfaces from fortuitous crystal-packing contacts. Here, we tackled this problem by developing strategies for aligning and comparing QS states across both homologs and data repositories. QS conservation across homologs proved remarkably strong at predicting biological relevance and is implemented in two methods, QSalign and anti-QSalign, for annotating homo-oligomers and monomers, respectively. QS conservation across repositories is implemented in QSbio (http://www.QSbio.org), which approaches the accuracy of manual curation and allowed us to predict >100,000 QS states across the Protein Data Bank. Based on this high-quality data set, we analyzed pairs of structurally conserved interfaces, and this analysis revealed a striking plasticity whereby evolutionary distant interfaces maintain similar interaction geometries through widely divergent chemical properties.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

References
Goodsell, D.S. & Olson, A.J. Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 29, 105–153 (2000).
Levy, E.D., Pereira-Leal, J.B., Chothia, C. & Teichmann, S.A. 3D complex: a structural classification of protein complexes. PLoS Comput. Biol. 2, e155 (2006).
Lukatsky, D.B., Shakhnovich, B.E., Mintseris, J. & Shakhnovich, E.I. Structural similarity enhances interaction propensity of proteins. J. Mol. Biol. 365, 1596–1606 (2007).
André, I., Strauss, C.E., Kaplan, D.B., Bradley, P. & Baker, D. Emergence of symmetry in homooligomeric biological assemblies. Proc. Natl. Acad. Sci. USA 105, 16148–16152 (2008).
Marsh, J.A. & Teichmann, S.A. Structure, dynamics, assembly, and evolution of protein complexes. Annu. Rev. Biochem. 84, 551–575 (2015).
Ahnert, S.E., Marsh, J.A., Hernández, H., Robinson, C.V. & Teichmann, S.A. Principles of assembly reveal a periodic table of protein complexes. Science 350, aaa2245 (2015).
Nooren, I.M. & Thornton, J.M. Diversity of protein-protein interactions. EMBO J. 22, 3486–3492 (2003).
Kühner, S. et al. Proteome organization in a genome-reduced bacterium. Science 326, 1235–1240 (2009).
Perica, T. et al. The emergence of protein complexes: quaternary structure, dynamics and allostery. Colworth Medal Lecture. Biochem. Soc. Trans. 40, 475–491 (2012).
Renatus, M., Stennicke, H.R., Scott, F.L., Liddington, R.C. & Salvesen, G.S. Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. USA 98, 14250–14255 (2001).
Tang, P. Hung M-C, & Klostergaard, J. Human pro-tumor necrosis factor is a homotrimer. Biochemistry 35, 8216–8225 (1996).
Pereira-Leal, J.B., Levy, E.D., Kamp, C. & Teichmann, S.A. Evolution of protein complexes by duplication of homomeric interactions. Genome Biol. 8, R51 (2007).
Berman, H.M. et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000).
Velankar, S. et al. PDBe: improved accessibility of macromolecular structure data from PDB and EMDB. Nucleic Acids Res. 44 D1, D385–D395 (2016).
Henrick, K. & Thornton, J.M. PQS: a protein quaternary structure file server. Trends Biochem. Sci. 23, 358–361 (1998).
Janin, J. Specific versus non-specific contacts in protein crystals. Nat. Struct. Biol. 4, 973–974 (1997).
Carugo, O. & Argos, P. Protein-protein crystal-packing contacts. Protein Sci. 6, 2261–2263 (1997).
Ponstingl, H., Henrick, K. & Thornton, J.M. Discriminating between homodimeric and monomeric proteins in the crystalline state. Proteins 41, 47–57 (2000).
Zhu, H., Domingues, F.S., Sommer, I. & Lengauer, T. NOXclass: prediction of protein-protein interaction types. BMC Bioinformatics 7, 27 (2006).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Bernauer, J., Bahadur, R.P., Rodier, F., Janin, J. & Poupon, A. DiMoVo: a Voronoi tessellation-based method for discriminating crystallographic and biological protein-protein interactions. Bioinformatics 24, 652–658 (2008).
Tsuchiya, Y., Nakamura, H. & Kinoshita, K. Discrimination between biological interfaces and crystal-packing contacts. Adv. Appl. Bioinform. Chem. 1, 99–113 (2008).
Bahadur, R.P., Chakrabarti, P., Rodier, F. & Janin, J. A dissection of specific and non-specific protein-protein interfaces. J. Mol. Biol. 336, 943–955 (2004).
Pal, A., Chakrabarti, P., Bahadur, R., Rodier, F. & Janin, J. Peptide segments in protein-protein interfaces. J. Biosci. 32, 101–111 (2007).
Tina, K.G., Bhadra, R. & Srinivasan, N. PIC: Protein Interactions Calculator. Nucleic Acids Res. 35, W473–W4766 (2007).
Liu, Q., Li, Z. & Li, J. Use B-factor related features for accurate classification between protein binding interfaces and crystal packing contacts. BMC Bioinformatics 15 (Suppl. 16), S3 (2014).
Elcock, A.H. & McCammon, J.A. Identification of protein oligomerization states by analysis of interface conservation. Proc. Natl. Acad. Sci. USA 98, 2990–2994 (2001).
Guharoy, M. & Chakrabarti, P. Conservation and relative importance of residues across protein-protein interfaces. Proc. Natl. Acad. Sci. USA 102, 15447–15452 (2005).
Schärer, M.A., Grütter, M.G. & Capitani, G. CRK: an evolutionary approach for distinguishing biologically relevant interfaces from crystal contacts. Proteins 78, 2707–2713 (2010).
Baskaran, K., Duarte, J.M., Biyani, N., Bliven, S. & Capitani, G. A PDB-wide, evolution-based assessment of protein-protein interfaces. BMC Struct. Biol. 14, 22 (2014).
Ashkenazy, H. et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 (2016).
Xu, Q. et al. Statistical analysis of interface similarity in crystals of homologous proteins. J. Mol. Biol. 381, 487–507 (2008).
Xu, Q. & Dunbrack, R.L. Jr. The protein common interface database (ProtCID)—a comprehensive database of interactions of homologous proteins in multiple crystal forms. Nucleic Acids Res. 39, D761–D770 (2011).
Shoemaker, B.A. et al. IBIS (Inferred Biomolecular Interaction Server) reports, predicts and integrates multiple types of conserved interactions for proteins. Nucleic Acids Res. 40, D834–D840 (2012).
Faure, G., Andreani, J. & Guerois, R. InterEvol database: exploring the structure and evolution of protein complex interfaces. Nucleic Acids Res. 40, D847–D856 (2012).
Levy, E.D. PiQSi: protein quaternary structure investigation. Structure 15, 1364–1367 (2007).
Sippl, M.J. & Wiederstein, M. Detection of spatial correlations in protein structures and molecular complexes. Structure 20, 718–728 (2012).
Koike, R. & Ota, M. SCPC: a method to structurally compare protein complexes. Bioinformatics 28, 324–330 (2012).
Mukherjee, S. & Zhang, Y. MM-align: a quick algorithm for aligning multiple-chain protein complex structures using iterative dynamic programming. Nucleic Acids Res. 37, e83 (2009).
Ritchie, D.W., Ghoorah, A.W., Mavridis, L. & Venkatraman, V. Fast protein structure alignment using Gaussian overlap scoring of backbone peptide fragment similarity. Bioinformatics 28, 3274–3281 (2012).
Zhang, Y. & Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005).
Perica, T., Chothia, C. & Teichmann, S.A. Evolution of oligomeric state through geometric coupling of protein interfaces. Proc. Natl. Acad. Sci. USA 109, 8127–8132 (2012).
Moal, I.H. & Fernández-Recio, J. SKEMPI: a structural kinetic and energetic database of mutant protein interactions and its use in empirical models. Bioinformatics 28, 2600–2607 (2012).
Andreani, J., Faure, G. & Guerois, R. Versatility and invariance in the evolution of homologous heteromeric interfaces. PLOS Comput. Biol. 8, e1002677 (2012).
Sudha, G., Singh, P., Swapna, L.S. & Srinivasan, N. Weak conservation of structural features in the interfaces of homologous transient protein-protein complexes. Protein Sci. 24, 1856–1873 (2015).
Shi, Z. & Moult, J. Structural and functional impact of cancer-related missense somatic mutations. J. Mol. Biol. 413, 495–512 (2011).
David, A. & Sternberg, M.J. The contribution of missense mutations in core and rim residues of protein-protein interfaces to human disease. J. Mol. Biol. 427, 2886–2898 (2015).
Garcia-Seisdedos, H., Empereur-Mot, C., Elad, N. & Levy, E.D. Proteins evolve on the edge of supramolecular self-assembly. Nature 548, 244–247 (2017).
Bloom, J.D., Drummond, D.A., Arnold, F.H. & Wilke, C.O. Structural determinants of the rate of protein evolution in yeast. Mol. Biol. Evol. 23, 1751–1761 (2006).
Minasov, G. et al. Functional implications from crystal structures of the conserved Bacillus subtilis protein Maf with and without dUTP. Proc. Natl. Acad. Sci. USA 97, 6328–6333 (2000).
Levy, E.D., Boeri Erba, E., Robinson, C.V. & Teichmann, S.A. Assembly reflects evolution of protein complexes. Nature 453, 1262–1265 (2008).
R Core Team. R. A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria https://www.R-project.org/ (2016).
Robin, X. et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12, 77 (2011).
Bahadur, R.P., Chakrabarti, P., Rodier, F. & Janin, J. Dissecting subunit interfaces in homodimeric proteins. Proteins 53, 708–719 (2003).
Duarte, J.M., Srebniak, A., Schärer, M.A. & Capitani, G. Protein interface classification by evolutionary analysis. BMC Bioinformatics 13, 334 (2012).
Levy, E.D. A simple definition of structural regions in proteins and its use in analyzing interface evolution. J. Mol. Biol. 403, 660–670 (2010).
Suzek, B.E., Wang, Y., Huang, H., McGarvey, P.B. & Wu, C.H. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015).
Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Pupko, T., Bell, R.E., Mayrose, I., Glaser, F. & Ben-Tal, N. Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 18 (Suppl. 1), S71–S77 (2002).
Dey, S., Pal, A., Chakrabarti, P. & Janin, J. The subunit interfaces of weakly associated homodimeric proteins. J. Mol. Biol. 398, 146–160 (2010).
Acknowledgements
We thank H. Greenblatt for valued help with operating the computer cluster, and we thank O. Dym and S. Rogotner for providing the photo of a protein crystal used in Figure 1. We thank J. Sussman for feedback on the work and D. Fass for comments on the manuscript. This work was supported by a VATAT fellowship to S.D. by the Israel Science Foundation and the I-CORE Program of the Planning and Budgeting Committee (grant nos. 1775/12 and 2179/14), by the Marie Curie CIG Program to E.D.L. (project no. 711715), by the HFSP Career Development Award to E.D.L. (award no. CDA00077/2015), and by a research grant from A.-M. Boucher. E.D.L. is incumbent of the Recanati Career Development Chair of Cancer Research.
Author information
Authors and Affiliations
Contributions
S.D. and E.D.L. designed and performed the experiments. D.W.R. adapted the Kpax algorithm to enable the calculations. S.D. and E.D.L. wrote the manuscript with input from D.W.R. All authors corrected and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Measuring quaternary structure similarity using global versus local measures.
(a) Structural similarity of two protein complexes can be inferred from a global superposition, which yields a global score, as was done in this work. (b) Structural similarity can also be assessed at the level of pairwise interfaces1–14, but such information would have to be integrated to infer a global similarity measure when complexes contain multiple interfaces. For example, in the case of a tetramer with four interfaces, four similarity measures will be obtained and this number would increase further when comparing complexes with more subunits.
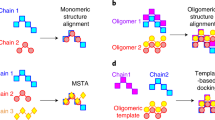
Supplementary Figure 2 Heuristic employed for superposing protein complexes.
The names of the chains in a PDB file are arbitrary. For example, considering the two tetramers depicted, chains may be labeled clockwise in one PDB file but counter-clockwise in another. Thus, although two structures can be similar structurally, differences in chain order can yield a false negative result when structures are being compared. To circumvent this problem, we must infer chain-chain correspondences among the structures being compared. This was achieved using a seed superposition of the two structures, which is based on chains from the first QS maximizing the TM-score with the second QS. If the QSs are similar, this seed superposition naturally places structurally equivalent chains in proximity, which made their identification possible by analysis of the aligned coordinates. We then used this mapping to re-write the coordinate files in matching chain order, and recalculated a global superposition of the complete QSs using the re-ordered coordinates. The latter provided us with the final TM-score.
Supplementary Figure 3 Procedure used to infer the biological significance of QSs.
Each symmetry group is considered iteratively. Within each group, each QS is used to search for structural homologs. If a homologue is found, both QSs are annotated to be “correct.” Once all the QSs of a symmetry group have been processed, each QS is used again to search for proteins identical in sequence but having different QSs. If found, we considered such QSs to be likely non-biological and annotated them as such.
Supplementary Figure 5 Integrating pairwise interface information to infer biological relevance of quaternary structures.
QSbio needs to compare QSs from PDB with predictions from PISA, EPPIC, and QSalign/anti-QSalign. Comparing QSs between PDB and PISA is achieved with the full QS superimposition approach described above (Figure S2). However, to compare QSs between PDB and EPPIC, we must employ a different strategy because EPPIC provides pairwise interface information (as opposed to assembly information). We therefore mapped pairwise information from EPPIC onto QSs from PDB using the following approach. First, each QS from PDB was decomposed into pairs of chains, using all pairs burying >90 Å2. Each pair was subsequently matched to an interface group from EPPIC by structural superposition. Each interface group in EPPIC is classified as being either biological (green) or non-biological (magenta). In the case where all subunits of the QS could be linked by biological contacts, the QS was deemed to match EPPIC (example 1) and otherwise it was inferred as non-matching (example 2).
Supplementary Figure 6 Protein interfaces are plastic.
(a) We compared interfaces of structurally similar protein complexes. We examined whether interface properties of one complex were predictive of the same property in its homologues, given different levels of sequence identity between them. (b) We first compared the interaction propensity of interfaces. Higher values indicate interfaces with a high fraction of residues normally enriched at interfaces while lower values correspond to interfaces chemically close to solvent-exposed surfaces. (c) We then compared the hydrophobicity of interface pairs, defined as the ratio of non-polar residues to the total number of interface residues. (d) Finally, we compared evolutionary conservation of interface residues relative to surface residues. Values below 1 correspond to complexes where the interface is more conserved than the surface. The right-most plot summarizes the squared correlation coefficient (R2) for each property considered, calculated for pairs of proteins binned by shared sequence identity: < 30%, 30-45%, 45-60%, 60-75% and 75-90%. All properties show very low correlation values for pairs sharing less than 30% identity, showing that despite being structurally similar, interfaces can differ dramatically in their chemistry and evolutionary properties. One thousand random data points were sampled for each plot to ease visualization.
Supplementary Figure 7 Annotating monomers with anti-Qsalign.
We annotated monomers based on the enrichment of monomeric homologs over oligomeric ones. This enrichment is used to derive probabilities by the formulae above. Proteins sharing at least 30% and at most 90% sequence identity and having an overlap of 60% or more were considered as homologs.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–7, Supplementary Tables 1–2 and Supplementary Note 1 (PDF 1050 kb)
Supplementary Data 1
Prediction details of PISA, EPPIC, QSalign/anti-QSalign and QSbio on the different datasets. (XLSX 118 kb)
Supplementary Data 2
QSbio results; for the most up-to-date information see www.QSbio.org. (XLSX 4929 kb)
Rights and permissions
About this article

Cite this article
Dey, S., Ritchie, D. & Levy, E. PDB-wide identification of biological assemblies from conserved quaternary structure geometry. Nat Methods 15, 67–72 (2018). https://doi.org/10.1038/nmeth.4510
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmeth.4510
This article is cited by
-
Mutational biases favor complexity increases in protein interaction networks after gene duplication
Molecular Systems Biology (2024)
-
Protein language models can capture protein quaternary state
BMC Bioinformatics (2023)
-
Annotating Macromolecular Complexes in the Protein Data Bank: Improving the FAIRness of Structure Data
Scientific Data (2023)
-
ScanNet: an interpretable geometric deep learning model for structure-based protein binding site prediction
Nature Methods (2022)
-
Attogram-level light-induced antigen-antibody binding confined in microflow
Communications Biology (2022)
