
Abstract
In read cloud approaches, microfluidic partitioning of long genomic DNA fragments and barcoding of shorter fragments derived from these fragments retains long-range information in short sequencing reads. This combination of short reads with long-range information represents a powerful alternative to single-molecule long-read sequencing. We develop Genome-wide Reconstruction of Complex Structural Variants (GROC-SVs) for SV detection and assembly from read cloud data and apply this method to Illumina-sequenced 10x Genomics sarcoma and breast cancer data sets. Compared with short-fragment sequencing, GROC-SVs substantially improves the specificity of breakpoint detection at comparable sensitivity. This approach also performs sequence assembly across multiple breakpoints simultaneously, enabling the reconstruction of events exhibiting remarkable complexity. We show that chromothriptic rearrangements occurred before copy number amplifications, and that rates of single-nucleotide variants and SVs are not correlated. Our results support the use of read cloud approaches to advance the characterization of large and complex structural variation.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


References
Weischenfeldt, J., Symmons, O., Spitz, F. & Korbel, J.O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 14, 125–138 (2013).
Yang, L. et al. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell 153, 919–929 (2013).
Stephens, P.J. et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011).
Baca, S.C. et al. Punctuated evolution of prostate cancer genomes. Cell 153, 666–677 (2013).
Chiang, C. et al. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat. Genet. 44, 390–397, S1 (2012).
Tupler, R. et al. A complex chromosome rearrangement with 10 breakpoints: tentative assignment of the locus for Williams syndrome to 4q33----q35.1. J. Med. Genet. 29, 253–255 (1992).
Sudmant, P.H. et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015).
Quinlan, A.R. & Hall, I.M. Characterizing complex structural variation in germline and somatic genomes. Trends Genet. 28, 43–53 (2012).
Amini, S. et al. Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing. Nat. Genet. 46, 1343–1349 (2014).
Peters, B.A. et al. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature 487, 190–195 (2012).
Voskoboynik, A. et al. The genome sequence of the colonial chordate, Botryllus schlosseri. eLife 2, e00569 (2013).
Bishara, A. et al. Read clouds uncover variation in complex regions of the human genome. Genome Res. 25, 1570–1580 (2015).
Zheng, G.X.Y. et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 34, 303–311 (2016).
Layer, R.M., Chiang, C., Quinlan, A.R. & Hall, I.M. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 15, R84 (2014).
Oliner, J.D., Kinzler, K.W., Meltzer, P.S., George, D.L. & Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 358, 80–83 (1992).
Newburger, D.E. et al. Genome evolution during progression to breast cancer. Genome Res. 23, 1097–1108 (2013).
Gao, R. et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 48, 1119–1130 (2016).
Greenman, C.D. et al. Estimation of rearrangement phylogeny for cancer genomes. Genome Res. 22, 346–361 (2012).
Garsed, D.W. et al. The architecture and evolution of cancer neochromosomes. Cancer Cell 26, 653–667 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013).
Peng, Y., Leung, H.C.M., Yiu, S.M. & Chin, F.Y.L. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012).
Rausch, T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012).
Weng, Z. et al. Cell-lineage heterogeneity and driver mutation recurrence in pre-invasive breast neoplasia. Genome Med. 7, 28 (2015).
Acknowledgements
We thank K. Giorda, S. Kyriazopoulou-Panagiotopoulou and M. Schnall-Levin for their assistance in preparing and analyzing the 10x data; and we thank D. Ramazzotti for analyzing mutation spectra. This work was supported by the Stanford Center for Computational, Evolutionary and Human Genomics (N.S.), R01CA183904 (NIH/NCI; R.B.W., S.B. and A.S.), and the BRCA Foundation (A.S.). Certain commercial equipment, instruments or materials are identified in this document. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the products identified are necessarily the best available for the purpose.
Author information
Authors and Affiliations
Contributions
N.S., Z.W., A.B., R.B.W., J.M.Z., M.S., S.B. and A.S. designed the experiments and/or analyses. N.S., Z.W., J.M. and D.C. conducted the experiments. N.S. wrote analysis software. N.S. and A.S. analyzed the data. N.S. and A.S. wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Fragment lengths for 10x libraries.
(a) Cumulative distribution functions for size-selected Sarcoma samples are quite tight, with most density concentrated between 30–60kb. (b) Non-size-selected HCC1143 breast cancer cell line libraries show much broader fragment length distributions, with more density at low and high fragment sizes.
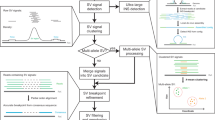
Supplementary Figure 2 Overview of GROC-SVs
(a), 10x Genomics platform begins by molecular partitioning of long DNA fragments into microfluidic droplets (each compartment is shown by a differently colored circle). Within each droplet, short reads are produced by amplification templated from the long fragments, incorporating a droplet-specific barcode. Short reads are then Illumina sequenced and aligned to the reference genome, resulting in clusters of reads all sharing the same barcode (indicated by color), allowing inference of the originating long fragment. In this example, cyan, dark brown, and beige are not involved in any structural variants, and are shown to illustrate the signal emanating from the pairwise comparison of barcodes at nearby coordinates (Diagonal, D). Because no fragments are shared between distant points, we observe a low background (B) between any two random positions in the genome. Structural variants then are identified as high barcode similarity between points that are distant within the reference genome, indicative of a genomic rearrangement. In this example, black, orange, and blue clouds span a translocation breakpoint resulting in similar barcode patterns between locus a and locus x and a characteristic off-diagonal signal (T). Green and light brown originate from the other allele. (b), Breakpoints showing barcode similarity are clustered together for the subsequent assembly and complex event reconstruction steps. All reads from all supporting barcodes are used to produce assemblies (C1, C2, C4) and these assemblies and the patterns of barcode similarity are used to produce a graph describing the order of breakpoints with each complex event. Letters (a, b, c, for chromosome 1; x, y for chromosome 2) indicate genomic segments, numbers are the breakpoint connections, in order. Breakpoint 3 illustrates that not all high-confidence breakpoints yield an interpretable sequence assembly, but that they are still part of the reconstruction.
Supplementary Figure 3 Cartoon explanation of barcode similarity histograms.
Barcode similarity histograms (for example, Figure 1a) summarize evidence for a structural variant in the regions around the breakpoint loci. Here, we show a cartoon example of a chr1/chr2 translocation, with the input fragments spanning the breakpoint shown on the left, and the resulting barcode similarity histogram shown on the right. Read clouds that share a barcode and occur in both the chr1 and chr2 regions are shown as rectangles in the histogram. Histogram signal is then proportional to the number of overlapping rectangles at a point.
Three fragments, tiling across the translocation breakpoint, are highlighted in red, blue and purple. The red fragment falls mostly on the chr1 side of the breakpoint, crosses the translocation breakpoint and extends just slightly into chr2. Hence, the corresponding red rectangle here has a long x-axis (chr1) length but a short y-axis (chr2) height. The purple fragment falls only slightly on chr1, spans the breakpoint junction and then extends a longer distance on chr2. Hence, the corresponding purple rectangle has a long height but a short width. And similarly for the blue fragment.
Because these fragments all have the same length, the height and width of the rectangles is constrained within the triangular shape; once a number of fragments support a breakpoint, they start to fill in the triangle. This triangular shape is cut off at the bottom in Figure 1e because there is another downstream breakpoint at the 93 mb locus (y-axis).
Note that all the rectangles from supporting fragments end at the same point of the histogram on the x-axis (here, on the right side) and the y-axis (at the bottom). In the main text figures, the highest signal occurs near the base of the triangle, closest to the breakpoint. Because coverage of each long fragment is sparse, we may not observe a read right at the breakpoint; thus many of the supporting read clouds extend close but not all the way up to the breakpoint location. This results in a peak of signal just upstream of the breakpoint location (particularly in the 1st generation GemCode data, where coverage is less even).
The background shown in Figure 1 and Supplementary Figure 4 corresponds to 0 (darkest blue) to 10 (white) shared barcodes. The signal (above background) is shown in shades of pink to red; because the copy number of these events is different, the highest signal value differs, so the scales are different only for the shades of pink to red. Note that there are 0 or at most 1 dark-blue rectangles representing background values in Figures 4a,b, representing the extremely low background of the 2nd generation Chromium data.
Supplementary Figure 4 Example translocation between chromosome 1 and chromosome 8 in Chromium HCC1143 breast cancer cell line
(a), tumor and (b) matched normal cell lines show essentially no background when sequenced with the 10x Genomics Chromium platform, due to the ~5-fold increase in molecular partitions (barcodes), resulting in fewer fragments per droplet (Compare to figure 1, prepared with 1st-generation 10x Genomics GemCode platform). (c) fragments extend >100 kb away from the breakpoint in either direction, providing extremely long-distance context information. (d) copy number profiles in the region of the translocation, calculated directly from the short-read coverage within the Chromium libraries (we found the Chromium coverage profiles were smoother than those from standard PCR-plus Illumina libraries prepared from the same cell lines for The Cancer Genome Atlas, not shown).
Supplementary Figure 5 Overview of structural variation in sarcoma.
(a) Circos plot of all high-confidence breakpoints found across all sarcoma samples, indicated by arcs. Blue, interchromosomal events; magenta, intrachromosomal. Otherwise, as in 2c. (b) scatterplot of copy number in the immediate vicinity of breakpoints. Each point is a breakpoint consisting of two breakends (X and Y, arbitrarily assigned) whose copy number estimates are plotted against each other. (c), location of the sampling sites from the sarcoma; 0 to 9 are from one cross-section, 10 is from a cross section parallel to it and separated by 3 cm.
Supplementary Figure 6 Comparison of support for SVs across different sequencing approaches.
(a) High correlation between the number of GROC-SVs 10x supporting barcodes (x-axis) and the number of high-quality mate-pairs (y-axis) supporting each SV. Each point represents a single breakpoint. Overall signal from mate-pair data is typically a bit higher than for 10x data. (b) Lower correlation between GROC-SVs 10x barcodes and number of short-fragment read-pairs. Note that overall signal is ~3-fold lower for short-fragment libraries, and many events are not supported by any high-quality short-fragment read-pairs. (c,d) Same as in (a) and (b), but only including subclonal SVs private to sarcoma 10. (c) Good concordance with mate-pair data demonstrates the accuracy of these calls. (d) The low number of short-fragment read-pairs supporting these events (y-axis, most events are supported by fewer than 10 read-pairs, red line) suggests that the read cloud approach can help identify subclonal SVs that are present even at relatively low allele frequency within a tumor sample. Note that only events called by GROC-SVs are shown.
Supplementary Figure 7 Mate-pair validation of SV calls.
(a) Validation of all SVs, including both germline and somatic events, called by GROC-SVs from the 10x read cloud data (orange curve), shown with decreasingly stringent confidence thresholds. A given point on the curve represents the n most confident SV calls (x-axis) and how many of these were validated by mate-pair data (y-axis). At the least stringent cutoff, 83% of events are corroborated by mate-pairs. The blue curve similarly represents SV calls performed using the ~35x standard PCR-free short-fragment Illumina libraries (Layer et al 2014). Note that the short-fragment line initially shows a steeper slope at the beginning, where the most confident events include supporting split-reads, before leveling off to the less confident events which are typically supported by only read-pairs. Overall sensitivity for short fragments was somewhat higher than for GROC-SVs, but at a much lower specificity. (b) Same as in (a), but only showing somatic events. Overall sensitivity for short fragments was slightly lower than for GROC-SVs, even at the lowest specificity, and regardless of whether an event was considered to be somatic if the control sample included 0 supporting reads (light blue) or if the control sample was allowed to include at most 1 supporting read (purple). Only events that GROC-SVs (red line) or short fragments (blue lines) called as present in one of the mate-pair-sequenced samples are included (thus some putatively subclonal variants that were only present in other samples are excluded).
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–7, Supplementary Notes 1–2 and Supplementary Table 1 (PDF 1089 kb)
Supplementary Software
GROC-SVs software. See https://github.com/grocsvs/grocsvs for most up to date version (ZIP 1806 kb)
Rights and permissions
About this article

Cite this article
Spies, N., Weng, Z., Bishara, A. et al. Genome-wide reconstruction of complex structural variants using read clouds. Nat Methods 14, 915–920 (2017). https://doi.org/10.1038/nmeth.4366
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmeth.4366
This article is cited by
-
Navigating bottlenecks and trade-offs in genomic data analysis
Nature Reviews Genetics (2023)
-
Most large structural variants in cancer genomes can be detected without long reads
Nature Genetics (2023)
-
Detection and genomic analysis of BRAF fusions in Juvenile Pilocytic Astrocytoma through the combination and integration of multi-omic data
BMC Cancer (2022)
-
Structural variant analysis of a cancer reference cell line sample using multiple sequencing technologies
Genome Biology (2022)
-
Structural variant detection in cancer genomes: computational challenges and perspectives for precision oncology
npj Precision Oncology (2021)
