
Abstract
High-throughput RNA sequencing is an increasingly accessible method for studying gene structure and activity on a genome-wide scale. A critical step in RNA-seq data analysis is the alignment of partial transcript reads to a reference genome sequence. To assess the performance of current mapping software, we invited developers of RNA-seq aligners to process four large human and mouse RNA-seq data sets. In total, we compared 26 mapping protocols based on 11 programs and pipelines and found major performance differences between methods on numerous benchmarks, including alignment yield, basewise accuracy, mismatch and gap placement, exon junction discovery and suitability of alignments for transcript reconstruction. We observed concordant results on real and simulated RNA-seq data, confirming the relevance of the metrics employed. Future developments in RNA-seq alignment methods would benefit from improved placement of multimapped reads, balanced utilization of existing gene annotation and a reduced false discovery rate for splice junctions.
Similar content being viewed by others

Main
Programs for aligning transcript reads to a reference genome address the challenging task of placing spliced reads across introns and correctly determining exon-intron boundaries. The advent of RNA-seq prompted the development of a new generation of spliced-alignment software, with several advances over earlier programs such as the BLAST-like alignment tool (BLAT)1,2. The tools GEM3, GSTRUCT, MapSplice4 and TopHat5,6 implement a two-step approach in which initial read alignments are analyzed to discover exon junctions; these junctions are then used to guide final alignment. Several programs can also use existing gene annotation to inform spliced-read placement5,6,7,8,9. Most RNA-seq aligners can further increase accuracy by prioritizing alignments in which read pairs map in a consistent fashion3,5,6,7,9,10. To place reads that match multiple genomic sequences, GSTRUCT examines the density of independent reads at those loci. Many algorithms also consider base-call quality scores and use sophisticated indexing schemes to decrease runtime.
Here we assess the performance of 26 RNA-seq alignment protocols on real and simulated human and mouse transcriptomes. We adopted a competitive evaluation model applied in other areas of bioinformatics11,12,13,14. Developers were invited to run their software and submit results for evaluation as part of the RNA-seq Genome Annotation Assessment Project (RGASP). Programs included six spliced aligners GSNAP7, MapSplice4, PALMapper8, ReadsMap, STAR9 and TopHat5,6) and four alignment pipelines (GEM3, PASS15, GSTRUCT and BAGET). GSTRUCT is based on GSNAP, whereas BAGET uses a contiguous DNA aligner to map reads to the genome as well as to exon junction sequences derived from reference gene annotation. For comparison, the contiguous aligner SMALT was also tested. SMALT can map reads in a split manner, but it lacks several features of dedicated spliced aligners, such as precise determination of exon-intron boundaries. We demonstrate that choice of alignment software is critical for accurate interpretation of RNA-seq data, and we identify aspects of the spliced-alignment problem in need of further attention.
Results
Alignment protocols were evaluated on Illumina 76-nucleotide (nt) paired-end RNA-seq data from the human leukemia cell line K562 (1.3 × 109 reads), mouse brain (1.1 × 108 reads) and two simulated human transcriptomes (8.0 × 107 reads each; Supplementary Table 1). Nine development teams contributed alignments for evaluation. We additionally included two versions of the widely used RNA-seq aligner TopHat5,6. Most development teams provided results from several alignment protocols, corresponding to different parameter choices and pipeline configurations (Fig. 1 and Supplementary Note).

Shown is the percentage of sequenced or simulated read pairs (fragments) mapped by each protocol. Protocols are grouped by the underlying alignment program (gray shading). Protocol names contain the suffix “ann” if annotation was used. The suffix “cons” distinguishes more conservative protocols from others based on the same aligner. The K562 data set comprises six samples, and the metrics presented here were averaged over them.
Alignment yield
There were major differences among protocols in the alignment yield (68.4–95.1% of K562 read pairs; mean = 91.5%, s.d. = 5.4), extent to which both reads from a pair were mapped, and frequency of ambiguous mappings (reads with several reported alignments) (Fig. 1 and Supplementary Tables 2 and 3). These trends were similar across data sets (Fig. 1). The fraction of pairs with only one read aligned was typically highest for TopHat, ReadsMap and PASS, whereas PALMapper output exhibited more complex discrepancies within read pairs. GEM results consistently included many ambiguous mappings (37% of sequenced reads per data set on average). Mapping ambiguities were also common with PALMapper, although these were reduced with the more conservative protocols that involve stringent filtering of alignments (Fig. 1 and Supplementary Fig. 1). To avoid introducing bias at later evaluation stages due to differences in the number of alignments per read, we instructed developer teams to assign a preferred (primary) alignment for each read mapped in their program output. The following results are based on these primary alignments unless otherwise noted.
Mismatches and basewise accuracy
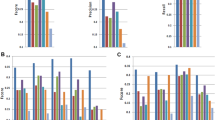
Compared to the other aligners, GSNAP, GSTRUCT, MapSplice, PASS, SMALT and STAR reported more primary alignments devoid of mismatches (Fig. 2a), partly because these methods can truncate read ends and thus output an incomplete alignment when they are unable to map an entire sequence (Fig. 2b). PASS and SMALT performed extensive truncation, suggesting that these programs often report alignments shorter than is optimal. MapSplice, PASS and TopHat displayed a low tolerance for mismatches (Fig. 2a). Consequently, a large proportion of reads with low base-call quality scores were not mapped by these methods (Supplementary Fig. 2). The mapping yield of TopHat was particularly low (mean yield of 84% on K562 data, compared to 90% for MapSplice; Fig. 2a and Supplementary Tables 2 and 3), likely owing to a lack of read truncation (Fig. 2b). Note that many aligners have options to increase mismatch tolerance beyond the settings used here, but this approach may negatively affect other performance aspects.

(a) Percentage of sequenced reads mapped with the indicated number of mismatches. (b) Percentage of sequenced reads truncated at either or both ends. Bar colors indicate the number of bases removed.
Polymorphisms and accumulated mutations distinguish the cancer cell line K562 from the human reference assembly, which itself is a consensus based on several individuals16. Conversely, mouse RNA samples were obtained from strain C57BL/6NJ, the genome of which is nearly identical to the mouse reference assembly17. Accordingly, high-quality reads from mouse were mapped at a greater rate and with fewer mismatches than those from K562 (Supplementary Fig. 3). Even so, differences among aligners in mismatch and truncation frequencies were consistent across data sets (Fig. 2 and Supplementary Fig. 4). Mapping properties are thus largely dependent on software algorithms even when the genome and transcriptome are virtually identical.
Consistent with real RNA-seq data, GSNAP, GSTRUCT, MapSplice and STAR outperformed other methods for basewise accuracy on simulated data (Supplementary Table 2). As expected, error rates were substantially lower for uniquely mapped reads than for primary alignments of multimapped reads (Supplementary Table 4). Notably, despite the many ambiguous mappings reported by GEM and PALMapper, the primary alignments were usually correct (Supplementary Table 4).
Differences among methods were most apparent for spliced reads (Supplementary Tables 5,6,7). On the first simulated data set, GSNAP, GSTRUCT, MapSplice and STAR mapped 96.3–98.4% of spliced reads to the correct locations and 0.9–2.9% to alternative locations (Fig. 3 and Supplementary Table 6). Although these mappers assigned nearly all spliced reads to the correct locus, the frequency of reads for which they aligned all bases correctly was substantially lower (60.3–89.3% of spliced reads from simulation 1; Fig. 3). In contrast, ReadsMap and the annotation-based TopHat2 protocol produced high rates of perfect spliced alignments and few partially correct ones (Fig. 3 and Supplementary Table 6), a behavior consistent with the aforementioned lack of read truncation. However, ReadsMap also assigned an exceptionally high proportion of bases to the wrong genomic positions, largely owing to a programmatic error that placed reads a few bases from their correct locations (Fig. 3 and Supplementary Table 5).

The second simulated data set was designed to be more challenging, with higher frequencies of insertions and deletions (indels), base-calling errors and novel transcript isoforms. MapSplice, PASS and TopHat showed a reduction in performance on this data set relative to the other methods (Fig. 3 and Supplementary Tables 5,6,7), results consistent with the low mismatch tolerance of these protocols (Fig. 2a).
Indel frequency and accuracy
GEM and PALMapper output included more indels than any other method (up to 115 indels per 1,000 K562 reads; Fig. 4a and Supplementary Fig. 5), but GEM preferentially reported insertions, and PALMapper, mostly deletions. Long deletions were most common with GSNAP and GSTRUCT, whereas TopHat2 called numerous long insertions. In contrast, PASS, ReadsMap and TopHat1 reported few long indels, and the conservative PALMapper protocols allowed only single-nucleotide indels.

(a) Bars show the size distribution of indels for the human K562 data set. Indel frequencies are tabulated (number of indels per 1,000 sequenced reads). (b) Precision and recall, stratified by indel size, for human simulated data set 1.
These results were corroborated by analysis of indel accuracy on simulated data (Fig. 4b), which demonstrated that GEM and PALMapper report many false indels (indel precision < 37% for all protocols except PALMapper cons; simulation 1), that GSNAP and GSTRUCT exhibit high sensitivity for deletions largely independent of size (recall > 68% for each length interval depicted in Fig. 4b), and that the annotation-based TopHat2 protocol is the most sensitive method for long insertions (recall = 87% for insertions ≥5 bp; simulation 1). The ability of GSNAP, GSTRUCT and TopHat2 to detect long indels was accompanied by high false discovery rates, however, and MapSplice achieved a better balance between precision and recall for long deletions than GSNAP (Fig. 4b; this balance can be quantified using the F-score, which for deletions ≥5 bp was 87% for MapSplice and 36% for GSNAP on simulation 1 when these programs were executed without provision of gene annotation). Supplementary Figure 6 illustrates alignments of two simulated reads that each contain a small insertion, resulting in erroneous mappings by several protocols.
Positioning of mismatches and gaps in reads
We determined the spatial distribution of mismatches, indels and introns over read sequences (Supplementary Fig. 7). All methods except MapSplice and PASS consistently reported an increasing frequency of mismatches along reads, in agreement with base-call quality-score distributions (Supplementary Figs. 2 and 8). BAGET, GEM, MapSplice, PALMapper and TopHat produced an excess of mismatches at read termini, whereas other methods avoided such a bias by truncating reads (Fig. 2b). Indels were preferentially placed near ends of reads by some methods, such as PALMapper and TopHat; others, such as MapSplice and STAR, tended to place them internally. GSTRUCT produced the most uniform distribution of indel frequency over the K562 data (coefficient of variation (CV) = 0.32), and TopHat produced the most variable (CV = 1.5 and 1.1 for TopHat1 and TopHat2, respectively). The positioning of splice junctions was generally more even, although several methods did not call junctions near read termini (Supplementary Fig. 7).
Coverage of annotated genes
We assessed how RNA-seq reads were placed in relation to annotated gene structures from the Ensembl database (Supplementary Note). Given the extensive annotation of the human and mouse genomes, the majority of reads would be expected to originate from known exons. Experimental data will also contain an unknown fraction of sequencing reads from unannotated transcripts and heterogeneous nuclear RNA. The simulated data sets were generated to recapitulate these features (Online Methods). Mapping trends were typically very similar between real and simulated data, a result indicating that simulation results reflect alignment performance in real RNA-seq experiments (Supplementary Figs. 9,10,11). The number of reads mapped to annotated exons were highest for GSNAP and GSTRUCT, on both real and simulated data, and close to the true number for the latter (Supplementary Figs. 9,10,11,12). However, all methods dispersed reads across too many genes: whereas reads from the first simulation should map to 16,554 Ensembl genes, all protocols reported primary alignments for more than 17,800 genes. This effect was largely due to the placement of reads at pseudogenes and was most severe for SMALT, BAGET and GEM (Supplementary Figs. 9,10,11).
Spliced alignment
In assessing spliced-alignment performance, we distinguish between detection of splices in individual reads and detection of unique splice junctions on the genomic sequence. The latter are often supported by multiple splices depending on expression level and sequencing depth. In general, GSNAP, GSTRUCT, ReadsMap, STAR and TopHat2 reported more (predicted) splices than other aligners (Fig. 5a and Supplementary Table 2). However, these results differed among protocol variants, such that GSNAP, STAR 1-pass and TopHat2 produced substantially fewer spliced mappings unless alignment was guided by known splice sites. SMALT, BAGET, PASS and the conservative PALMapper protocols inferred the fewest splices from the data (Fig. 5a and Supplementary Fig. 13). Several methods reported numerous splices not corresponding to known introns, particularly ReadsMap and PALMapper, and, to a lesser extent, SMALT, GSTRUCT and STAR 2-pass (Fig. 5a). These novel splice junctions were typically supported by few alignments, and many featured noncanonical splice signals, which suggests that they may be incorrect (Fig. 5b and Supplementary Figs. 14 and 15). A substantial proportion were exclusive to particular methods. For example, 52–54% of the novel junctions reported by GSNAP/GSTRUCT on K562 whole-cell RNA were absent from the output of all other mappers (Supplementary Table 8).

(a) Frequency and accuracy of splices in primary alignments. Splice frequency was defined as the number of reported splices divided by the number of sequenced reads. For simulated data (center and right), splice recall and false discovery rate (FDR) is presented. Insets show details of the dense upper-left areas (gray rectangles). (b) Number of annotated and novel junctions reported at different thresholds for the number of supporting mappings. In the rightmost plot, filled symbols depict the number of junctions with at least one supporting mapping, and lines demonstrate the result of thresholding. (c) Junction discovery accuracy for simulated data set 1 (top) and 2 (bottom). Counts of true and false junctions were computed at increasing thresholds for the number of supporting mappings, and results were depicted as in b to obtain receiver operating characteristic–like curves. Gray horizontal lines indicate the number of junctions supported by true simulated alignments. (d) Accuracy for the subset of junctions contained in the Ensembl annotation. (e) Accuracy for junctions absent from the Ensembl annotation.
Analysis of splice-detection performance on simulated data confirmed a substantial false discovery rate for ReadsMap, PALMapper and SMALT, whereas the highest accuracy was achieved by protocols based on GSNAP, GSTRUCT, MapSplice and STAR (Fig. 5a). Splices near the ends of reads can be particularly difficult to align, as a minimum amount of sequence is needed to confidently identify exon boundaries. Accuracy improved when the assessment was restricted to splices located between positions 20 and 57 in the 76-nt reads, but the same four methods still performed best (Supplementary Fig. 16). The use of simulated data further allowed us to measure the rate at which splices were detected in individual reads as a function of true coverage at corresponding junctions. Most protocols displayed decreased sensitivity at junctions covered by <5 reads (Supplementary Fig. 17). This reflects the reliance on junction coverage by alignment algorithms to increase precision. Accordingly, the trend was absent for methods that align each read independently (BAGET, GSNAP, PASS, SMALT and STAR 1-pass). Notably, the annotation-based GSNAP protocol achieved high sensitivity irrespective of junction coverage (Supplementary Fig. 17).
The number of false junction calls was considerable for most protocols but was greatly reduced if junctions were filtered by supporting alignment counts (Fig. 5c). At a threshold of two alignments, GSTRUCT outperformed most other methods on both simulated data sets when assessed by numbers of true and false junction calls (Fig. 5c and Supplementary Tables 2 and 9). MapSplice displayed similar performance on the first simulated data set, but only if used without annotation.
The simulated transcriptomes contain a subset of splice junctions in the Ensembl annotation as well as junctions from other gene catalogs and those created by simulating alternate isoforms of known genes. This corresponds to a realistic scenario wherein a subset of known transcripts are expressed in the assayed sample and knowledge of the transcriptome is incomplete. Protocols using annotation recovered nearly all of the known junctions in expressed transcripts, but most of these protocols also aligned reads at thousands of annotated junctions that were not expressed the simulated transcriptomes (Fig. 5d). This effect was particularly severe for TopHat2, PALMapper and STAR. For novel-junction discovery, GSTRUCT and MapSplice outperformed other methods (Fig. 5e).
Most programs could detect three or more splices per read, but PASS and PALMapper rarely reported more than two, and BAGET and SMALT never reported more than one (Supplementary Fig. 18 and Supplementary Table 10). In general, ReadsMap, STAR and the annotation-based TopHat2 protocol produced the most primary alignments with at least three splices. The last protocol was also the most sensitive for recovering such multi-intron alignments from the simulated reads (recall = 79.3% for simulation 1; Supplementary Table 11). Among the protocols run without annotation, ReadsMap exhibited the best recall for alignments spanning three or more introns (72.1%), followed by the 2-pass version of STAR (70.7%) and GSTRUCT (65.8%). However, ReadsMap also exhibited exceptionally low precision for such alignments (Supplementary Table 11).
Influence of aligners on transcript reconstruction
To assess the impact of alignment methodology on exon discovery and transcript reconstruction, we applied the transcript assembly program Cufflinks to the alignments. Exon detection results based on K562 data were similar for GEM, GSNAP, GSTRUCT, MapSplice, STAR and TopHat (Fig. 6a). With the K562 whole-cell RNA primary alignments from these methods, up to 69% of the exons reported by Cufflinks matched Ensembl annotation, and up to 51% of all exons from annotated protein-coding genes were recovered. Performance was substantially lower with output from the other alignment programs (Fig. 6a). Inclusion of secondary alignments negatively affected transcript reconstruction for methods that reported numerous such alignments (GEM and PALMapper) but typically had a small effect for other methods (Supplementary Fig. 19).

(a,b) Cufflinks performance was assessed by measuring precision and recall for individual exons (a) and spliced transcripts (b). For K562 data, precision was defined as the fraction of predicted exons matching Ensembl annotation, and recall as the fraction of annotated protein-coding gene exons that were predicted.
The six aligners noted above also enabled highly accurate exon detection on the first simulated data set, with recall reaching 84% and precision 83% (Fig. 6a). On the second, more challenging simulated data set, the TopHat2 protocol using annotation outperformed other methods, followed by GSNAP (with annotation) and GSTRUCT (with or without annotation) (Fig. 6a). The same protocols gave the best Cufflinks accuracy for the more complex task of reconstructing spliced transcripts (Fig. 6b).
It should be noted that the advantage of the annotation-based TopHat2 protocol was apparent only for reconstruction of exons and transcripts present in the annotation provided to aligners (Supplementary Table 12). This observation is consistent with the unique approach of TopHat2 involving read alignment to full-length annotated transcript sequences. It may seem paradoxical that several methods exhibiting relatively poor precision for junction alignments (Fig. 5c–e) produced high-quality input for transcript reconstruction. However, the Cufflinks algorithm is able to discard erroneous exon junctions in the input data at a high rate. For example, on the data from the first simulation, 71% of true junctions identified by the annotation-based TopHat2 protocol were incorporated into transcripts by Cufflinks, compared to 5% of false junctions (Supplementary Table 13).
Discussion
In general, GSNAP, GSTRUCT, MapSplice and STAR compared favorably to the other methods, consistent with an earlier evaluation that included a subset of these tools18. Our assessment shows MapSplice to be a conservative aligner with respect to mismatch frequency, indel and exon junction calls. Conversely, the most significant issue with GSNAP, GSTRUCT and STAR is the presence of many false exon junctions in the output. This can be ameliorated by filtering junctions on the number of supporting alignments. It should be noted that both GSNAP and GSTRUCT require considerable computing time when parameterized for sensitive spliced alignment7, and the GSTRUCT pipeline has not yet been released. A recent runtime comparison found GSNAP and MapSplice to perform similarly, whereas TopHat2 and STAR were about 3 and 180 times faster, respectively9.
RNA-seq aligners use gene annotation to achieve better placement of spliced reads, and the resulting improvement was apparent on several metrics, particularly for GSNAP and the 1-pass version of STAR. Notably, these programs align each read independently, and the effect of using annotation was generally less pronounced for tools that carry out splice-junction discovery before final alignment, such as GEM, MapSplice, GSTRUCT and STAR 2-pass. TopHat also belongs to this class of programs, but provision of annotation still had a major effect on TopHat2 results, most likely because of the unique strategy whereby reads are aligned directly against annotated transcripts. This approach is clearly effective in several respects but may be suitable only for genomes with near-complete annotation.
Remaining challenges include exploiting gene annotation without introducing bias, correctly placing multimapped reads, achieving optimal yet fast alignment around gaps and mismatches, and reducing the number of false exon junctions reported. Ongoing developments in sequencing technology will demand efficient processing of longer reads with higher error rates and will require more extensive spliced alignment as reads span multiple exon junctions. We expect performance of the aligners evaluated here to improve as current shortfalls are addressed. Differential treatment of these issues will enhance and expand the range of RNA-seq aligners suited to varied computational methodologies and analysis aims.
Methods
RNA-seq data.
The human K562 data used here correspond to the K562 poly(A)+ RNA samples produced at Cold Spring Harbor Laboratory for the ENCODE project19 and can be accessed at http://www.encodeproject.org/. RNA-seq libraries were sequenced using a strand-specific protocol and comprise two biological replicates each of whole-cell, cytoplasmic and nuclear RNA. The mouse RNA-seq data set was produced at the Wellcome Trust Sanger Institute as part of the Mouse Genomes Project using brain tissue from adult mice of strain C57BL/6NJ. The library was constructed using the standard Illumina protocol that does not retain strand information. These data have been previously described20 and are available from the European Nucleotide Archive (http://www.ebi.ac.uk/ena/) under accessions ERR033015 and ERR033016. All of the data used in this study have been consolidated as a single experimental record in the ArrayExpress repository (http://www.ebi.ac.uk/arrayexpress/) under accession E-MTAB-1728.
Simulated RNA-seq data were generated using the BEERS toolkit (http://cbil.upenn.edu/BEERS/), and additional modeling of base-call errors and quality scores was done with simNGS (http://www.ebi.ac.uk/goldman-srv/simNGS/). BEERS has been previously described18. Briefly, the simulator takes as input a database of transcript models and a quantification file that specifies expression levels for each transcript and intron in the database. A transcriptome is simulated by sampling a specified number of transcript models from the database at random and creating additional alternative splice forms from each model. Polymorphisms (indels and substitutions) are introduced into the exons according to independent rates. Reads are then produced from the transcriptome in an iterative manner. In each iteration, a transcript is chosen with probability proportional to its expression level in the quantification file. An intron may be left in, with probability based on the intronic expression levels in the quantification file. A fragment of normally distributed length is sampled from the transcript, and the L bases from each end of this fragment are reported, where L is the read length.
Here, the simulator was executed using the transcript database and quantification file previously described18. This database comprises 538,991 transcript models merged from 11 annotation tracks available from the UCSC Genome Browser (AceView, Ensembl, Geneid, Genscan, NSCAN, Other RefSeq, RefSeq, SGP, Transcriptome, UCSC and Vega), and expression levels were derived from a human retina RNA-seq data set. In each of the two simulations, 25,000 transcripts were randomly chosen from the database, and two additional alternative isoforms were generated for each sampled transcript. The proportion of signal originating from novel isoforms was 20% and 35% for simulation 1 and 2, respectively. Substitution variants were introduced into exons at rates of 0.001 (simulation 1) and 0.005 (simulation 2) events per base pair, and indel polymorphisms at rates of 0.0005 (simulation 1) and 0.0025 (simulation 2). The simulated transcriptomes included 136,226 (simulation 1) and 134,717 (simulation 2) unique splice junctions, of which 90% and 92%, respectively, were represented in the simulated reads (Supplementary Table 9).
The option to simulate sequencing errors was disabled. Instead, the program simNGS was used to add noise to the simulated reads. simNGS recreates observations from Illumina sequencing machines using the statistical models underlying the AYB base-calling software21. Here, base-call errors and quality scores were simulated by applying simNGS version 1.5 with a paired-end simulation model. The model was trained on intensity data released by Illumina from a sequencing run on the HiSeq 2000 instrument using TruSeq chemistry. The resulting quality-score distributions are shown in Supplementary Figure 8, and the correct alignments of simulated data have been deposited in ArrayExpress under accession E-MTAB-1728.
Alignment protocols making use of gene annotation were provided with annotation from Ensembl only (Supplementary Note), whereas the simulated transcriptomes were based on Ensembl as well as several additional gene catalogs. In addition, novel transcript isoforms and retained introns were simulated, as detailed above. This reflects a realistic scenario where knowledge of the transcriptome is incomplete even for well-studied organisms, and a proportion of transcripts captured by RNA-seq correspond to pre-spliced mRNAs.
Read alignment.
Developer teams were provided with RNA-seq data, human and mouse reference genome sequences, and transcript annotations from the Ensembl database. So that we avoided potential biases, teams were not informed of the final evaluation criteria and were not given the true results for simulated data. Developers providing alignments for evaluation could not access submissions from other teams and were prohibited from participating in the analysis phase as part of the study design. Details of alignment protocols are provided in the Supplementary Note.
Evaluation of alignments.
Developer teams provided alignments in BAM format. These files were processed to ensure compliance with the SAM specification22 and eliminate formatting discrepancies that otherwise could have affected the evaluation. Mismatch information (NM and MD tags) was stripped from the files and recomputed using the SAMtools command “calmd” to ensure that mismatches were counted in the same manner for all protocols22. The resulting alignment files have been deposited in ArrayExpress under accession E-MTAB-1728.
With inspiration from earlier benchmarking studies9,18,23, we devised several performance metrics to assess attributes ranging from fundamental (for example, proportion of mapped reads and base-level alignment characteristics) to advanced, including splice junction detection, read placement around indels and suitability of alignments for transcript reconstruction. A detailed description of evaluation metrics is provided in the Supplementary Note, and key results are summarized in Supplementary Table 2. Unless otherwise noted, evaluation metrics for alignments of K562 RNA-seq data were averaged over the six K562 data sets (Supplementary Table 1). A subset of K562 samples were not processed by PALMapper and ReadsMap (Supplementary Table 3). Comparisons with gene annotation were performed using the Ensembl annotation that was provided to aligners (Supplementary Note).
Treatment of alignment gaps.
In the BAM format, alignment gaps in read sequences can be described as either deletions or introns. Small gaps are typically labeled deletions and longer gaps considered introns, but the exact criteria differ among aligners. To prevent the introduction of bias from such differences, we reclassified deletions and introns where appropriate. Specifically, for the indel results presented in Figure 4 and Supplementary Figure 5 and the evaluation of splice accuracy on simulated data, an alignment gap in the read sequence was considered a deletion if shorter than 19 bp and otherwise counted as an intron. We aimed to select a threshold that would minimize relabeling of gaps in the read sequence, and we observed that only three methods (BAGET, GSNAP and GSTRUCT) reported a substantial frequency of deletions longer than 18 bp from any data set. Up to 2.0% of the deletions in the output from GSNAP and GSTRUCT exceeded 18 bp, compared to 0.16% for BAGET and <0.001% for all other methods. The adjustment noticeably affected the results for GSNAP and GSTRUCT only.
For alignments of simulated RNA-seq data, accuracy metrics were computed by comparison with the alignments produced by the simulator. For computation of basewise and indel accuracy, ambiguity in indel placement was accounted for18. For example, in an alignment of the sequences ATTTA and ATTA, there are three equivalent gap placements in the latter sequence (A-TTA, AT-TA and ATT-A), all of which were considered correct. A general strategy was implemented to handle positional ambiguity for indels of any size.
Transcript reconstruction.
Transcript assembly was conducted with Cufflinks version 2.0.2. The option library-type was set to fr-firststrand for the K562 data, which are strand specific, and to fr-unstranded for the simulated data, which are not. Default values were used for other parameters.
Cufflinks requires spliced alignments to have a SAM format tag (XS) indicating the genomic strand (plus or minus) on which the transcript represented by the read is likely to be encoded. Alignment programs such as TopHat can set the XS tag by using information about the library construction protocol (for strand-specific libraries) or by inspecting sequence at exon-intron boundaries. Five of the methods evaluated here (BAGET, GEM, ReadsMap, SMALT and STAR) did not provide XS tags; we therefore post-processed the alignment output from these methods to add them. For the strand-specific K562 data, XS tags were set on the basis of alignment orientation and read number (first or second in pair), as done by TopHat. For alignments of simulated reads, we set XS tags according to the initial and terminal dinucleotides of the inferred introns, which are expected to be GT/AG, GC/AG or AT/AC for plus-strand transcripts and CT/AC, CT/GC or GT/AT for minus-strand transcripts24. For the XS tag to be added to an alignment, at least one exon junction was required to have these signals, and conflicting signals among junctions were not allowed.
We noted that the annotation-based TopHat2 protocol uses the annotation provided to set the XS tag for unspliced alignments that overlap annotated exons. As this is a unique feature of TopHat2 that might confer an advantage in the evaluation of transcript reconstruction, we investigated the effect of removing the XS tag from unspliced alignments in the TopHat2 output before running Cufflinks. This modification had a negligible effect on the Cufflinks accuracy metrics presented here (data not shown), demonstrating that provision of XS tags for unspliced alignments cannot explain why the annotation-based TopHat2 protocol resulted in better Cufflinks performance than other protocols.
For K562 data, exon precision was defined as the fraction of predicted exons matching GENCODE annotation, and exon recall as the fraction of annotated exons that were predicted. Only exons from protein-coding genes were considered when computing recall, as some noncoding RNA classes are likely to be underrepresented in the RNA-seq libraries. Results on simulated data were benchmarked against simulated gene models, using analogous definitions of precision and recall, such that exon precision measures the proportion of predicted exons matching an exon in the simulated transcriptome, and transcript precision is the fraction of predicted spliced transcripts matching a simulated spliced transcript. To stratify recall by expression, we divided simulated transcripts into three groups of equal size according to expression level (Fig. 6b). Internal exons were required to be recovered with exact boundaries, first and terminal exons were required to have correctly predicted internal borders only, and exons constituting unspliced transcripts were scored as correct if covered to at least 60% by a predicted unspliced transcript. For the simulated data, only exons of spliced transcripts were required to be placed on the correct strand, as the orientation of single-exon transcripts cannot be reliably predicted unless RNA-seq libraries are strand specific. Spliced transcripts were considered to be correctly assembled if the strand and all exon junctions matched.
Program availability.
Source code for the evaluations performed in this study can be obtained from https://github.com/RGASP-consortium/.
Accession codes
References
Kent, W.J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Fonseca, N.A., Rung, J., Brazma, A. & Marioni, J.C. Tools for mapping high-throughput sequencing data. Bioinformatics 28, 3169–3177 (2012).
Marco-Sola, S., Sammeth, M., Guigó, R. & Ribeca, P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods 9, 1185–1188 (2012).
Wang, K. et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 38, e178 (2010).
Trapnell, C., Pachter, L. & Salzberg, S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Wu, T.D. & Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881 (2010).
Jean, G., Kahles, A., Sreedharan, V.T., De Bona, F. & Rätsch, G. RNA-Seq read alignments with PALMapper. Curr. Protoc. Bioinformatics 32, 11.6 (2010).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Guigó, R. et al. EGASP: the human ENCODE Genome Annotation Assessment Project. Genome Biol. 7 (suppl. 1), S2 (2006).
Moult, J., Fidelis, K., Kryshtafovych, A. & Tramontano, A. Critical assessment of methods of protein structure prediction (CASP)—round IX. Proteins 79 (suppl. 10), 1–5 (2011).
Earl, D. et al. Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome Res. 21, 2224–2241 (2011).
Marbach, D. et al. Wisdom of crowds for robust gene network inference. Nat. Methods 9, 796–804 (2012).
Campagna, D. et al. PASS: a program to align short sequences. Bioinformatics 25, 967–968 (2009).
International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004).
Keane, T.M. et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294 (2011).
Grant, G.R. et al. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM). Bioinformatics 27, 2518–2528 (2011).
Djebali, S. et al. Landscape of transcription in human cells. Nature 489, 101–108 (2012).
Danecek, P. et al. High levels of RNA-editing site conservation amongst 15 laboratory mouse strains. Genome Biol. 13, R26 (2012).
Massingham, T. & Goldman, N. All Your Base: a fast and accurate probabilistic approach to base calling. Genome Biol. 13, R13 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Langmead, B. & Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Iwata, H. & Gotoh, O. Comparative analysis of information contents relevant to recognition of introns in many species. BMC Genomics 12, 45 (2011).
Acknowledgements
This work was supported by the European Molecular Biology Laboratory, US National Institutes of Health/NHGRI grants U54HG004555 and U54HG004557, Wellcome Trust grant WT09805, and grants BIO2011-26205 and CSD2007-00050 from the Ministerio de Educación y Ciencia.
Author information
Authors and Affiliations
Consortia
Contributions
P.B., R.G., J.H., T.J.H. and N.G. conceived of and organized the study. G.R.G. and B.S. created the simulated RNA-seq data. Consortium members provided alignments for evaluation. P.G.E., T.S., B.S. and G.R.G. analyzed the data. P.G.E. and P.B. coordinated the analysis and wrote the paper with input from the aforementioned authors. A.K. and G.R. carried out preliminary analysis and metric development based on earlier RNA-seq and alignment data but did not evaluate the alignments described herein.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–19, Supplementary Tables 1–13 and Supplementary Note (PDF 7988 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Engström, P., Steijger, T., Sipos, B. et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods 10, 1185–1191 (2013). https://doi.org/10.1038/nmeth.2722
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmeth.2722
This article is cited by
-
c-Kit signaling potentiates CAR T cell efficacy in solid tumors by CD28- and IL-2-independent co-stimulation
Nature Cancer (2023)
-
ReadZS detects cell type-specific and developmentally regulated RNA processing programs in single-cell RNA-seq
Genome Biology (2022)
-
Blocking ActRIIB and restoring appetite reverses cachexia and improves survival in mice with lung cancer
Nature Communications (2022)
-
Genomic and transcriptomic analysis of a library of small cell lung cancer patient-derived xenografts
Nature Communications (2022)
-
Zea mays RNA-seq estimated transcript abundances are strongly affected by read mapping bias
BMC Genomics (2021)
