
Abstract
We evaluated 25 protocol variants of 14 independent computational methods for exon identification, transcript reconstruction and expression-level quantification from RNA-seq data. Our results show that most algorithms are able to identify discrete transcript components with high success rates but that assembly of complete isoform structures poses a major challenge even when all constituent elements are identified. Expression-level estimates also varied widely across methods, even when based on similar transcript models. Consequently, the complexity of higher eukaryotic genomes imposes severe limitations on transcript recall and splice product discrimination that are likely to remain limiting factors for the analysis of current-generation RNA-seq data.
Similar content being viewed by others


Main
High-throughput sequencing instruments necessitate a shotgun approach for all but the shortest target molecules. Full-length representation of most cellular RNAs from sequencing data requires computational reconstruction of transcript structures. The majority of such programs infer transcript models from the accumulation of read alignments to the genome1,2,3,4; some take the alternative approach of de novo reconstruction, in which contiguous transcript sequences are assembled without the use of a reference genome5,6,7.
Here we present a detailed evaluation of computational methods for transcript reconstruction and quantification from RNA-seq data, in a framework based on the Encyclopedia of DNA Elements (ENCODE) Genome Annotation Assessment Project (EGASP)8. Developers of leading software programs were invited to participate in a consortium effort, the RNA-seq Genome Annotation Assessment Project (RGASP), to benchmark methods to predict and quantify expressed transcripts from RNA-seq data. Results were evaluated from methods based on genome alignments (Augustus9, Cufflinks3, Exonerate10, GSTRUCT, iReckon2, mGene11, mTim, NextGeneid12, SLIDE4, Transomics, Trembly and Tromer13) as well as de novo assembly (Oases5 and Velvet14). Our results identify aspects of RNA-seq analysis in which current approaches are relatively adept, along with more challenging areas for future improvement.
Results
We evaluated a total of 25 transcript reconstruction protocols, basing our analysis on alternate parameter usage of 14 software packages on RNA-seq data sets for three species (Supplementary Fig. 1, Supplementary Table 1 and Supplementary Note). Programs were run by the original developers, with the exception of Cufflinks, iReckon and SLIDE. So that we could assess the ability of each method to interpret transcript expression from RNA-seq data without prior knowledge of gene content, programs were run without genome annotation, aside from iReckon and SLIDE, which require such information. Performance was benchmarked relative to the subset of annotated exons to which RNA-seq reads mapped (coverage of ≥1 read pair per 100 bp) and their corresponding transcripts (Online Methods).
Identification of annotated features
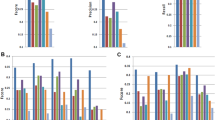
We first assessed the degree to which gene components reported by each algorithm matched the reference annotation at the nucleotide level. From the Caenorhabditis elegans data, the methods Augustus, mGene and Transomics displayed excellent performance in detecting exonic bases but also reported the expression of substantial proportions of genomic sequence outside of reference exons (Fig. 1 and Supplementary Table 2). Recall (sensitivity) was generally lower for Drosophila melanogaster, although most protocols exceeded 75% for both model organisms. Performance decreased for Homo sapiens data, for which trade-offs between precision and recall were more apparent. SLIDE and iReckon must be provided with gene annotation and therefore outperformed most other methods. Even so, iReckon attained low precision at the nucleotide level, primarily owing to the prediction of transcript isoforms with retained introns. Augustus, Exonerate, GSTRUCT, NextGeneid, Trembly and Velvet attained both precision and recall above 60% on the human data. The highest recall for methods without annotation was observed for Tromer and Cufflinks, albeit at the cost of low precision. These programs consistently displayed high sensitivity across the three species, but the precision rates for Tromer in particular indicate a tendency for overprediction.

The plots show performance at detecting exonic nucleotides. Sensitivity (blue) indicates the proportion of known exon sequence in each genome covered by assembled transcripts, and precision (orange) indicates the proportion of reported expressed sequence confined to known exons. Some protocol variants considered all expressed transcripts (all) or excluded those of low abundance (high). Programs run with gene annotation are grouped separately. iReckon was run with complete reference annotation (full) and with transcript boundaries only (ends). Transcript reconstruction methods are described in the Supplementary Note.
Exon identification from RNA-seq data
We assessed the ability of each method to identify individual exons from RNA-seq data relative to the reference annotation (Fig. 2). Inaccurate determination of transcription start and end sites is a known shortcoming of RNA-seq and, together with biological variation, impairs the identification of transcript boundaries15,16,17,18,19. To mitigate this, we allowed the 5′ ends of first exons and 3′ ends of terminal exons to differ from the reference coordinates (Online Methods and Supplementary Fig. 2). Without these relaxed criteria, agreement in transcription start site and polyadenylation site positioning between predicted and annotated exons was extremely rare (Supplementary Fig. 3). Similarly, prediction accuracy for translation start and stop sites was lower than for internal exon boundaries, which can be inferred from spliced alignments (Supplementary Fig. 3 and Supplementary Table 3). Allowing for variable transcript boundaries led to substantial improvements (Supplementary Tables 4 and 5). Although most protocols exhibited the lowest precision for the human RNA-seq data, for all three species, performance approached that of iReckon and SLIDE, despite the latter two benefiting from the use of high-quality gene annotation.

The plots show performance at detecting individual exons as the percentage of reference exons with a matching feature in the submission (sensitivity, blue) and the proportion of reported exons that agree with annotation (precision, orange).
Coding exons can be identified directly from genomic sequence by the presence of translation start and stop sites and of splice acceptors and donors. Programs such as Augustus, Exonerate, mGene, NextGeneid, Tromer and Transomics use these features to improve exon discovery. Of these programs, Augustus, mGene and Transomics identified a greater proportion of annotated coding exons than did Exonerate, mTim, NextGeneid and Tromer (Supplementary Fig. 4). These methods augment data-driven transcript reconstruction with ab initio gene prediction, leading us to conclude that higher sensitivity measures are due to more extensive utilization of the underlying genomic sequence, which reduces the need for support from RNA-seq data.
We investigated the impact of sequencing depth on exon detection rates (Fig. 3a). Through the use of ab initio prediction, Augustus, mGene and Transomics were able to detect exons from protein-coding transcripts present at very low abundance. All other methods required a minimum average read depth to identify exons. Exon detection increased with sampling coverage at a roughly linear rate until reaching a plateau. One exception was Tromer, which often reported short exon fragments of 50–75 bp flanking introns without extending them to full exons (Supplementary Fig. 5). With increasing coverage, Tromer showed a tendency to predict very long exons spanning multiple annotated features. To a lesser extent, Oases and Velvet also showed reduced performance for high-coverage exons (Supplementary Fig. 6).

(a) Sensitivity for detection of annotated exons stratified by read depth. (b) Annotated introns were binned on length, and sensitivity was calculated separately for each bin.
The ab initio prediction advantage of Augustus, mGene and Transomics was lost for noncoding transcripts, which lack the sequence features exploited by these methods (Supplementary Fig. 7). Nevertheless, detection rates were similar to those of other protocols (Supplementary Fig. 8). Noncoding RNAs tend to be expressed at lower levels than protein-coding genes (Supplementary Fig. 9) and were detected with lower sensitivity even when we controlled for differences in sequencing coverage (Fig. 3a and Supplementary Fig. 7). Exons of long intergenic noncoding RNAs were usually identified with lower frequency than those from pseudogenes and unclassified processed transcripts (Supplementary Fig. 10).
Intron detection from RNA-seq data
The relative number and size of introns differ markedly between the three species used for this study (Supplementary Table 6). Overall Augustus, mGene and Transomics showed the highest intron detection rates (Fig. 3b). However, Transomics exhibited a sharper decline with increased intron length. This trend was apparent for all methods except Tromer, for which a markedly lower detection rate was observed for introns shorter than 300 bp. To better characterize the differences in intron detection between methods, we classified reported introns on the basis of overlap with known splice sites (Fig. 4). Most protocols predominantly detected known introns; several, however, also predicted a substantial number of introns with one or two novel splice sites. The highest frequencies of novel junctions were predicted by mGene, Transomics, Tromer, Velvet and the Augustus protocol that used only genomic sequence.

Reported introns were classified by overlap with splice sites annotated in the reference gene sets.
To explain this trend, we note that intron detection is highly dependent on the underlying read alignments and that some aligners are more conservative than others20. For example, PALMapper21 was used as the alignment component in the mGene and mTim protocols. This aligner places more reads across unannotated splice sites than do GEM22, GSNAP23 and TopHat24,25; the latter programs form part of the NextGeneid, GSTRUCT and Cufflinks protocols, respectively.
Assembly of exons into transcript isoforms
We next evaluated the performance of each method in linking exons into defined splice products. We initially determined the gene loci for which any expression was reported, regardless of whether a valid transcript was identified, followed by those consistent with at least one annotated isoform (Supplementary Fig. 11). Most algorithms detect transcription at over 80% of gene loci where expression is supported by RNA-seq reads. However, performance decreased substantially when genes were considered for which at least one annotated transcript had been identified. For unguided transcript reconstruction, valid isoforms were assembled for roughly half of expressed genes on average (H. sapiens mean 41%, maximum 61%; D. melanogaster mean 55%, maximum 73%; C. elegans mean 50%, maximum 73%), and for those only one isoform was typically identified (Supplementary Fig. 12).
A substantial reduction in sensitivity was also observed from the gene to transcript level, even when the flexible evaluation mode was used for first and terminal exons (Fig. 5a and Supplementary Table 5). The best-performing methods identified at most 56–59% of spliced protein-coding transcripts from C. elegans (Augustus, mGene and Transomics), 43% from D. melanogaster (Augustus) and merely 21% from H. sapiens (Trembly). Sensitivity increased by roughly 10% when partial isoform matches were considered, as did precision when partial predictions consistent with annotated isoforms were included (Fig. 5a).

(a) Reference transcripts with a matching submission entry (transcript-level sensitivity, blue) and reported transcripts that match the reference (transcript-level precision, orange). (b) Transcripts for which various subsets of constituent exons have been reported.
Greater sequencing depth improved transcript assembly for D. melanogaster and C. elegans (Supplementary Fig. 13a), whereas in H. sapiens transcript detection remained low despite sequencing coverage in excess of 4,000 read pairs per kilobase in exonic regions. Generally, at least one consistent isoform was identified for highly expressed genes: >50% in D. melanogaster and C. elegans and >35% in H. sapiens (Supplementary Fig. 13b). Detection rates were even lower for noncoding RNAs (Supplementary Fig. 14). Pseudogenes were reported with similar frequency to that of protein-coding genes by Augustus, mGene, NextGeneid and Transomics, as pseudogenes retain partially intact coding sequences that can be identified by these methods (Supplementary Fig. 15).
The dramatic differences between species is further due to the tendency of methods to assign one splice product per gene (Supplementary Table 1). Whereas fewer than 25% of genes in C. elegans and D. melanogaster give rise to more than two transcript isoforms, human genes are annotated with an average of five, and it is unclear how many are simultaneously expressed. Assigning a single transcript model per gene will therefore impede the detection of multiple isoforms expressed in a given sample.
To identify the limiting factors in this process, for each method we calculated the number of known transcripts for which (i) all exons were identified, (ii) exactly one exon was missing, (iii) more than one exon was missing and (iv) no exons were detected at all (Fig. 5b). The results clearly show that missing exons severely compromised transcript identification. For a substantial percentage of transcripts, not all exons were identified, ranging from 30% in C. elegans to greater than 60% in H. sapiens. Interestingly, although Trembly did not perform as well as Augustus, mGene and Transomics at the exon level, this method reported the highest number of transcripts for which all exons were represented from H. sapiens data. In contrast, Augustus, mGene and Transomics detected at least one exon for most transcripts. The remaining methods failed to identify any exons for nearly 20% of all transcripts expressed in the RNA-seq data. SLIDE exhibited the same trend despite the provision of annotated exon coordinates.
We then examined the topology of transcript structures to determine how well each method was able to link exons into complete isoforms. Even in cases in which all exons of an annotated transcript had been identified, full isoforms were often not assembled (Supplementary Fig. 16). For C. elegans and D. melanogaster, most methods were able to reconstruct 60% of transcripts from the RNA-seq data. However, from the H. sapiens data, less than 40% of known transcripts were assembled. Tromer stands out as an exception: the program identified all exons for relatively few genes; but once accounted for, these were frequently linked into annotated transcript structures. Further inspection showed that these tended to be short isoforms comprising 2–3 exons on average and thus represent a more tractable subset of the transcriptome.
Provision of transcript start and end sites gave iReckon an advantage for the more complex human transcriptome, as evidenced by increased accuracy in assembling partial transcripts. In contrast, SLIDE consults exon coordinates but ignores their connectivity, performing at a level similar to methods without any prior transcript-level information. Reported transcript structures often differed substantially (Fig. 6a), and few were consistent across all methods (Supplementary Fig. 17). Pairwise agreement (Fig. 6b) was markedly higher for the model organisms than for human (median 25%), reflecting the number of partial isoforms identified as a function of transcriptome complexity.

(a) The upper tracks show RNA-seq read coverage (from STAR alignments; see Online Methods) and annotated genes. Exon predictions from the ten methods that quantified transcripts are illustrated below the annotated gene by colored boxes. Exons predicted to belong to the same transcript isoform are connected. Original and median-scaled RPKM values are presented to the right and left, respectively, of the transcript models. For the gene RPF2, all methods reported different isoforms and expression levels. Where multiple overlapping isoforms were identified, that with the higher RPKM was selected for visualization, and spliced isoforms were prioritized over unspliced ones. The noncoding RNA U6 is not expressed. (b) Heat maps illustrate pairwise agreement between reported transcript isoforms for H. sapiens (left), D. melanogaster (center) and C. elegans (right). (c) Correlation between reported RPKM values and NanoString counts (Pearson r of log-transformed values). NanoString counts were compared to the highest RPKM value reported for transcript isoforms consistent with the probe design (correlation rc) or for any isoform from the locus (correlation ra).
Quantification of expression levels from RNA-seq data
A common feature of transcript reconstruction software is the estimation of expression levels from transcribed genes. These are given as digital read counts normalized by transcript length and sequencing depth (reads per kilobase of exon model per million mapped reads, RPKM)26. RPKM values were reported at the transcript level from a subset of methods. A range of expression-level distributions was evident (Supplementary Fig. 18), but generally there was strong agreement among Augustus, iReckon, mGene and Trembly for all three RNA-seq data sets (Supplementary Figs. 19,20,21,22). One source of variation arises from gene loci where divergent or incomplete transcript models have been computed (Fig. 5a and Supplementary Figs. 23 and 24). However, expression-level estimates can vary considerably even where concordant transcript structures are reported (Supplementary Fig. 17). Such differences were also apparent after we scaled the RPKM distributions to equalize median expression values.
To establish independent expression-level quantification, we assayed a set of human genes using the NanoString nCounter amplification-free detection system27 (Supplementary Tables 7,8,9). Correlation between NanoString counts and RNA-seq RPKMs ranged from 0.34 for Transomics to 0.68 for Cufflinks (Fig. 6c and Supplementary Fig. 25). Many methods failed to report numerous targeted exons or junctions that were expressed according to NanoString counts. Read support at those loci was typically sparse, with 19 probes having no corresponding alignments from the RNA-seq data. These were, however, represented by low NanoString counts, which indicated that the nCounter assay exhibits higher sensitivity for low-abundance transcripts than RNA-seq (Supplementary Fig. 26). For ten of the unsupported NanoString probes, consistent isoforms were still reported by either Augustus, iReckon, mGene, SLIDE or Transomics. Thus, although the expression levels of these genes reflect the lower limits of detection for both technologies, sequencing reads dispersed over the gene body can allow for adequate transcript identification where ab initio methods or gene annotation were applied.
In general, all methods displayed higher identification rates for exons and junctions with higher NanoString counts, and reliable detection from RNA-seq data was dependent on read depth (Supplementary Fig. 27). Nonetheless, each failed to report a subset of exons and junctions despite the availability of adequate RNA-seq alignments (Supplementary Fig. 27b). Comparing NanoString counts with RPKM values of the predominant isoform reported for each gene (irrespective of whether the targeted exon or junction was identified) improved correlation for most methods and did so substantially for mTim and Transomics (Fig. 6c and Supplementary Fig. 28).
Discussion
Technical limitations imposed by short-read sequencing lead to a number of computational challenges in transcript reconstruction and quantification. Methods that combine ab initio prediction with experimental data were more effective at detecting genes expressed at low abundance or genes from samples with low sequencing coverage. Even so, the benefits of this approach lessened with increased transcriptome complexity.
These results underscore the difficulty of transcript assembly. For most transcripts, automated methods failed to identify all constituent exons, and in cases in which all exons were reported, the protocols tested often failed to assemble the exons into complete isoforms. Whereas methods using ab initio prediction retained an advantage in detecting individual exons, others performed better at linking them together. No single protocol excelled at all metrics. Comparing the performance of Augustus with and without RNA-seq data as input revealed that using experimental evidence only slightly improved exon-level detection but increased transcript-level precision. Transomics featured enhanced precision for high-abundance transcripts, but expression-level differences had little impact on detection sensitivity. Precision was a consistent strength of GSTRUCT, whereas mGene exhibited diminished performance on human RNA-seq data, a result underscoring that choice of method can depend on the organism under study.
Considerable variation was observed in the range of expression-level estimates reported for transcripts arising from the same gene loci. This was exacerbated by nonuniform exon detection and linkage between methods but was also apparent when similar or identical transcript structures were reported. Thus, it may be unreliable to directly compare gene-based RPKM values from sample data processed independently with different software tools. RNA-seq data to be compared from disparate sources should be treated in an identical manner from the initial processing steps. When this is not possible, care should be taken to ensure that similar gene models have been identified, and RPKM distributions should be inspected before expression-level thresholds are applied in downstream analyses. Alternatively, uniform quantification of predicted transcripts can be performed with dedicated software28,29,30.
The potential for noncoding RNA discovery and characterization is a distinct advantage of RNA-seq over gene-based expression profiling. However, this remains a challenging area for automated analysis methods. Performance is often impaired by lower expression levels of noncoding transcripts relative to that of many protein-coding genes, coupled with the inherent lack of translational features at the sequence level. The presence of open reading frames and translation start and stop signals allowed some methods to identify protein-coding transcripts even at very low expression levels, whereas the detection of noncoding RNAs at high confidence required much greater read depth. Sequencing coverage thus appears to be crucial for accurate noncoding RNA analysis.
The methods evaluated here can be applied to a range of analysis strategies, largely dependent on the state of the reference genome assembly and associated gene annotation for the target species (Supplementary Table 10 and Supplementary Note). To improve the accuracy of existing annotation using RNA-seq data, both Cufflinks and iReckon consult known gene structures during the transcript assembly process and may be useful in refining the coordinates of exon and transcript boundaries. Where a finished genome and high-quality annotation are available, Cufflinks and rQuant (part of the mGene protocol) can be applied solely for transcript quantification, which can be further improved by correcting for fragment bias. Gene prediction algorithms such as Augustus and mGene can be used to automate the annotation of novel genomes, whereas RNA-seq experiments based on partial or low-quality genome builds can be approached with a de novo assembler such as Oases. This last application is expected to receive increasingly wider attention with the continued sequencing of new genomes.
RNA-seq offers the potential to refine existing gene annotation through the discovery of novel exons and junction sites. However, unannotated transcript isoforms assembled from RNA-seq data should be interpreted with care, and those critical to an experimental study should be subjected to independent validation. The expression of multiple transcript isoforms and novel splice variants presents a major obstacle to accurate transcriptome reconstruction. Both exon identification and novel RNA discovery can improve with increased read depth, but the benefits of additional sampling to transcript assembly are inherently limited by the library construction requirements of current high-throughput sequencing platforms. Ultimately, the evolution of RNA-seq will move toward single-pass determination of intact transcripts. Third-generation instruments will realize that potential and inspire new computing approaches to meet the next wave of innovation in transcriptome analysis.
Methods
RNA-seq data.
RNA-seq data were generated as part of the ENCODE31 and modENCODE projects32, along with a third data set of compatible sequencing format and read depth, and represent three widely studied species: H. sapiens (liver hepatocellular carcinoma cell line HepG2)33, D. melanogaster (L3 stage larvae)34, and C. elegans (L3 stage larvae)35. These were chosen to reflect realistic examples of varying transcriptome complexity and where high-quality annotated reference genomes are available. Libraries were prepared for the Illumina platform and sequenced in 76-nt paired-end format to obtain approximately 100 million read pairs per sample.
H. sapiens RNA-seq data correspond to ENCODE33 HepG2 whole-cell long poly(A)+ RNA CALTECH replicate 2, available from http://www.encodeproject.org/. The D. melanogaster data set comprised a total of five sequencing runs from the modENCODE project34 for three L3 stage larval samples and can be obtained from the Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) under accession numbers SRR023546, SRR023608, SRR023505, SRR027108 and SRR026433. The C. elegans RNA-seq data have previously been described35 and are available under accession SRR065719. All of the data used in this study have been consolidated as a single experimental record in the ArrayExpress repository (http://www.ebi.ac.uk/arrayexpress/) under accession E-MTAB-1730.
Reference gene annotation.
As not all genes are expressed in the samples used in the study, benchmarking methods against the entire set of annotated genes would underestimate transcript detection sensitivity. Therefore, we processed the genome annotations (H. sapiens: GENCODE31 v.15 (Ensembl release 70), D. melanogaster: FB2013_01, C. elegans: WS200) to include only exons and transcripts with sufficient support in the RNA-seq data. Reads were mapped to the reference genomes using STAR version 2.2.0c, an independent RNA-seq aligner that is not a component in any of the evaluated transcript assembly methods36. To improve spliced alignment, STAR was provided with exon junction coordinates from the reference annotations. Default alignment parameters were used for the human data. For D. melanogaster and C. elegans, the intron size limit was reduced to 100,000 and 15,000 respectively (using options -alignIntronMax and -alignMatesGapMax). For each annotated exon, the read coverage (number of uniquely mapped read pairs divided by exon length) was computed, and exons with a value below 0.01 fragments per base pair were excluded from further analysis. Only transcripts for which all exons satisfied this criterion were included in transcript-level assessments. The threshold was determined by examining the exonic read coverage distribution, which consisted of three main features: a small peak at the low end (coverage < 0.01 fragments per base pair), a dominant peak (coverage > 0.1) and a shoulder in between. Inspection of read alignments suggested that spurious reads are overrepresented in the minor peak, whereas the shoulder region comprises low-abundance transcripts and was therefore included in the analysis. To rule out potential bias imparted by the choice of alignment program, we calculated sensitivity and precision metrics for expressed genes using several different spliced aligners (GSNAP, STAR and TopHat2), with no substantial change to the results (Supplementary Figs. 29,30,31).
Transcript prediction and assembly.
Developer teams were provided with RNA-seq data and reference genome sequences for each species. So that we avoided potential biases, teams were not informed of the final evaluation criteria and were not provided with gene annotation unless otherwise noted (for example, iReckon and SLIDE). Developers providing transcript models for evaluation could not access submissions from other teams and were prohibited from participating in the analysis phase as part of the study design. Details of transcript reconstruction protocols are provided in the Supplementary Note.
Data processing for Cufflinks, iReckon and SLIDE.
RNA-seq reads were aligned with TopHat version 2.0.3 using parameters suited to each species. The genomes of D. melanogaster and C. elegans contain a high percentage of small introns (Supplementary Table 2); examining their size distributions led us to set the parameters -i, -min-coverage-intron and -min-segment-intron to 30 for C. elegans, 40 for D. melanogaster and 50 for H. sapiens.
Cufflinks was run with default settings except for the parameter -min-intron-length, which was set to 30 for C. elegans, 40 for D. melanogaster and 50 for H. sapiens, consistent with the TopHat alignments. So that we maintained the greatest compatibility with submitted results that were computed without annotation. The protocol iReckon ends was run with the minimum annotation requirements, i.e., start and end sites of all annotated transcripts (not filtered by read coverage), whereas iReckon full was provided with the complete reference annotation. SLIDE was run in discovery mode and provided with the full unfiltered annotation for each genome.
Evaluation of prediction sets.
Feature predictions were evaluated against the filtered reference annotation sets at four structural levels: nucleotide, exon, transcript and gene. The nucleotide-level metrics measure the ability of methods to identify exonic regions, ignoring the strand and exact boundaries of features. Nucleotide-level precision was computed as the number of genomic base pairs within both annotated and predicted exons, divided by the number of genomic base pairs within predicted exons. Similarly, nucleotide recall was computed as the number of genomic base pairs shared between annotated and predicted exons, divided by the number of genomic base pairs within annotated exons.
The exon-level metrics measure the ability of the different algorithms to identify the correct strand and boundaries of exons. Precision was calculated as the percentage of reported exons with an annotated counterpart, and recall denotes the percentage of annotated exons that were correctly assembled. Annotated exons were classified as first, internal, terminal and those comprising unspliced transcripts (single exons). Unless stated otherwise, a flexible evaluation mode was employed for first, terminal and single exons. Specifically, first and terminal exons were required to have correctly predicted internal borders only, and exons constituting unspliced transcripts were scored as correct if covered to at least 60% by a predicted transcript. Exons shared between different transcript isoforms were counted once. For comparison, certain analyses were also carried out using a fixed evaluation mode, where annotated and predicted exons were required to match exactly.
Transcript-level precision was computed as the percentage of reported spliced transcripts matching an annotated transcript, and recall as the percentage of annotated spliced transcripts with a counterpart in the transcript reconstruction output. Consistent with the flexible evaluation mode for exons (see above), transcript start and end sites were allowed to differ between reference and prediction, but splice sites were required to match exactly. Genes were scored as correctly predicted if at least one annotated transcript isoform in a given gene locus was correct. To estimate the degree of similarity between transcript predictions, we calculated a pairwise agreement score. The score a[i,j] denotes the fraction of transcription products predicted by protocol i consistent with those from protocol j. Methods were ordered by hierarchical clustering based on the distance metric 1 – (a[i,j] + a[j,i])/2.
Evaluation of transcript quantification.
To compare transcript quantification results between methods, we identified for each annotated gene the corresponding predominant transcript reported; this was defined as the transcript with the highest reported RPKM value among those isoforms intersecting annotated exons of the gene. A subset of human transcripts was quantified independently by NanoString assays. Genes of at least 1 kb in length, for which annotated exon-intron structures have been manually curated, and having at least two transcripts satisfying these criteria were selected. A total of 109 genes were targeted by 141 distinct probes, designed against specific exons or splice junctions.
NanoString counts were compared to the highest RPKM value reported for transcript isoforms consistent with the probe design (correlation rc) or for any isoform from the locus (correlation ra). Predicted transcripts were required to contain the exon or junction targeted by the NanoString probe. Where multiple such transcripts were reported for the same gene, the highest RPKM value was used. Where no such transcript was reported, an RPKM of 0 was assigned. Percentages reflect the probes for which transcripts satisfying these criteria were reported. Pearson's r was calculated on the basis of the log-transformed NanoString counts and RNA-seq RPKM values. Expression values were incremented by 1 before transformation to avoid infinite numbers.
Software availability.
Source code for the evaluations performed in this study can be obtained from https://github.com/RGASP-consortium/.
References
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Mezlini, A.M. et al. iReckon: simultaneous isoform discovery and abundance estimation from RNA-seq data. Genome Res. 23, 519–529 (2013).
Roberts, A., Pimentel, H., Trapnell, C. & Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 27, 2325–2329 (2011).
Li, J.J., Jiang, C.-R., Brown, J.B., Huang, H. & Bickel, P.J. Sparse linear modeling of next-generation mRNA sequencing (RNA-Seq) data for isoform discovery and abundance estimation. Proc. Natl. Acad. Sci. USA 108, 19867–19872 (2011).
Schulz, M.H., Zerbino, D.R., Vingron, M. & Birney, E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28, 1086–1092 (2012).
Grabherr, M.G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Robertson, G. et al. De novo assembly and analysis of RNA-seq data. Nat. Methods 7, 909–912 (2010).
Guigó, R. et al. EGASP: the human ENCODE Genome Annotation Assessment Project. Genome Biol. 7 (suppl. 1), S2 (2006).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Slater, G.S.C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Schweikert, G. et al. mGene: accurate SVM-based gene finding with an application to nematode genomes. Genome Res. 19, 2133–2143 (2009).
Blanco, E., Parra, G. & Guigó, R. Using geneid to identify genes. Curr. Protoc. Bioinformatics 18, 4.3 (2007).
Sperisen, P. et al. trome, trEST and trGEN: databases of predicted protein sequences. Nucleic Acids Res. 32, D509–D511 (2004).
Zerbino, D.R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Lenhard, B., Sandelin, A. & Carninci, P. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nat. Rev. Genet. 13, 233–245 (2012).
Di Giammartino, D.C., Nishida, K. & Manley, J.L. Mechanisms and consequences of alternative polyadenylation. Mol. Cell 43, 853–866 (2011).
Tian, B., Hu, J., Zhang, H. & Lutz, C.S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 33, 201–212 (2005).
Batut, P., Dobin, A., Plessy, C., Carninci, P. & Gingeras, T.R. High-fidelity promoter profiling reveals widespread alternative promoter usage and transposon-driven developmental gene expression. Genome Res. 23, 169–180 (2013).
Shepard, P.J. et al. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA 17, 761–772 (2011).
Engström, P.G. et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods 10.1038/nmeth.2722 (3 November 2013).
Jean, G., Kahles, A., Sreedharan, V.T., De Bona, F. & Rätsch, G. RNA-Seq read alignments with PALMapper. Curr. Protoc. Bioinformatics 32, 11.6 (2010).
Marco-Sola, S., Sammeth, M., Guigo, R. & Ribeca, P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods 9, 1185–1188 (2012).
Wu, T.D. & Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881 (2010).
Trapnell, C., Pachter, L. & Salzberg, S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Mortazavi, A., Williams, B.A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Kulkarni, M.M. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr. Protoc. Mol. Biol. 94, 25B.10 (2011).
Li, B. & Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Katz, Y., Wang, E.T., Airoldi, E.M. & Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 7, 1009–1015 (2010).
Bohnert, R. & Rätsch, G. rQuant.web: a tool for RNA-Seq-based transcript quantitation. Nucleic Acids Res. 38, W348–W351 (2010).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
The modENCODE Consortium et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330, 1787–1797 (2010).
Djebali, S. et al. Landscape of transcription in human cells. Nature 489, 101–108 (2012).
Graveley, B.R. et al. The developmental transcriptome of Drosophila melanogaster. Nature 471, 473–479 (2011).
Mortazavi, A. et al. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res. 20, 1740–1747 (2010).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Acknowledgements
This work was supported by European Molecular Biology Laboratory, US National Institutes of Health/NHGRI grants U54HG004555 and U54HG004557, Wellcome Trust grant WT098051, and grants BIO2011-26205 and CSD2007-00050 from the Ministerio de Educación y Ciencia.
Author information
Authors and Affiliations
Consortia
Contributions
J.H., R.G. and T.J.H. conceived of and organized the study. Consortium members provided transcript models for evaluation. J.H. and P.B. coordinated the analysis, which was carried out by T.S., J.F.A., P.G.E. and F.K. T.S., P.B. and P.G.E. wrote the manuscript with input from the other authors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–31, Supplementary Tables 1–10 and Supplementary Note (PDF 9770 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Steijger, T., Abril, J., Engström, P. et al. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods 10, 1177–1184 (2013). https://doi.org/10.1038/nmeth.2714
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmeth.2714
This article is cited by
-
ASAPA: a bioinformatic pipeline based on Iso-Seq that identifies the links among alternative splicing, alternative transcription initiation and alternative polyadenylation
Functional & Integrative Genomics (2024)
-
Detection of Novel BEST1 Variations in Autosomal Recessive Bestrophinopathy Using Third-generation Sequencing
Current Medical Science (2024)
-
Long-read sequencing reveals the landscape of aberrant alternative splicing and novel therapeutic target in colorectal cancer
Genome Medicine (2023)
-
Comprehensive analysis of neoantigens derived from structural variation across whole genomes from 2528 tumors
Genome Biology (2023)
-
Long read isoform sequencing reveals hidden transcriptional complexity between cattle subspecies
BMC Genomics (2023)
