
Abstract
Faithful segregation of replicated chromosomes is essential for maintenance of genetic stability and seems to be monitored by several mitotic checkpoints. Various components of these checkpoints have been identified in mammals, but their physiological relevance is largely unknown. Here we show that mutant mice with low levels of the spindle assembly checkpoint protein BubR1 develop progressive aneuploidy along with a variety of progeroid features, including short lifespan, cachectic dwarfism, lordokyphosis, cataracts, loss of subcutaneous fat and impaired wound healing. Graded reduction of BubR1 expression in mouse embryonic fibroblasts causes increased aneuploidy and senescence. Male and female mutant mice have defects in meiotic chromosome segregation and are infertile. Natural aging of wild-type mice is marked by decreased expression of BubR1 in multiple tissues, including testis and ovary. These results suggest a role for BubR1 in regulating aging and infertility.
Similar content being viewed by others


Main
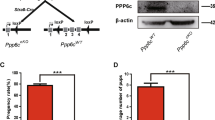
In humans, aueuploidy is a hallmark of spontaneous abortions, birth defects and most cancers1,2,3. Accurate separation of duplicated chromosomes is ensured by the spindle assembly checkpoint, which prevents anaphase onset until each kinetochore has attached to mitotic spindle microtubules4,5,6. To determine the physiological role of the spindle assembly checkpoint protein BubR1, encoded by Bub1b, we produced a series of mice in which expression of BubR1 is reduced in graded fashion from normal levels to zero by the use of wild-type (Bub1b+), knockout (Bub1b−) and hypomorphic (Bub1bH) alleles (Fig. 1a,b and Supplementary Methods and Supplementary Fig. 1 online). As previously described7, complete loss of BubR1 caused early embryonic lethality (data not shown). Bub1b−/H mice died within several hours of birth, seemingly due to respiratory insufficiency, whereas Bub1bH/H mice survived to adulthood. Bub1bH/H mice were normal in appearance and size at birth but had slow postnatal growth (Fig. 1c). Bub1b+/− and Bub1b+/H mice had no overtly abnormal phenotypes (Supplementary Fig. 2 online). Decreased expression of BubR1 was confirmed by western-blot analysis (Fig. 1d). BubR1 signals from Bub1b+/H, Bub1b+/−, Bub1bH/H and Bub1b−/H mouse embryonic fibroblasts (MEFs) were 42% (± 15%), 29% (± 9%), 11% (± 3%) and 4% (± 2%), respectively, of those from Bub1b+/+ MEFs.

(a) Schematic representation of the Bub1b gene-targeting strategy. Part of the Bub1b locus (+), the targeting vector with loxP sites (open triangles), the hypomorphic (H) and knockout (−) Bub1b alleles, BamHI restriction sites (B) and the Southern blot probe are indicated. (b) Southern-blot analysis of mice with indicated Bub1b genotypes. The 15-kb and 10-kb fragments represent the wild-type and hypomorphic alleles, respectively. (c) Growth curves of Bub1b+/+, Bub1b+/H and Bub1bH/H mice. (d) Western-blot analysis of MEFs, testis and thymus from mice carrying the indicated Bub1b alleles with antibodies against BubR1 (α-tubulin and β-actin were used for loading control). (e–h) Photographs of Bub1bH/H and Bub1b+/+ mice at indicated ages. (i) Survival analysis of Bub1bH/H (n = 212), Bub1b+/H (n = 108, two animals died before 15 months), Bub1b+/− (n = 43) and Bub1b+/+ (n = 50) mice.
We monitored 50 Bub1b+/+, 108 Bub1b+/H, 43 Bub1b+/− and 230 Bub1bH/H mice to 15–16 months of age. One Bub1bH/H mouse developed a life-threatening tumor. Six of 116 moribund or deceased Bub1bH/H mice had solitary tumors at autopsy. Bub1bH/H mice had a normal appearance until 2–3 months of age (Fig. 1f; compare with wild-type mouse in Fig. 1e) but typically developed cachexia and lordokyphosis at 3–6 months of age (Fig. 1h and Supplementary Fig. 2 online; compare with wild-type mouse in Fig. 1g). The median lifespan of Bub1bH/H mice was ∼6 months, compared with more than 15 months for Bub1b+/−, Bub1b+/H and Bub1b+/+ mice (Fig. 1i). At age 2 months and older, Bub1bH/H mice developed progressive bilateral cataracts with features reminiscent of age-related human cataracts (Fig. 2a). No cataracts were observed in Bub1b+/−, Bub1b+/H or Bub1b+/+ mice. Histological analysis of skin showed that the dermis and subcutaneous fat cell layers were significantly thinner in 12-month-old Bub1bH/H mice than in control mice (Fig. 2b,c and Supplementary Fig. 2 online). Skinning of Bub1bH/H mice confirmed substantial loss of subdermal adipose tissue and highlighted their severe spinal kyphosis and muscle atrophy (Fig. 2d). Dual-energy X-ray absorptiometry measurements confirmed that total body fat of Bub1bH/H mice declined prematurely (Fig. 2e). Bub1bH/H mice also had less ability to repair wounds at an early age (Fig. 2f). Taken together, these results (Supplementary Table 1 online) suggest that multiple aging-associated phenotypes develop early in mice expressing BubR1 below a threshold level.

(a) Cross-sections of lenses from Bub1bH/H and Bub1b+/+ mice at various ages stained with hematoxylin and eosin. Bub1bH/H lenses were normal at day 10 but showed Morgagnian globules at day 60 (arrowheads), overt cataracts at day 120 and advanced stage nuclear cataracts at day 270. (b) Dermal layer thickness of Bub1bH/H and Bub1b+/+ mice at indicated ages (n = 3 mice per group). (c) Subcutaneous adipose layer thickness of Bub1bH/H and Bub1b+/+ mice at indicated ages (n = 3 mice per group). (d) Skinned 9-month-old Bub1b+/+ (top) and Bub1bH/H (bottom) females. The Bub1bH/H mouse had little subcutaneous fat (arrows) and pronounced kyphosis (arrowheads). (e) Analysis of total body fat of Bub1b+/+ and Bub1bH/H females at indicated ages (n = 3 mice per genotype and age group). (f) Closure of 3-mm punch biopsy wounds in Bub1b+/+ and Bub1bH/H mice at 2 months (left panel) and 12 months (right panel) of age.
To investigate whether the progeroid phenotypes could be due to a defective spindle assembly checkpoint, we measured the ability of Bub1b-mutant MEFs to induce a sustained preanaphase arrest in the presence of nocodazole. Approximately 11–12% of Bub1b+/+, Bub1b+/H and Bub1b+/− MEFs were arrested by 12 h (Fig. 3a). In contrast, only 2.5% of Bub1bH/H MEFs were arrested by 12 h, suggesting that spindle assembly checkpoint function was severely compromised. We confirmed this result by flow cytometry (Supplementary Fig. 3 online). Next, we synchronized MEFs in G1 and analyzed their cyclin B–associated Cdc2 kinase activity after releasing them into medium with nocodazole. Bub1bH/H cells had high Cdc2 kinase activity by 24–30 h after release, similar to Bub1b+/+ cells (Fig. 3b). Bub1b+/+ cells sustained high levels of Cdc2 activity until 42 h after restimulation, but Bub1bH/H cells did not maintain activity after 30 h, consistent with a spindle assembly checkpoint defect8,9.

(a) Mitotic index (defined as the percentage of mitotic cells) of nocodazole-treated MEF lines of indicated genotypes (n = 3 for each genotype). The growth rates of these MEF lines were similar (growth curves not shown). (b) Cyclin B–associated Cdc2 kinase activity of synchronized MEF cells at indicated time points after release into medium with high serum and nocodazole. The asterisk marks an aspecific spot on the autoradiogram. (c) Spectral karyotype of a numerically normal Bub1bH/H metaphase with a gain of chromosome 3 and a loss of chromosome 14. (d) Spectral image of a Bub1bH/H metaphase (MEF) that shows PMSCS. (e) Bub1bH/H MEF cell with a lagging chromosome in anaphase. DNA was stained with Hoechst. The arrowhead highlights a lagging chromosome. (f) The average percentage of anaphases with lagging chromosomes per genotype.
The average percentage of aneuploid metaphases was much higher in passage 5 (P5) Bub1bH/H MEFs than in P5 Bub1b+/−, Bub1b+/H and Bub1b+/+ MEFs (Table 1a). We observed even more profound aneuploidy in P5 Bub1b−/H MEFs. We also carried out spectral karyotype analysis on metaphase spreads from Bub1b+/+ and Bub1bH/H MEFs. Bub1b+/+ metaphases (n = 9) were karyotypically normal. In contrast, five of ten Bub1bH/H metaphases had numerical abnormalities. Furthermore, two numerically normal Bub1bH/H metaphases showed loss of one chromosome and gain of another (Fig. 3c). We observed premature sister chromatid separation (PMSCS), a hallmark of a defective spindle assembly checkpoint8,9, in 38% and 15% of mitotic figures from Bub1b−/H and Bub1bH/H MEFs, respectively, but in only 1–3% of Bub1b+/−, Bub1b+/H and Bub1b+/+ MEFs (Table 1a and Fig. 3d). As expected, we observed more anaphase figures with lagging chromosomes in Bub1b−/H and Bub1bH/H cells than in Bub1b+/−, Bub1b+/H or Bub1b+/+ cells (Fig. 3e,f). Together, these data suggest that the accuracy of chromosome segregation is greatly affected when BubR1 levels drop below a certain level.
Bub1b−/H pups showed substantial aneuploidy at birth, but Bub1bH/H mice did not (Table 1b). At age 2 months, however, Bub1bH/H mice had developed mild aneuploidy that increased in both degree and severity as mice aged further. Bub1b+/− and Bub1b+/H mice did not have detectable aneuploidy. We observed no genome maintenance defects other than chromosome number instability in Bub1bH/H and Bub1b−/H cells (Table 1a and Supplementary Fig. 4 online). The strong correlation between the onset and progression of the aging-associated phenotypes and the degree and the severity of the chromosome number instability in Bub1bH/H mice are suggestive of a role for aneuploidy stemming from BubR1 dysfunction in the development of progeroid features.
We next determined whether BubR1 deficiency triggers apoptosis or cellular senescence, two responses that have been linked to aging10,11. We found similar numbers of apoptotic cells in kidney, liver and lung sections from 1-year-old Bub1bH/H and Bub1b+/+ mice, and in Bub1bH/H and Bub1b+/+ MEF cultures (data not shown). Senescence-associated β-galactosidase activity was high in kidney sections from 5-month-old Bub1bH/H mice, but not in those from age-matched Bub1b+/+ mice and 2-month-old Bub1bH/H mice (Fig. 4a). P3 Bub1bH/H MEFs had comparable growth rates (Fig. 4d) and numbers of cells positive for senescence-associated β-galactosidase activity to those of Bub1b+/+ MEFs (Fig. 4b). At P7, however, Bub1bH/H MEF cultures had substantially slower growth rates and more cells positive for senescence-associated β-galactosidase activity than Bub1b+/+ cultures (Fig. 4b,d). Bub1b−/H MEFs had even more profound growth inhibition and senescence-associated β-galactosidase activity (Fig. 4b,d), suggesting that the rate of senescence correlates with the degree of aneuploidy. Bub1bH/H MEFs senesced quickly in both 20% and 3% oxygen (Fig. 4c), indicating that oxidative DNA damage12 is an unlikely cause of the senescence phenotype. Bub1bH/H MEFs showed early accumulation of the senescence markers p53, p21, p16 and p19 (Fig. 4e). Taken together, these results suggest that aneuploidy in cells from BubR1-insufficient mice might elicit signals that drive them into a senescent state and cause early aging-related phenotypes as they accumulate.

(a) Senescence-associated β-galactosidase activity in kidney sections of 2- and 5-month-old Bub1bH/H and Bub1b+/+ mice, and in P8 Bub1bH/H and Bub1b+/+ MEFs. (b) Percentages of Bub1b−/H, Bub1bH/H and Bub1b+/+ MEF cells cultured in 20% oxygen that were positive for senescence-associated β-galactosidase activity. Passages at which measurements were made are indicated. (c) Percentages of MEF cells of indicated genotypes cultured in 3% oxygen that were positive for senescence-associated β-galactosidase activity. (d) Growth curves of P3 (left panel) and P7 (right panel) MEF cells of indicated genotypes. (e) Expression of senescence-associated markers in Bub1bH/H and Bub1b+/+ MEF cells at P2–P5.
Bub1bH/H mice did not produce any pregnancies. Testicular weight of Bub1bH/H males (0.088 g ± 0.009; n = 8 testes) was slightly below that of Bub1b+/+ males (0.106 g ± 0.006; n = 8 testes). Testes of 4-month-old Bub1bH/H mice were histologically normal (data not shown), but sperm counts of Bub1bH/H mice (9.3 × 106 ± 2.6 × 106; n = 3) were about four times lower than those of Bub1b+/+ mice (37.1 × 106 ± 8 × 106; n = 3). Bub1bH/H spermatozoa had normal motility and morphology (data not shown) and were able to attach to and fertilize Bub1b+/+ eggs in vitro, but they produced 2-cell-stage embryos at 13 times less frequently than Bub1b+/+ spermatozoa (Fig. 5a). Chromosome counts on metaphases of spermatocytes in meiosis II showed that 5% of Bub1bH/H karyotypes had abnormal chromosome numbers compared with 0% of Bub1b+/+ karyotypes (Fig. 5b). Thus, reduced expression of BubR1 seems to affect male fertility at the levels of meiotic chromosome segregation, sperm number and fertilization.

(a) In vitro fertilization experiments with eggs from Bub1b+/+ females and sperm from Bub1b+/+ or Bub1bH/H males. (b) Karyotype analysis of Bub1b+/+ and Bub1bH/H spermatocytes at prometaphase of meiotic division II. Abnormal Bub1bH/H karyotypes showed either a gain or loss of one duplicated chromosome. (c) Oocytes from Bub1bH/H mice arrested at metaphase II stained with α-tubulin and Hoechst. Normal metaphase configuration (left panel), metaphase with misaligned chromosomes (middle panel) and metaphase with an extra spindle (arrow) and misaligned chromosomes (arrowheads; right panel) are shown. (d) Western-blot analyses of BubR1 levels in testis of young and old Bub1b+/+ mice. (e) Western-blot analyses of BubR1 levels in ovary of young and old Bub1b+/+ mice. (f) Cross-sections through ovary from young and old Bub1b+/+ mice immunostained for BubR1 (green) and DNA (blue). Representative corpora lutea are shown.
Ovaries from 4-month-old Bub1bH/H mice appeared histologically normal (data not shown) and were capable of producing mature eggs that arrested at metaphase of meiosis II. But we observed highly abnormal metaphase II configurations in 9 of 13 arrested Bub1bH/H oocytes (Fig. 5c). Oocytes from age-matched Bub1b+/+ mice (n = 14) yielded normal metaphase II configurations. We conclude that infertility in female mice expressing low levels of BubR1 is caused, at least in part, by meiotic chromosome segregation defects.
To determine whether reduced expression of BubR1 might have a role in natural aging, we measured BubR1 levels in wild-type mice of various ages. BubR1 was high in testes of 4-month-old mice but gradually decreased to very low levels as mice aged up to 32 months (Fig. 5d). In contrast, the spindle assembly checkpoint proteins Bub3 and Rae1 remained highly expressed as mice aged (Fig. 5d). Reductions in BubR1 expression were also seen in ovaries and spleens of 22-month-old mice, but not in lungs (Fig. 5e,f and data not shown). Overall, these data are consistent with the idea that BubR1 might have a role in normal aging.
The finding that very few Bub1bH/H mice had detectable tumors when they died, despite substantial chromosome number instability, is unexpected because aneuploidy is a hallmark of most human cancers3,13,14, and because Bub1b is mutated or expressed at low levels in a subset of colorectal carcinomas with chromosomal instability15,16. Instead, BubR1-deficient mice have early onset of several aging-associated phenotypes and have severely shortened lifespans. This, together with the demonstration that BubR1 expression declines in several tissues as wild-type mice age, suggests that this checkpoint protein may be a key regulator of normal aging. Reproductive aging in female mammals occurs relatively early in life and is characterized by the production of increasing numbers of aneuploid oocytes. In humans, this leads to increased abortions and birth defects, such as Down syndrome1. Chromosomal segregation defects associated with reproductive aging are reminiscent of those seen in oocytes of mutant mice expressing low levels of BubR1. Given this age-dependent decline in ovarian BubR1, we propose that downregulation of BubR1 might be a mechanism that contributes to age-related female infertility and certain birth defects. Paternal fertility also declines with advancing age17,18, and a regulatory role for BubR1 is certainly conceivable, given the decline in BubR1 levels in testes of aging normal mice and the negative impact of BubR1-deficiency on fertility of male mutant mice.
Methods
Generation of BubR1 mutant mice.
We used a previously reported gene targeting strategy to create a hypomorphic Bub1b allele in mouse embryonic stem cells19 (for details, see Supplementary Methods and Supplementary Fig. 1 online). Bub1bH/H mice obtained from four independently targeted embryonic stem cell clones had identical phenotypes. We monitored body weight in ten females of each genotype for 70 d. All mice were housed in a pathogen-free barrier environment for the duration of the study. Experimental procedures involving laboratory mice were reviewed and approved by the Institutional Animal Care and Use Committee of the Mayo Clinic.
Western-blot analysis.
We carried out western-blot analyses as previously described20. We used affinity-purified rabbit antibody against mouse BubR1 (382–420) to detect BubR1. We quantified BubR1 signals (n = 6 per genotype) by the use of Bio-Rad Quantity One Software (version 4.1.0) and normalized them to β-actin (AC-151, Sigma; 1:40,000 dilution). We also probed some blots with α-tubulin (T-9026, Sigma; 1:2,000 dilution) as a loading control. We used antibody against human BubR1 (1–350) to exclude production of truncated BubR1 products by Bub1bH and Bub1b− alleles. To quantify senescence-associated proteins in various MEF lysates, we used the following antibodies at 1:200 dilution (purchased from Santa Cruz unless noted otherwise): p16 (M-156; sc-1207), p19 (NB200-106; Novus Biologicals), p21 (M-19; sc-471), p53 (Fl-393-G, sc-6243-G). We detected Bub3 and Rae1 as previously described8.
Histopathology.
We screened all major organs for overt tumors using a dissection microscope and processed the collected tumors routinely for histopathological confirmation. We fixed dissected tissues for histology in 10% formalin, processed them and embedded them in paraffin. We cut 5-μm sections of all tissues and stained them with hematoxylin and eosin using standard procedures. We stained dorsal skin sections and determined the thickness of the dermal and adipose layers by taking 40 random measurements of each mouse for each genotype and age group (n = 3). Calculations were done using a calibrated computer program (Spot Advanced by BioSpot). We stained ovary sections with affinity-purified antibody against BubR1 (382-420) as previously described21.
Bone and fat analyses by dual-energy X-ray absorptiometry.
We analyzed bone mineral content, bone mineral density and total body adipose using a LUNAR PIXI-mus small animal densitometer (LUNAR Corporate Headquarters) as described22 in three anesthetized mice (with avertin) of each genotype and age.
Wound healing analysis.
We analyzed the ability of mice to repair wounds as previously described23. We introduced 3-mm punch biopsy wounds into dorsal skin of anesthetized mice. For a period of 6 d, we measured wound diameters using a digital caliper. Bub1bH/H mice did not survive at a standard anesthetic dose of 375 μg of avertin per g body weight but did survive with half that dose.
Generation and culture of MEFs.
We intercrossed Bub1b+/H mice to derive Bub1b+/+, Bub1b+/H and Bub1bH/H MEFs from individual 13.5-d-old fetuses. We intercrossed Bub1b+/− and Bub1b+/+ mice to generate Bub1b+/+ and Bub1b+/− MEFs, and Bub1b+/− and Bub1b+/H mice to generate Bub1b−/H MEFs. They were cultured in 20% oxygen, frozen at P2 and P3 and used for experimentation at indicated passages. For the studies described, we examined at least three Bub1bH/H, Bub1b+/+ and Bub1b+/− clones. We generated growth curves using P3 and P7 MEFs. At day 0, we plated 1.5 × 105 MEFs per 35-mm dish and counted duplicate cultures at 24-h intervals thereafter (n = 3 MEF lines per genotype). In certain experiments, we wanted to limit senescence due to oxidative stress12, and so we used MEFs that were generated and cultured in 3% oxygen. MEFs were synchronized as previously described8. We washed confluent cultures three times with phosphate-buffered saline and then cultured them in Dulbecco's modified Eagle medium containing 0.1% fetal bovine serum and 0.04% bovine serum albumin for 18 h. We treated quiescent MEFs with trypsin and reseeded them in Dulbecco's modified Eagle medium with 10% fetal bovine serum to allow their reentry into the cell cycle.
Spindle assembly checkpoint analyses.
We measured mitotic index (n = 3 MEF lines per genotype) and carried out FACS-based analysis of spindle assembly checkpoint activity as previously described8. We carried out Cdc2-kinase assays as previously described24.
Karyotype analyses.
We prepared metaphase spreads from MEFs and splenocytes and analyzed them for aneuploidy and PMSCS as previously described8. For chromosome number analysis of spermatocytes, we minced testes between two microscope slides and instantly prepared metaphase spreads from the resulting cell suspensions. We carried out spectral karyotypic analysis of MEFs using the protocol, reagents, instrumentation and software from Applied Spectral Imaging.
Analysis of DNA repair functions.
We carried out DNA damage MEF survival experiments as described25,26. For mitomycin C (Sigma) and paraquat (Sigma) survival experiments, we seeded 104 P2 MEFs in triplicate in a 96-well flat-bottom tissue culture dish. The next day, we replaced the medium with drug-containing medium and allowed cells to grow for 72 h. For ultraviolet-B and γ-irradiation experiments, we exposed 2 × 104 P2 MEFs to various doses of ultraviolet-B or ionizing irradiation, seeded them in triplicate and cultured them for 72 h. We measured cell survival by the use of the MTS assay (Promega). Data represent three independent MEF lines of each genotype. For γ-irradiation colony forming assays, we seeded 4 × 104 MEF cells in duplicate in 10-cm tissue culture dishes and exposed them to various doses of ionizing radiation at P2. We grew cells in 3% oxygen for 14 d and counted colonies. In the colony-forming assay after treatment with mitomycin C, we seeded 4 × 104 P2 MEF cells in duplicate in 10-cm tissue culture dishes and allowed them to grow for 24 h. The indicated concentration of medium containing mitomycin C was added and cells were allowed to grow for 14 d in 3% oxygen. For the highest doses of γ-irradiation and mitomycin C, we used five times as many cells. We counted colonies containing >20 cells after staining with Coomassie blue.
Senescence-associated β-galactosidase staining.
We stained cryosections of mouse kidney for senescence-associated β-galactosidase activity according to manufacturer's protocol (Cell Signaling Technology). To quantify MEFs stained for senescence-associated β-galactosidase activity, we counterstained cells with Hoechst to visualize nuclei. The percentage of senescent cells was the total number of senescent cells divided by the total number of cells counted using immunofluorescence (n = 3 MEF lines per genotype).
Collection and analysis of metaphase II oocytes and in vitro fertilization.
We collected ovulated oocytes and instantly fixed them as previously described27. We stained fixed oocytes with α-tubulin (1:1,000 dilution of T-9026, Sigma) and Hoechst and analyzed them by confocal microscopy. We carried out in vitro fertilization experiments as previously described28. We obtained aged testes from the aged rodent tissue bank maintained by the National Institute of Aging (Bethesda).
Note: Supplementary information is available on the Nature Genetics website.
References
Hassold, T. & Hunt, P. To err (meiotically) is human: the genesis of human aneuploidy. Nat. Rev. Genet. 2, 280–291 (2001).
Thomas, N.S. et al. Maternal sex chromosome non-disjunction: evidence for X chromosome-specific risk factors. Hum. Mol. Genet. 10, 243–250 (2001).
Jallepalli, P.V. & Lengauer, C. Chromosome segregation and cancer: cutting through the mystery. Nat. Rev. Cancer 1, 109–117 (2001).
Wassmann, K. & Benezra, R. Mitotic checkpoints: from yeast to cancer. Curr. Opin. Genet. Dev. 11, 83–90 (2001).
Yu, H. Regulation of APC-Cdc20 by the spindle checkpoint. Curr. Opin. Cell Biol. 14, 706–714 (2002).
Cleveland, D.W., Mao, Y. & Sullivan, K.F. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 112, 407–421 (2003).
Wang, Q. et al. BUBR1 deficiency results in abnormal megakaryopoiesis. Blood 103, 1278–1285 (2004).
Babu, J.R. et al. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J. Cell Biol. 160, 341–353 (2003).
Michel, L.S. et al. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 409, 355–359 (2001).
Hasty, P., Campisi, J., Hoeijmakers, J., van Steeg, H. & Vijg, J. Aging and genome maintenance: lessons from the mouse? Science 299, 1355–1359 (2003).
Campisi, J. Cancer and ageing: rival demons? Nat. Rev. Cancer 3, 339–349 (2003).
Parrinello, S. et al. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 5, 741–747 (2003).
Lengauer, C., Kinzler, K.W. & Vogelstein, B. Genetic instabilities in human cancers. Nature 396, 643–649 (1998).
Li, R., Sonik, A., Stindl, R., Rasnick, D. & Duesberg, P. Aneuploidy vs. gene mutation hypothesis of cancer: recent study claims mutation but is found to support aneuploidy. Proc. Natl. Acad. Sci. USA 97, 3236–3241 (2000).
Cahill, D.P. et al. Mutations of mitotic checkpoint genes in human cancers. Nature 392, 300–303 (1998).
Shichiri, M., Yoshinaga, K., Hisatomi, H., Sugihara, K. & Hirata, Y. Genetic and epigenetic inactivation of mitotic checkpoint genes hBUB1 and hBUBR1 and their relationship to survival. Cancer Res. 62, 13–17 (2002).
Ford, W.C. et al. Increasing paternal age is associated with delayed conception in a large population of fertile couples: evidence for declining fecundity in older men. The ALSPAC Study Team (Avon Longitudinal Study of Pregnancy and Childhood). Hum. Reprod. 15, 1703–1708 (2000).
Eskenazi, B. et al. The association of age and semen quality in healthy men. Hum. Reprod. 18, 447–454 (2003).
Meyers, E.N., Lewandoski, M. & Martin, G.R. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat. Genet. 18, 136–141 (1998).
Kasper, L.H. et al. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol. Cell. Biol. 19, 764–776 (1999).
Feldmann, K.A., Pittelkow, M.R., Roche, P.C., Kumar, R. & Grande, J.P. Expression of an immediate early gene, IEX-1, in human tissues. Histochem. Cell Biol. 115, 489–497 (2001).
Nagy, T.R. & Clair, A.L. Precision and accuracy of dual-energy X-ray absorptiometry for determining in vivo body composition of mice. Obes. Res. 8, 392–398 (2000).
Tyner, S.D. et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 415, 45–53 (2002).
Wassmann, K. & Benezra, R. Mad2 transiently associates with an APC/p55Cdc complex during mitosis. Proc. Natl. Acad. Sci. USA 95, 11193–11198 (1998).
Koomen, M. et al. Reduced fertility and hypersensitivity to mitomycin C characterize Fancg/Xrcc9 null mice. Hum. Mol. Genet. 11, 273–281 (2002).
de Boer, J. et al. Premature aging in mice deficient in DNA repair and transcription. Science 296, 1276–1279 (2002).
Woods, L.M. et al. Chromosomal influence on meiotic spindle assembly: abnormal meiosis I in female Mlh1 mutant mice. J. Cell Biol. 145, 1395–1406 (1999).
Kang-Decker, N., Mantchev, G.T., Juneja, S.C., McNiven, M.A. & van Deursen, J.M. Lack of acrosome formation in Hrb-deficient mice. Science 294, 1531–1533 (2001).
Acknowledgements
We thank J. Harden, R. Babu, M. Gazi, S. Guyse, M. Pittelkow, R. Goorha, J. Davenport, D. Pearson, K. Hafner (Cytogenetics Shared Resource), J. van Ree and S. Kaufman for supporting this project; R. Bram for critically reviewing the manuscript and for discussions; and T. Yen for providing BubR1 antibody. This work was supported by a grant from the US National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Fig. 1
Alternative RNA splicing mechanism underlying hypomorphic BubR1 expression. (PDF 108 kb)
Supplementary Fig. 2
Phenotypic analyses of Bublb+/− and Bub1bH/H mice. (PDF 1504 kb)
Supplementary Fig. 3
Analysis of spindle assembly checkpoint activity by flow cytometry. (PDF 142 kb)
Supplementary Fig. 4
Analysis of the fidelity of distinct DNA damage repair pathways by measuring cell survival and colony formation ability after exposing early-passage MEFs to various kinds of DNA damaging agents. (PDF 84 kb)
Supplementary Table 1
Summary of aging-related phenotypes. (PDF 7 kb)
Supplementary Methods
Generation of Bub1b mutant mice. (PDF 50 kb)
Rights and permissions
About this article
Cite this article
Baker, D., Jeganathan, K., Cameron, J. et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet 36, 744–749 (2004). https://doi.org/10.1038/ng1382
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1382
This article is cited by
-
Missing checkpoints in premature aging
Nature Aging (2023)
-
Hyperphosphorylated PTEN exerts oncogenic properties
Nature Communications (2023)
-
Unique progerin C-terminal peptide ameliorates Hutchinson–Gilford progeria syndrome phenotype by rescuing BUBR1
Nature Aging (2023)
-
Chromosome instability and aneuploidy in the mammalian brain
Chromosome Research (2023)
-
Tracking footprints of artificial and natural selection signatures in breeding and non-breeding cats
Scientific Reports (2022)
