
Abstract
Gene targeting is accomplished using embryonic stem cells in the mouse but has been successful, only using primary somatic cells followed by embryonic cloning, in other species. Gene targeting in somatic cells versus embryonic stem cells is a challenge; consequently, there are few reported successes and none include the targeting of transcriptionally silent genes or double targeting to produce homozygotes. Here, we report a sequential gene targeting system for primary fibroblast cells that we used to knock out both alleles of a silent gene, the bovine gene encoding immunoglobulin-μ (IGHM), and produce both heterozygous and homozygous knockout calves. We also carried out sequential knockout targeting of both alleles of a gene that is active in fibroblasts, encoding the bovine prion protein (PRNP), in the same genetic line to produce doubly homozygous knockout fetuses. The sequential gene targeting system we used alleviates the need for germline transmission for complex genetic modifications and should be broadly applicable to gene functional analysis and to biomedical and agricultural applications.
Similar content being viewed by others


Main
Gene targeting by homologous recombination is a powerful method of specifically modifying a gene of interest used extensively for gene functional analysis in mice1,2,3. Gene targeting is accomplished in the mouse using embryonic stem (ES) cells, but in essentially all other species, ES cells suitable for gene targeting are not available. The few reports on gene targeting in other mammalian species used primary somatic cells followed by embryonic cloning4,5,6,7; in some instances, the embryos were then used to produce cloned offspring. Gene targeting in primary somatic cells is a challenge8,9,10,11,12 because somatic cells have a relatively short lifespan, which limits selection of properly targeted cell colonies, and a low frequency of homologous recombination11 compared with mouse ES cells. Because of these limitations, success in somatic cell gene targeting has been achieved for only a couple of genes that were transcriptionally active in the cell line used for targeting and only in sheep and pig. Transcriptionally active genes are more amenable to gene targeting than silent genes, because they have a higher frequency of homologous recombination5,8 and correctly targeted cells can be easily selected by having the targeted gene promoter drive expression of a selection marker. Application of this 'promoter-less' positive selection4,5,6,7 is limited to transcriptionally active genes in the somatic cells.
To fully evaluate the consequences of a genetic modification, both alleles of the gene must be targeted. In mice, this is generally done by breeding heterozygous knockout founders to produce a homozygous knockout inbred line. But breeding to homozygosity is severely impeded in species that have a long generation interval, such as cows, sheep and pigs, and that are negatively impacted by the consequences of inbreeding. In pigs, two innovative approaches have been used to circumvent the long generation interval and low rate of homologous recombination for targeting the second allele of the gene encoding α-(1,3)-galactocyltransferase. Heterozygous knockout fibroblasts were selected in vitro for lacking enzymatic activity resulting either from a spontaneous point mutation in the second allele of the gene13 or from mitotic recombinants14. Unfortunately, these approaches are neither useful for silent genes nor widely applicable for active genes.
In this study, we developed a broadly applicable and rapid method for generating multiple gene targeting events in cattle. The method consists of sequential application of gene targeting by homologous recombination and rejuvenation of cell lines by production of cloned fetuses (Fig. 1). We used this procedure to demonstrate the first successful targeting of a transcriptionally silent gene and production of both heterozygous and homozygous knockout calves. We also targeted a second gene, resulting in doubly homozygous knockout bovine fetuses and cell lines.

Holstein fetal fibroblasts (6939) were targeted and wells containing targeted cells were then selected and cloned to generate IGHM+/− fetuses. The IGHM+/− cell line (3287) was then used to produce calves and to target the second allele of IGHM. Once again, cells were selected and regenerated by production of fetuses. Fetuses were collected to produce IGHM−/− cell lines, analyze IGHM expression and produce calves. An IGHM−/− cell line (4658) was transfected with a Cre-recombinase expression plasmid to remove both neo and puro genes simultaneously. A third round of embryonic cloning then generated cloned fetuses and cell lines in which both neo and puro selection marker genes were excised. One Cre-excised IGHM−/− fibroblast cell line (1404) was used for a third round of gene targeting to produce triply targeted Cre-IGHM−/− PRNP+/− fetuses and cell lines. One cell line (8334) was subjected to the fourth round of gene targeting to produce doubly homozygous knockout (Cre-IGHM−/− PRNP−/−) fetuses and cell lines and to analysis of PRNP expression. A representative time line for each step is indicated. KO, knockout.
Results
Targeting the first allele of IGHM
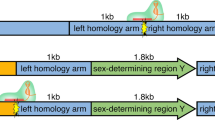
We chose to target IGHM, which is transcriptionally silent in fibroblasts. We characterized this gene in a male Holstein fetal fibroblast cell line (6939) to identify a polymorphic marker DNA sequence, outside the knockout vector sequence, that could be used to distinguish the two alleles (allele A and allele B; Fig. 2a). We constructed the first knockout vector using IGHM genomic fragments from around the constant μ exon 2 region, which was derived from a nonisogenic Holstein genomic library. The knockout vector used to target the first allele contained a diphtheria toxin A (DT-A) gene15 as a negative selection marker and a puro selection marker driven by a mouse PGK promoter, flanked by loxP sequences and followed by a transcriptional and translational STOP16 cassette (pBCμΔKOpuro; Fig. 2a). We electroporated fetal fibroblasts from cell line 6939 with the first knockout vector to produce 446 wells resistant to puromycin. We split the wells on day 14 and screened half the cells by PCR (primer pairs puroF2 × puroR2; Fig. 2a) to identify wells containing correctly targeted cells. Initially, six wells seemed to contain correctly targeted cells. To exclude wells giving a false positive result, we subjected all the PCR products to bidirectional sequencing analysis with the puroF2 and puroR2 primers. Two wells (147 and 384; 0.45%) were correctly targeted and contained heterozygous IGHM knockout (IGHM+/−) cells. On the basis of polymorphic differences identified by sequence analysis, we determined that the knockout vector was integrated into allele A in well 384 and into allele B in well 147.

(a) Structure of IGHM constant region locus in cell line 6939, the puro and neo vectors used for the first and second round of targeting, respectively, and the genomic PCR assay used for the first and second targeting events. In fibroblasts of cell line 6939, polymorphic sequences were found to distinguish allele A and allele B, as indicated. (b) Identification of IGHM+/− fetuses by genomic PCR. N, negative control; P, positive control. Cell lines 2184-1, 2184-2 and 3287 were IGHM+/−. (c) Genotyping of IGHM+/− calves by genomic PCR. N, negative control; P, positive control. Five IGHM+/− calves were genotyped and all contained correctly targeted cells from the first targeting event. (d) Identification of IGHM−/− fetuses and fibroblasts by genomic PCR. N, negative control; P, positive control. 6939 is the original fibroblast cell line. Cell lines 4658, 3655, 5109, 5139 and 4554 contained correctly targeted cells from both targeting events but no wild-type alleles. (e) RT-PCR analysis of IGHM expression in mRNA extracted from spleen in 90-d-old fetuses. Clear expression was detected from a positive control (P) and the wild-type (6939) fetuses but not from IGHM−/− fetuses. (f,g) Genotyping of IGHM−/− calves by genomic PCR. N, negative control; P, positive control. (f) Two IGHM−/− calves were genotyped and contained correctly targeted cells from targeting events at both alleles but (g) no wild-type alleles.
Generating IGHM+/− fetuses and calves
We used the remaining cells from the two wells for embryonic cloning to generate fetuses and rejuvenate the cell lines. Pregnancy rate at 40 days of gestation was 50% (15 of 30, two embryos per recipient; Table 1), and at 60 days of gestation, we collected six fetuses and re-established fibroblasts. Three of six fetuses (2184-1, 2184-2 and 3287) were IGHM+/− (Fig. 2b) as confirmed by the PCR (primer pairs puroF2 × puroR2) and sequence analysis. Nontargeted fetuses probably resulted either from nontargeted cells that coexisted with the targeted cells in the wells or from loss of the transgene due to lack of selection pressure during fetal development. Both fetuses 2184-1 and 2184-2 were derived from well 384, where the knockout vector was integrated into allele A, and fetus 3287 was from well 147, where the knockout vector was integrated into allele B. We produced cloned IGHM+/− embryos from all three regenerated cell lines and transferred them to 153 recipients to produce 13 (8%, Table 1) healthy IGHM+/− calves, whose genotypes were confirmed by PCR (Fig. 2c) and sequence analysis (data not shown).
Targeting the second allele of IGHM
To target the second allele of IGHM, we prepared a second knockout vector in which the puro selection marker was replaced with a neo gene driven by an ST (SV40 promoter and thymidine kinase enhancer) promoter. In attempting to target the second allele of a gene, there is the possibility that the targeting vector will undergo homologous recombination with the integrated targeting vector, resulting in replacement of the knockout vector in the previously targeted allele rather than disruption of the intact allele. This is a problem particularly if the first targeting vector has a strong bias for one allele. This was not observed with our first, nonisogenic, knockout vector, indicating either that the two alleles had similar sequences or that polymorphisms had an equal effect on targeting efficiency. We assumed the latter and determined whether the frequency of targeting of allele A could be enhanced by constructing a second knockout vector in which the short homologous arm was replaced with a PCR-derived sequence amplified directly from allele A of the cell line 6939 (this vector was designated pBCμΔNKOneo).
We used all three IGHM+/− cell lines (2184-1 and 2184-2, targeted in allele A; 3287, targeted in allele B) for targeting with the second knockout vector (Fig. 2a). In cell lines 2184-1 and 2184-2, we screened 1,211 wells resistant to G418 by PCR (primer pairs neoF3 × neoR3; Fig. 2a) and then carried out sequence analysis. Five wells contained correctly targeted cells. In two of them (0.17%), the vector was integrated into the intact allele B, producing homozygous knockout (IGHM−/−) cells, and in three wells, the targeting vector in allele A was replaced. In cell line 3287, we screened 569 wells resistant to G418 by PCR (primer pairs neoF3 × neoR3; Fig. 2a) and then carried out sequence analysis. Seven wells contained correctly targeted cells. In six of them (1.1%), the vector was integrated into the intact allele A, producing IGHM−/− cells, and in one well, the targeting vector in allele B was replaced. Overall, the vector had a bias of 6:1 for intact allele A to allele B and was more efficient for homozygous targeting when used with cell line 3287 in which allele B was first targeted, as expected.
Generating IGHM−/− fetuses and calves
We selected two IGHM−/− wells (76 and 91) derived from cell line 3287 for embryonic cloning to generate fetuses and rejuvenate the cell lines. Overall pregnancy rate for IGHM−/− fetuses at 40–50 days of gestation was 45% (40 of 89; Table 1). At 45 days of gestation, we collected and evaluated 5 fetuses derived from well 76 and 15 fetuses from well 91. All 5 from well 76 (Fig. 2d) and 3 of 15 from well 91 (data not shown) contained correctly targeted cells specific for the first and second targeting events (primer pairs puroF2 × puroR2 and neoF3 × neoR3), as shown by PCR. PCR results were confirmed by sequence analyses and negative PCR17 results (primer pairs bCμf × bCμr; Fig. 2a) for the wild-type alleles (Fig. 2d). We confirmed functional knockout by generating 90-day fetuses from regenerated IGHM−/− fibroblasts and evaluating IGHM expression in spleen cells. Absence of expression was confirmed by RT-PCR (primers pairs bCμf × bCμr; Fig. 2e). We created cloned embryos from five IGHM−/− cell lines and transferred them to recipients for development to term. Eight calves (6%; Table 1) were born recently and were confirmed to be IGHM−/− by PCR (Fig. 2f) and sequence analyses (data not shown), verifying that sequential gene targeting and successive rounds of cell rejuvenation are compatible with full-term development of healthy homozygous knockout calves (Fig. 2g).
Excising neo and puro in IGHM−/− fibroblasts
Sequential gene targeting requires a strategy for antibiotic selection of a newly integrated targeting vector in a cell line that already contains one or multiple antibiotic selection markers. The simplest approach is to use a different selection marker gene for each targeting event, but this approach limits the number of targeting events that may take place in a cell line. Another approach is to remove the selection markers using a Cre-loxP recombination system, as has been done in mouse ES cells18. Unexpectedly, the selection marker genes were not expressed in our regenerated IGHM-targeted fibroblasts, probably because reprogramming of the fibroblasts after embryonic cloning silenced the newly integrated sequence as part of the silent IGHM locus. Although selection marker removal was not necessary for further targeting in our IGHM−/− fibroblasts, we evaluated whether it was possible to remove the selection markers by transfection with a Cre recombinase expression plasmid. Because we intended Cre recombinase to be expressed transiently, we used a circular plasmid and restricted antibiotic selection to the first 3 days of culture. We used bovine IGHM−/− cell line 4658 for transfection and evaluated 24 selected wells by PCR for excision of the antibiotic selection genes from the targeted alleles (Fig. 3a). Multiple wells showed evidence of excision of both puro and neo genes, and we chose one for fetal cloning and regeneration of cell lines. Pregnancy rate at 40–50 days of gestation was 35% (21 of 60; Table 1). We recovered five fetuses, all of which had both selection markers removed (Fig. 3b), but all except fetus 1404 had the Cre recombinase plasmid integrated into the genome (data not shown). These results indicate that Cre-loxP recombination can be used to remove selection markers in somatic cells. Routine use in this system, however, will require improvements to reduce the integration frequency of the Cre expression plasmid.

(a) Structure of alleles of IGHM−/− cell line 4658 and the genomic PCR assay for Cre-loxP-mediated removal of selection marker genes. (b) Identification of Cre-IGHM−/− fetuses and fibroblasts by genomic PCR. Before introduction of Cre, 2.5-kb (puro) and 4.3-kb (neo) PCR products were detected in cell line 4658. A 0.4-kb band is detected in five Cre-excised fetuses.
Targeting the first allele of PRNP
To evaluate the possibility of sequentially targeting a second gene, we subjected Cre-excised IGHM−/− (Cre-IGHM−/−) fibroblasts (cell line 1404) to a third round of targeting to disrupt PRNP. We first characterized this gene to identify a polymorphic sequence, outside the knockout vector sequence, to distinguish the two alleles (allele C and allele D; Fig. 4a). The vector comprised nonisogenic sequences derived from the region around exon 3 of PRNP and the DT-A gene, the neo selection marker driven by the ST promoter, flanked by loxP sequences and followed by the STOP cassette (pBPrP(H)KOneo; Fig. 4a). We transfected cells with the third knockout vector and screened 203 G418-resistant wells by PCR. We identified 13 (6.4%) wells with cells that had a heterozygous knockout in PRNP on the Cre-IGHM−/− background (Cre-IGHM−/− PRNP+/−; primer pairs neoF7 × neoR7; Fig. 4a and data not shown). Sequence analysis showed that the third knockout vector was integrated into allele C of PRNP in all the positive wells. We used some wells for cloning to generate 28 pregnancies at 45 days of gestation (71%; Table 1). We collected five fetuses, all of which contained correctly targeted cells with the vector integrated into allele C of PRNP, as confirmed by PCR (primer pairs neoF7 × neoR7; Fig. 4b) and sequencing analyses (data not shown). Furthermore, we detected no amplification of wild-type IGHM alleles (primer pairs bCμf × bCμr; Fig. 4b), as expected. Targeting efficiency for PRNP, which is transcriptionally active in bovine fibroblasts, was substantially higher than for IGHM (6.4% versus 0.63%, respectively), which is not expressed in fibroblast cells.

(a) Structure of the PRNP locus in Cre-IGHM−/− cell line 1404, neo and puro vectors used for the third and fourth rounds of targeting and the genomic PCR assay. (b) Identification of the triply targeted fetuses and fibroblast cell lines by positive and negative genomic PCR. P, positive control; cell line 1404, negative control. Cell lines derived from fetuses 8103, 1661, 8375, 8112 and 8443 contained correctly targeted cells from PRNP targeting and no wild-type IGHM alleles. (c) Identification of doubly homozygous knockout fibroblasts by genomic PCR. P, positive control; N, negative control. IGHM−/− fetal cell line 4658 and Cre-IGHM−/− PRNP+/− cell line 8443 are indicated. Cell lines 8454, 8400 and 6397 contained correctly targeted cells from the third and fourth targeting events but no wild-type alleles. (d) RT-PCR analysis of doubly homozygous knockout fetuses. Expression was observed in fetuses 4658 and 8443 but not in doubly homozygous knockout fetuses.
Targeting the second allele of PRNP
To examine the feasibility of quadruple targeting to produce doubly homozygous knockout fetuses and cell lines, we transfected the triply targeted cell line (8443, Cre-IGHM−/− PRNP+/−) with a fourth knockout vector for the remaining allele of PRNP. We constructed the vector by replacing the neo gene with the puro gene (pBPrP(H)KOpuro; Fig. 4a) in the PRNP targeting vector used for the first allele. After selection and PCR screening (primer pairs puroF14 × puroR14; Fig. 4a), 17 (5.2%) wells contained targeted cells. Sequence analysis confirmed that the fourth knockout vector was integrated into allele D of PRNP, creating doubly homozygous knockout (Cre-IGHM−/− PRNP−/−) cells, in 16 wells. In the remaining well, the targeted sequence in allele C was replaced. We used cells from correctly targeted Cre-IGHM−/− PRNP−/− wells for cloning to produce fetuses. The pregnancy rate derived from these embryos at 45 days of gestation was 68% (Table 1). We collected 18 fetuses, which were Cre-IGHM−/− PRNP−/−, as confirmed by PCR analysis using the targeting event-specific primer pairs puroF14 × puroR14 and neoF7 × neoR7 (Fig. 4c). Sequencing analyses confirmed integration of the third (neo) and fourth (puro) PRNP targeting vectors into alleles C and D, respectively. Furthermore, we carried out a negative PCR analysis to confirm the absence of wild-type PRNP alleles (primer pairs BPrPex3F × BPrPex3R; Fig. 4c) and IGHM alleles (primer pairs bCμf × bCμr; data not shown); as expected, all four knockouts were confirmed. To evaluate PRNP mRNA expression, we examined fibroblasts from one IGHM−/− fetus, one Cre-IGHM−/− PRNP+/− fetus and three Cre-IGHM−/− PRNP−/− fetuses by RT-PCR. Functional disruption of PRNP expression was confirmed (Fig. 4d). These results indicate that multiple rounds of gene targeting, both for transcriptionally active and silent genes, were readily accomplished in a single somatic cell line using a cell rejuvenation approach.
Discussion
In this study we demonstrate, for the first time, a sequential gene targeting strategy for primary somatic cells, which can be used for targeting multiple alleles of a gene or for targeting multiple genes. The system proved effective for targeting both transcriptionally silent and active genes, demonstrating broad application, and was compatible with development of healthy calves through at least two rounds of gene targeting. There was no indication that additional rounds of gene targeting compromised development of cloned embryos, as judged from pregnancy rates at 45–60 days of gestation (Table 1). Pregnancies with the doubly homozygous knockout fetuses are in progress and pregnancy rates are consistent with the results obtained in this study.
One advantage of the sequential gene targeting system is that the time required to produce an animal with multiple genetic modifications is greatly reduced compared with traditional breeding strategies. With sequential gene targeting, each targeting event required ∼2.5 months from transfection to establishment of regenerated cell lines; therefore, homozygous targeted calves could be created in 14 months (5 months for targeting two alleles and 9 months of gestation) and doubly homozygous targeted calves, including Cre-mediated excision of selection genes, could be created in 21.5 months (Fig. 1). In contrast, for cattle, breeding a heterozygous founder to produce homozygous calves would require ∼5 years and generation of double homozygotes from two heterozygous founders is impractical.
Several factors were important for maximizing targeting efficiency and for successfully producing rejuvenated cell lines and calves. Overall, frequency of homologous recombination at each targeting step was sufficiently high (0.4–6.4%) to produce at least a couple of targeted colonies from ∼500 selected colonies that were screened by PCR in each experiment. The efficiency might be attributed to several conditions that were optimized specifically for bovine fibroblast targeting, including using appropriate promoters to maximize expression of positive selection marker genes, using the DT-A gene for negative selection19, using contiguous regions of homology in the targeted gene loci, optimizing electroporation conditions5 and cloning immediately after PCR selection with a modified system to facilitate reprogramming of the donor cells20.
Using this sequential targeting strategy, complex genetic modifications, in large animal species, are not only feasible but relatively straightforward and should be useful for many applications. Targeting of multiple genes in large animals may be useful for producing new models for human disease, for producing various therapeutic proteins, for producing organs or tissues for transplantation into humans and for improving the efficiency of agricultural production. Gene targeting has many useful applications in science, medicine and industry and may be one of the most useful applications of somatic cell cloning technology. Currently, gene targeting using ES cells has been successful only in mice, but somatic cell cloning has been successful for many species21,22,23,24,25. The results obtained in this study indicate that complex genetic modifications can now be readily made for a wide variety of genes in many species.
Methods
Constructing knockout vectors.
We obtained a bovine genomic fragment around exon 2 of the IGHM constant region locus from nonisogenic Holstein genomic library by probing with a 32P-labeled PCR fragment. We analyzed one genomic clone further by restriction mapping. We subcloned 7.2 kb of the BglII-XhoI genomic fragment (5′ homologous arm) and 2.0 kb of the BamHI-BglII fragment (3′ homologous arm) around exon 2 into pBluescript II SK(-) (Stratagene) and then inserted puro, STOP cassettes (pBS302, Stratagene) and DT-A genes (pBCμΔKOpuro vector). To construct the second targeting vector, we carried out genomic PCR on cell line 6939. After digestion with BamHI-BglII, this fragment replaced the 3′ short arm of the pBCμΔKOpuro vector. By sequencing, we confirmed that the BamHI-BglII fragment was amplified from allele A. We replaced the puro gene with a neo gene (pBCμΔNKOneo vector). We obtained bovine genomic fragment around exon 3 of PRNP locus by screening the same Holstein genomic λ phage library with a 32P-labeled DNA fragment amplified by PCR. We analyzed one genomic clone further by restriction mapping. We subcloned 8.3 kb of the BamHI genomic fragment (3′ homologous arm) and 1.2 kb of the BamHI-BglII fragment (5′ homologous arm) containing exon 3 into pBluescript II SK(-) and inserted both neo and STOP cassettes at the BamHI site, which is behind the initial ATG codon. We also subcloned the DT-A gene (pBPrP(H)KOneo vector). Similarly, we constructed another knockout vector containing the puro gene (pBPrP(H)KOpuro vector). Primer sequences are available on request.
Cell culture and transfection.
We cultured Holstein fetal male fibroblasts as previously described26 and electroporated them with 30 μg of each targeting vector at 550 V and 50 μF by using a GenePulser II (Bio-rad). After 48 h, we selected the cells under 500 μg ml−1 of G418 or 1 μg ml−1 of puromycin for 2 weeks, picked the drug-resistant colonies and transferred them to replica plates, one for genomic DNA extraction (24-well plates) and the other for embryonic cloning (48-well plates).
Genomic PCR analyses.
From the replica 24-well plates, we extracted fetus or ear biopsy genomic DNA from calves using a Puregene DNA extraction kit (GentraSystem). To identify each homologous recombination event that occurred at the IGHM locus, we used primer pairs puroF2, puroR2, neoF3 and neoR3 (Fig. 2a). PCR was done in 30 cycles of 98 °C for 10 s and 68 °C for 8 min. For negative PCR, we used primer pairs BCμf and BCμr (Fig. 2a) in 40 cycles of PCR composed of 98 °C for 10 s, 62 °C for 30 s and 72 °C for 1 min. In the case of the PRNP locus, we used primer pairs neoF7, neoR7, puroF14 and puroR14 (Fig. 4a). PCR was done in 30 cycles of 98 °C for 10 s and 68 °C for 5 min. For negative PCR, we used primer pairs BPrPexF and BPrPexR (Fig. 4a) in 40 cycles of PCR composed of 98 °C for 10 s, 62 °C for 30 s and 72 °C for 1 min. To detect the Cre-mediated excision, we carried out PCR with primer pair CreExF and CreExR (Fig. 3a) in 40 cycles of PCR composed of 98 °C for 10 s and 68 °C for 7 min. All the PCR products were separated on 0.8% agarose gels. Primer sequences are available on request.
Sequencing analysis of the PCR products.
To confirm whether homologous recombination correctly occurred at each targeting step, we sequenced the amplified PCR products. We purified the PCR products through CHROMA SPIN-TE400 column (BD Biosciences Clontech) and sent them to ACGT for sequencing. Bidirectional sequencing was done with both the forward and reverse primers that were used for PCR. The allele into which each knockout vector was integrated was determined by polymorphisms in the sequence of the PCR products.
Embryonic cloning.
We produced cloned fetuses and calves as described previously20. We enucleated in vitro matured oocytes 20 h after maturation. We permeabilized correctly targeted clones by incubating ∼50–100,000 cells in suspension with 31.2 U Streptolysin O (Sigma) in 100 μl of Hank's balanced salt solution for 30 min in a water bath at 37 °C. Permeabilized cells were sedimented, washed and incubated with 40 μl of mitotic extract containing an ATP-generating system (1 mM ATP, 10 mM creatine phosphate and 25 μg ml−1 of creatine kinase) for 30 min at 38 °C. At the end of the incubation, we diluted the reaction mix, sedimented the cells and washed them. We fused these cells to enucleated oocytes, activated 28 h after maturation with 5 μM calcium ionophore for 4 min followed by 10 μg ml−1 of cycloheximide and 2.5 μg ml−1 of cytochalasin D for 5 h. After activation, we washed the embryos and cultured them with mouse fetal fibroblasts to the blastocyst stage in vitro. We selected grade 1 and 2 blastocysts and transferred them into synchronized recipients. All animal work was done following a protocol approved by the Transova Genetics Institutional Animal Care and Use Committee.
RT-PCR.
We extracted RNA from spleens of wild-type (6939) and IGHM−/− fetuses using an RNeasy mini kit (Qiagen) and carried out first-strand cDNA synthesis using the Superscript first-strand synthesis system for RT-PCR (Invitrogen). We carried out PCR using primers BCμf and BCμr in 40 cycles composed of 98 °C for 10 s, 62 °C for 30 s and 72 °C for 1 min. We also extracted RNA from 4658 (IGHM−/−), 8443 (IGHM−/− PRNP+/−) and doubly homozygous knockout (IGHM−/− PRNP−/−) fibroblasts and carried out first-strand cDNA synthesis as above. PCR was done using primers PrPmF3 and PrPmR3 in 40 cycles of 98 °C for 10 s, 62 °C for 30 s and 72 °C for 1 min. To detect expression of bovine β-actin mRNA, we used primers bBAF and bBAR in the same PCR condition (data not shown). To exclude the possibility of genomic DNA contamination, we carried out another RT-PCR without reverse transcriptase (data not shown). The PCR products were separated on 0.8% agarose gel. Primer sequences are available on request.
References
Hooper, M., Hardy, K., Handyside, A., Hunter, S. & Monk, M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature 326, 292–295 (1987).
Capecchi, M.R. Altering the genome by homologous recombination. Science 244, 1288–1292 (1989).
Thompson, S., Clarke, A.R., Pow, A.M., Hooper, M.L. & Melton, D.W. Germ line transmission and expression of corrected HPRT gene produced by gene targeting in embryonic stem cells. Cell 56, 313–321 (1989).
McCreath, K.J. et al. Production of gene-targeted sheep by nuclear transfer from cultured somatic cells. Nature 405, 1066–1069 (2000).
Denning, C. et al. Deletion of the α-(1,3)galactosyl transferase (GGTA1) gene and the prion protein (PrP) gene in sheep. Nat. Biotechnol. 19, 559–562 (2001).
Lai, L. et al. Production of α-1,3-galactosyltransferase knockout pigs by nuclear transfer cloning. Science 295, 1089–1092 (2002).
Yifan, D. et al. Targeted disruption of the α-1,3-galactosyltransferase gene in cloned pigs. Nat. Biotechnol. 20, 251–255 (2002).
Thomson, A.J., Marques, M.M. & McWhir, J. Gene targeting in livestock. Reprod. Suppl. 61, 495–508 (2003).
Denning, C. & Priddle, H. New frontiers in gene targeting and cloning: success, application and challenges in domestic animals and human embryonic stem cells. Reproduction. 126, 1–11 (2003).
Piedrahita, J.A. Targeted modification of the domestic animal genome. Theriogenology 53, 105–116 (2000).
Sedivy, J.M. & Dutriaux, A. Gene targeting and somatic cell genetics – a rebirth or a coming of age? Trends Genet. 15, 88–90 (1999).
Wang, B. & Zhou, J. Specific genetic modifications of domestic animals by gene targeting and animal cloning. Reprod. Biol. Endocrinol. 1, 103–111 (2003).
Phelps, C.J. et al. Production of alpha 1,3-galactosyltransferase-deficient pigs. Science 299, 411–414 (2003).
Sharma, A. et al. Pig cells that lack the gene for alpha1-3 galactosyltransferase express low levels of the gal antigen. Transplantation 75, 430 (2003).
Yagi, T. et al. Homologous recombination at c-fyn locus of mouse embryonic stem cells with use of diphtheria toxin A-fragment gene in negative selection. Proc. Natl. Acad. Sci. USA 87, 9918–9922 (1990).
Lakso, M. et al. Targeted oncogene activation by site-specific recombination in transgenic mice. Proc. Natl. Acad. Sci. USA 89, 6232–6236 (1992).
Valenzuela, D.M. et al. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat. Biotechnol. 21, 652–659 (2003).
Abuin, A. & Bradley, A. Recycling selectable markers in mouse embryonic stem cells. Mol. Cell. Biol. 16, 1851–1856 (1996).
Mansour, S.L., Thomas, K.R. & Capecchi, M.R. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature 336, 348–352 (1988).
Sullivan, E.J. et al. Cloned calves from chromatin remodeled in vitro. Biol. Reprod. 70, 146–153 (2004).
Wilmut, I., Schnieke, A.E., McWhir, J., Kind, A.J. & Campbell, K.H.S. Viable offspring derived from fetal and adult mammalian cells. Nature 385, 810–813 (1997).
Zhou, Q. et al. Generation of fertile cloned rats by regulating oocyte activation. Science 302, 1179 (2003).
Chesne, P. et al. Cloned rabbits produced by nuclear transfer from adult somatic cells. Nat. Biotechnol. 20, 366–369 (2002).
Cibelli, J.B. et al. Cloned transgenic calves produced from nonquiescent fetal fibroblasts. Science 280, 1256–1258 (1998).
Polejaeva, I.A. et al. Cloned pigs produced by nuclear transfer from adult somatic cells. Nature 407, 86–90 (2000).
Kuroiwa, Y. et al. Cloned transchromosomic calves producing human immunoglobulin. Nat Biotechnol. 20, 889–894 (2002).
Acknowledgements
We thank J. Pommer, J. Koster, J. Molina and D. Faber for their assistance in embryo transfer, fetal recovery, calf delivery and sample collection and M. Nichols, J. Griffin, M. Bien, T. King, M. Ahlers, R. Paulson, S. Viet and C. Voss for their assistance in gene targeting and embryo cloning.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
Y.K., M.K. and I.I. are employed by Kirin Brewery, which may gain or lose financially owing to publication of this paper. P.K., H.M., J.S., E.J.S. and J.M.R. are employed by Hematech, which may gain or lose financially owing to publication of this article. J.M.R. has a substantial financial interest in Hematech.
Rights and permissions
About this article
Cite this article
Kuroiwa, Y., Kasinathan, P., Matsushita, H. et al. Sequential targeting of the genes encoding immunoglobulin-μ and prion protein in cattle. Nat Genet 36, 775–780 (2004). https://doi.org/10.1038/ng1373
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1373
This article is cited by
-
Versatile generation of precise gene edits in bovines using SEGCPN
BMC Biology (2023)
-
Proposed U.S. regulation of gene-edited food animals is not fit for purpose
npj Science of Food (2019)
-
Combinations of chromosome transfer and genome editing for the development of cell/animal models of human disease and humanized animal models
Journal of Human Genetics (2018)
