
Abstract
Hirschsprung disease (HSCR) is a multigenic, congenital disorder that affects 1 in 5,000 newborns and is characterized by the absence of neural crest–derived enteric ganglia in the colon1. One of the primary genes affected in HSCR encodes the G protein–coupled endothelin receptor-B (EDNRB)2,3. The expression of Ednrb is required at a defined time period during the migration of the precursors of the enteric nervous system (ENS) into the colon4. In this study, we describe a conserved spatiotemporal ENS enhancer of Ednrb. This 1-kb enhancer is activated as the ENS precursors approach the colon, and partial deletion of this enhancer at the endogenous Ednrb locus results in pigmented mice that die postnatally from megacolon. We identified binding sites for SOX10, an SRY-related transcription factor associated with HSCR5, in the Ednrb ENS enhancer, and mutational analyses of these sites suggested that SOX10 may have multiple roles in regulating Ednrb in the ENS.
Similar content being viewed by others


Main
Mice and individuals with HSCR with mutations in the EDNRB-mediated pathway have megacolon because of the absence of enteric neurons in the distal gut1. This regional specificity of aganglionosis could be explained by a temporal requirement for Ednrb between embryonic day (E) 11 and E12.5 (ref. 4), when vagal neural crest–derived ENS progenitors are populating the hindgut during mouse embryogenesis, such that in the absence of EDNRB the migratory wavefront is delayed near the ileocecal junction (Fig. 1a)6,7,8. To elucidate the molecular mechanisms for Ednrb expression in the ENS, we dissected the Ednrb genomic region. We isolated a 78-kb P1 genomic clone encompassing Ednrb (Fig. 1b) and used it to create four independent transgenic lines. When we crossed the individual transgenic lines into the Ednrb-null mice, all the lines rescued postnatal death from megacolon (Fig. 1c). Although three of the lines did not rescue the melanocyte defect in the Ednrb-null mice (Fig. 1c), one line (when homozygous with respect to the transgene) partially rescued the pigmentation defect that resembles the hypomorphic Ednrbs allele9 (data not shown). These results suggested that the P1 clone contained the necessary information for expression of Ednrb in ENS progenitors. In addition, we mapped a main transcription start site ∼210 bp 5′ of the initiator ATG using an RNase protection assay (Fig. 1d), consistent with the mouse RefSeq transcript.

(a) Migration pattern of ENS precursors in guts from representative E12.5 Ednrb+/LacZ or EdnrbLacZ/LacZ embryos, showing the positions of cells expressing Ednrb. Mg, midgut; Ce, cecum; Co, colon; ICJ, ileocecal junction (red arrow). Scale bar, 100 μm. (b) Diagram of Ednrb genomic region and genomic clones. The arrow indicates the transcription initiation site. Sn, SnaBI; K, KpnI; S, SphI; B, BamHI. (c) Two independent P1 transgenic (Tg) lines in Ednrb-null (EdnrbLacZneo) background. (d) Mapping of the predominant transcription initiation site by RNase protection assay. Lanes 1 and 4, undigested antisense RNA probes; lanes 2 and 5, probes hybridized to yeast RNA and treated with RNase; lanes 3, 6 and 7, probes hybridized to poly(A)+ RNA and treated with RNase. The red asterisks indicate the positions of Ednrb-specific protected bands. M, RNA ladder.
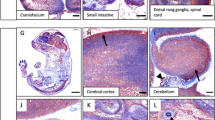
To identify cis-regulatory elements for directing Ednrb expression in the developing ENS within the 78-kb P1 clone, we isolated overlapping λ genomic clones containing exon 1 (Fig. 1b). We inserted the reporter gene β-galactosidase (LacZ) at the initiator ATG and made a series of constructs containing different 5′ genomic fragments. We observed LacZ expression in the ENS precursors from all transgenes except the −0.6-kb LacZ construct (Fig. 2a,b). In the developing gut, the enhancer activity of these constructs was limited to the neural crest–derived ENS precursors as confirmed by overlapping expression with another ENS precursor marker (Fig. 2b). Most cells expressing Ednrb, including melanoblasts, craniofacial ganglia and peripheral nervous system7, were not marked by these transgenic constructs (Fig. 2e).

(a) Schematic diagram of 5′ Ednrb-LacZ constructs. The fractions of embryos expressing LacZ in the ENS precursors (ENS) and the neural tube (NT) of the total number of transgenic embryos are shown. (b) Representative transgenic embryos expressing LacZ in the ENS precursors (arrows) and section of the developing gut hybridized with SOX10 antibody. (c) Deletion series of Ednrb enhancer linked to heterologous hsp68 promoter. The fractions of embryos expressing LacZ in the ENS precursors (ENS) and the neural tube (NT) of the total number of transgenic embryos are shown. (d) Representative E11.5 embryos expressing or not expressing LacZ in the ENS precursors. Black boxes indicate the developing guts and the corresponding enlarged views. (e) Temporal expression of LacZ in ENS precursors in Ednrb+/LacZ and transgenic (Tg) −1.2kb Ednrb-LacZ stable embryos. Red boxes indicate the developing gut and the corresponding enlarged views. Red asterisks indicate the cecum. Fg, foregut; Mg, midgut; Hg, hindgut.
To delineate the minimal ENS enhancer, we isolated a genomic fragment containing −1.2 kb to −160 bp and linked it to the heterologous hsp68 minimal promoter driving LacZ (Fig. 2c). This 1-kb Ednrb fragment contained the lineage-specific enhancer to direct hsp68 promoter in the ENS precursors (Fig. 2d). Additional 5′, 3′ and internal deletions in the hsp68-LacZ constructs showed that the region between −1.2 kb and −250 bp was required for ENS activity (Fig. 2c). Next, we wanted to determine whether the minimal ENS enhancer could recapitulate the endogenous expression patterns in the ENS precursors. Ednrb is expressed as early as E9.5 in ENS precursors, and expression is maintained in the developing gut during embryogenesis (Fig. 2e)7. We analyzed four stable lines from the −1.2-kb Ednrb-LacZ (Fig. 2a) construct and found that none of the transgenic embryos expressed LacZ in the ENS precursors before E10.5. The transgene was activated around E11, when ENS precursors are approaching the cecum, and was fully active by E11.5 and at later stages in the developing ENS precursors (Fig. 2e).
We determined the in vivo activity of the putative ENS enhancer in previously described EdnrbtetOΔHy mice4. We observed pigmented EdnrbtetOΔHy/− mice but did not address the effect of this mutation on ENS development. The EdnrbtetOΔHy allele was created by deleting a genomic fragment from −550 bp of the transcription initiation site to 240 bp 3′ of Ednrb exon 1 and replacing this region with a cassette containing the multimerized tetracycline responsive elements linked to a minimal CMV promoter driving expression of the Ednrb cDNA (Fig. 3a). This allele contains a 300-bp deletion at the 3′ end of the putative ENS enhancer that resembles the mutations generated in the hsp68-LacZ transgenic constructs (Fig. 2c). When Ednrb+/tetOΔHy mice were mated to the Ednrb+/LacZ mice7, pigmented EdnrbtetOΔHy/LacZ offspring were born (Fig. 3b). These mice eventually developed megacolon, and the embryonic defects were identical to those of Ednrb-null mice in which the ENS precursors did not enter the cecum and hindgut (Fig. 3c). These results suggest that the ENS enhancer but not the melanocyte enhancer was disrupted in Ednrb+/tetOΔHy mice (Fig. 3d). Deleting 240 bp of intron 1 probably did not cause the ENS defect, as the same deletion was created previously with no effect on ENS expression4,7. It is possible, however, that the deletion of the endogenous promoter and 5′ untranslated region contributed to the ENS defect. Hypopigmentation in the EdnrbtetOΔHy/LacZ mice could be a result of the heterologous minimal CMV promoter mediating transcription less efficiently than the endogenous Ednrb promoter in melanoblasts, because EdnrbtetOΔHy/tetOΔHy mice are fully pigmented (data not shown).

(a) Schematic diagram of the EdnrbtetOΔHy allele. The horizontal red bar indicates the putative ENS enhancer located between −1,200 and −250 bp relative to the transcription initiation site. The blue ovals indicate the relative positions of the SOX10 binding sites (Fig. 4c). The green bar indicates the tetO-CMV promoter. The vertical red bar is the relative position of the single loxP site remaining in the locus after excision of the pgk-Hygro cassette. Stu, StuI; Bg, BglII. (b) One-week-old Ednrb+/tetOΔHy and EdnrbtetOΔHy/LacZ mice. EdnrbtetOΔHy/LacZ mice are >95% pigmented and die from megacolon. (c) Isolated guts from E15.5 Ednrb+/LacZ and EdnrbtetOΔHy/LacZ littermates. The red arrows indicate the migratory path of ENS precursors into the hindgut. Ce, cecum; Hg, hindgut. (d) A model of transcription activation at EdnrbtetOΔHy locus. The 300-bp deletion of the ENS enhancer (ENS-E) results in Ednrb expression in melanocytes, mediated by unidentified melanocyte-specific enhancer (MEL-E), but not in the ENS precursors.
Sox10 encodes an SRY-related HMG domain protein that is widely expressed in most neural-crest precursors. Its absence results in multiple neural crest–derived defects, including aganglionosis of the whole gut10,11,12. Haploinsufficiency of SOX10 in mice and individuals with HSCR results in aganglionosis of the hindgut10,11,12,13,14,15. Both Sox10 and Ednrb are expressed in migrating ENS precursors (Fig. 4a), and these progenitor cells had migratory defect in the distal ileum, rostral to the cecum, around E11.5 in Ednrb−/− and Sox10+/− embryos (Fig. 4b)7,8,15. The decrease in the number of cells expressing Ednrb in Sox10+/− embryos (Fig. 4b) is probably due to the loss of ENS precursors, as expression of other ENS markers is lost (data not shown). But this raises the possibility that the loss of Ednrb expression in Sox10+/− embryos resulted in loss of ENS precursors. Therefore, we looked for SOX10 recognition sites in the Ednrb ENS enhancer to determine whether SOX10 could regulate Ednrb. We identified three putative SOX10 binding sites, which matched the consensus recognition site (5′-(A/T)(A/T)CAA(A/T)-3′; ref. 5) and contained at least five identical nucleotides between mouse and human (Fig. 4c). Binding assays with GST-SOX10 fusion protein showed that these sites were capable of binding SOX10, and the specificity of these interactions was confirmed by competition and supershift assays (Fig. 4d).

(a) Coexpression of EDNRB and SOX10 in the developing ENS of an Ednrb+/LacZ embryo. Fg, foregut; Hg, hindgut. (b) Phenotypic overlap between E12 Sox10 and Ednrb mutants during ENS development. Ce, cecum; Co, colon. Red arrows indicate the ileocecal junction. (c) Three conserved SOX10 binding sites in the Ednrb ENS enhancer. M, mouse; H, human (d) Gel-shift assay with GST-SOX10 (1–189). Probes for sites I–III were incubated with GST-SOX10 (lane 1), GST-SOX10 and SOX10 antibody (lane 2, 3,4) GST-SOX10 and 100× or 200× molar excess of SOX10 oligos (lanes 3 and 4), GST-SOX10 and 100× or 200× molar excess of mutant SOX10 oligos (lanes 5 and 6). Red arrow indicates the GST-SOX10–DNA complex. Black arrows indicate the α-SOX10–GST-SOX10–DNA supershift complexes.
To determine the functional relevance of these putative SOX10 binding sites, we created mutations at sites I–III in the −1.2-kb Ednrb-LacZ construct (Fig. 5a). We observed LacZ activity in the foregut, midgut and colon of wild-type transgenic E12.5–E13.5 embryos (Fig. 5b). LacZ expression was abolished when site II was mutated, suggesting that this site was necessary for the enhancer activity in the ENS precursors (Fig. 5a,b). When sites I and/or III were mutated, LacZ expression was observed in the colon in ∼50% of the embryos (12 of 26; Fig. 5a). In the other ∼50% of the embryos that expressed the mutated transgenes, LacZ expression was mostly limited to the gastrointestinal tract rostral to the cecum at E12.5–E13.5 (14 of 26; Fig. 5a,b). This latter result is notable because the transgenes were expressed in the wild-type embryos and the ENS precursors had migrated through the cecum and into the colon (Fig. 5c). The pattern of expression by mutant transgenes suggests that, in addition to temporal or tissue-specific activation mediated by site II, SOX10 regulates spatial expression of Ednrb through the cecum and hindgut (Fig. 5d). Thus, depending on the sites of SOX10 occupation in the Ednrb enhancer, individual Sox10+/− cells may respond to varying degree of activation of the Ednrb that result in the absence of migrating ENS precursors in the distal ileum or the colon (Fig. 5d). This stochastic response in the ENS enhancer could explain the variable penetrance and expression of megacolon observed in Sox10Dom/+ mice14,15,16.

(a) Schematic diagram of transgenes containing mutant SOX10 binding sites in the −1.2-kb Ednrb-LacZ construct. The fraction of embryos expressing LacZ in the ENS precursors of the total number of transgenic embryos is presented in the first column. The fraction of transgenic embryos expressing LacZ in the colon of the number of ENS expressors is presented on the second column. WT, wild-type. (b) E12.5–E13.5 embryos with wild-type (WT) or mutated (M) transgenes. Magnified views show that LacZ is expressed in the ENS precursors of transgenic embryos. Arrows indicate the rostral-caudal migratory direction of ENS precursors and red asterisks indicate the cecum. (c) Expression of LacZ and c-Ret in wild-type (WT) and transgenic embryos with mutated site I (M I). Hg, hindgut. (d) A model of SOX10 requirement for Ednrb expression in ENS precursors. Red box indicates the SOX10- and EDNRB-dependent region from the distal ileum to proximal colon. Site II is required for temporal activation in the distal ileum (blue arrow) or maintenance of expression in the ENS precursors (blue line). Sites I and III are required for expression in the colon. In Sox10+/− or cis mutants at the Ednrb enhancer, partial and stochastic occupancy of SOX10 sites result in absence of Ednrb in different regions of the distal gut. Ce, cecum; Fg, foregut; Hg, hindgut; WT, wild-type.
In this study, we identified an ENS enhancer element in the Ednrb locus that is spatiotemporally activated as ENS precursors are populating the distal gut. This temporal requirement is essential for EDNRB function and may be needed to upregulate EDNRB in response to the specific environmental cues encountered by the ENS precursors near the cecum4,8,17,18,19,20. The amount of signaling mediated by the receptor tyrosine kinase RET, another locus associated with HSCR, is also crucial for the colonization of the hindgut by ENS precursors and seems to modulate EDNRB function21,22,23. Sox10 and Ednrb are highly expressed in gut neural crest stem cells, and both pathways may regulate pluripotency or migration of the neural crest progenitors24,25,26,27,28,29. Future studies will determine if the individual SOX10 sites identified in this study are required for both tissue-specific and spatiotemporal regulation of the endogenous Ednrb. Finally, our results suggest that the corresponding region of the human EDNRB locus is a prime target for identifying potential regulatory mutations in individuals with HSCR and may provide insight into how transcription factor haploinsufficiency causes human disorders30.
Methods
Genomic clones.
We obtained the Ednrb P1 clone (Genome Systems) with specific PCR primers against the first and last exons of Ednrb. Primer sequences are available on request. The isolation of Ednrb λ genomic clones was described previously4.
Ribonuclease protection assay.
We generated a 775-bp genomic fragment located 5′ of the translational initiation codon, ATG, by PCR and cloned it into PCRII (Invitrogen). We synthesized radioactively labeled antisense RNA probes containing either 265 bp or 385 bp located immediately 5′ of the ATG by in vitro transcription. Each probe contained an additional 90 bp from the PCRII vector. We isolated total RNAs with Tri Reagent (Sigma) from either E11.5 embryos or adult tissues and processed them through the PolyATtract mRNA Isolation System (Promega). We hybridized poly(A)+ RNAs (0.6 μg) to the antisense probes and digested them with RNase (RPA III, Ambion) to map the predominant transcription initiation start site. We separated the samples on 5% acrylamide gels, dried them and autoradiographed them.
Constructs for transgenic studies.
We isolated the P1 DNA with a plasmid kit according to the manufacturer's recommendations (Qiagen). We subcloned the λ genomic clones into KSII+ (Stratagene) and inserted the LacZ cassette downstream of the Ednrb 5′ untranslated region to generate the −14 kb and −4 kb Ednrb-LacZ constructs. We prepared the other Ednrb-LacZ constructs by digesting either −14 kb or −4 kb constructs with specific restriction enzymes (Fig. 1a). For heterologous promoter experiments, we generated the different genomic fragments by PCR, sequenced them and fused them to the minimal mouse hsp68 promoter driving LacZ expression. To mutate sites I, II and III, we replaced the SOX10 binding sites (Fig. 4c) with the mutant sequence (5′-CCGCGG-3′) by site-directed mutagenesis. We isolated the constructs for transgenic injections by agarose gel electrophoresis and purified them with NucleoSpin Extraction Kit (Clontech).
Transgenics and analyses.
We established the P1 transgenic lines and crossed them into the EdnrbLacZneo line7. We intercrossed the Ednrb+/LacZneo transgenic P1 mice to generate EdnrbLacZneo/LacZneo transgenic P1 mice. We injected the LacZ transgenic constructs into fertilized C57BL6/C3H hybrids (Taconic) and isolated the F0 embryos at different stages of development. We processed the embryos and stained them for LacZ expression with either X-gal (Sigma-Aldrich) or Salmon-gal (Biosynth) as described previously7. We then hybridized some embryos with c-Ret RNA probe (provided by V. Pachnis; MRC National Institute for Medical Research) or processed them for immunohistochemistry. For immunohistochemistry, we stained the guts with LacZ, embedded them in OCT and cryosectioned them. We hybridized the sections with antibody against SOX10 (Santa Cruz Biotechnology) and then with secondary antibodies labeled with either Alexa Fluor 488 or Alexa Fluor 568 (Molecular Probe). We identified the transgenic embryos by PCR with LacZ-specific primers from yolk sac DNA. We generated the −1.2-kb Ednrb-LacZ stable lines by crossing the founders to C57BL/6J.
Mice.
We mated Ednrb+/tetO mice to EIIACre mice to obtain Ednrb+/tetOΔHy mice as described previously4. We then mated Ednrb+/tetOΔHy mice to Ednrb+/LacZ mice, isolated embryonic guts and stained them for β-galactosidase activity7. We hybridized the Sox10Dom embryos to an Ednrb RNA probe from pWP40 (provided by A. McCallion; Johns Hopkins University School of Medicine). Mice were housed and handled in accordance with the protocol approved by the Institutional Animal Care and Use Committee of Fox Chase Cancer Center.
SOX10 gel-shift assay.
We generated the N-terminal (amino acids 1–189) SOX10, containing the HMG domain, by PCR from pCMV5-Sox10 (provided by M. Wegner; Universität Erlangen-Nürnberg) and cloned it into pGex-2T (Amersham Biosciences). We expressed GST-SOX10 (1–189) and isolated it with Bulk GST Purification Module (Amersham Biosciences). We synthesized and labeled the oligonucleotides for SOX10 binding sites I, II and III. Oligonucleotide sequences are available on request. We incubated the labeled DNA with purified protein in 20 μl of reaction buffer (10 mM HEPES buffer (pH 7.9), 50 mM KCl, 1 mM EDTA, 5% glycerol, 1 mM dithiothreitol and 100 ng of poly-dGdC) for 15 min at room temperature. For competition, we added 100× or 200× molar excess of unlabeled SOX10 wild-type or mutant oligonucleotides to the reactions, in which the labeled probes were preincubated for 5 min with GST-SOX10 (1–189). For supershift assay, we added 1:10 dilution of SOX10 antibody (Santa Cruz Biotechnology) after 5 min of preincubation. We separated the samples on 0.25× TBE, dried them and exposed them on X-ray film overnight.
GenBank accession number.
Ednrb mRNA, NM_007904.
Accession codes
References
Chakravarti, A. & Lyonnet, S. Hirschsprung Disease. in The Metabolic and Molecular Bases of Inherited Disease (eds. Scriver, C.R. et al.) 6231–6255 (McGraw-Hill, New York, 2001).
Hosoda, K. et al. Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell 79, 1267–1276 (1994).
Puffenberger, E.G. et al. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung's disease. Cell 79, 1257–1266 (1994).
Shin, M.K., Levorse, J.M., Ingram, R.S. & Tilghman, S.M. The temporal requirement for endothelin receptor-B signalling during neural crest development. Nature 402, 496–501 (1999).
Mollaaghababa, R. & Pavan, W.J. The importance of having your SOX on: role of SOX10 in the development of neural crest-derived melanocytes and glia. Oncogene 22, 3024–3034 (2003).
Kapur, R.P., Sweetser, D.A., Doggett, B., Siebert, J.R. & Palmiter, R.D. Intercellular signals downstream of endothelin receptor-B mediate colonization of the large intestine by enteric neuroblasts. Development 121, 3787–3795 (1995).
Lee, H.O., Levorse, J.M. & Shin, M.K. The endothelin receptor-B is required for the migration of neural crest-derived melanocytes and enteric neurons. Dev. Biol. 259, 162–175 (2003).
Newgreen, D.F. & Hartley, L. Extracellular matrix and adhesive molecules in the early development of the gut and its innervation in normal and spotting lethal rat embryos. Acta Anat. (Basel) 154, 243–260 (1995).
Shin, M.K., Russell, L.B. & Tilghman, S.M. Molecular characterization of four induced alleles at the Ednrb locus. Proc. Natl. Acad. Sci. USA 94, 13105–13110 (1997).
Southard-Smith, E.M., Kos, L. & Pavan, W.J. Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model. Nat. Genet. 18, 60–64 (1998).
Herbarth, B. et al. Mutation of the Sry-related Sox10 gene in Dominant megacolon, a mouse model for human Hirschsprung disease. Proc. Natl. Acad. Sci. USA 95, 5161–5165 (1998).
Britsch, S. et al. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 15, 66–78 (2001).
Pingault, V. et al. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat. Genet. 18, 171–173 (1998).
Lane, P.W. & Liu, H.M. Association of megacolon with a new dominant spotting gene (Dom) in the mouse. J. Hered. 75, 435–439 (1984).
Kapur, R.P. et al. Abnormal microenvironmental signals underlie intestinal aganglionosis in Dominant megacolon mutant mice. Dev. Biol. 174, 360–369 (1996).
Southard-Smith, E.M. et al. The Sox10(Dom) mouse: modeling the genetic variation of Waardenburg-Shah (WS4) syndrome. Genome Res. 9, 215–225 (1999).
Rothman, T.P., Goldowitz, D. & Gershon, M.D. Inhibition of migration of neural crest-derived cells by the abnormal mesenchyme of the presumptive aganglionic bowel of ls/ls mice: analysis with aggregation and interspecies chimeras. Dev. Biol. 159, 559–573 (1993).
Rothman, T.P. et al. Increased expression of laminin-1 and collagen (IV) subunits in the aganglionic bowel of ls/ls, but not c-ret −/− mice. Dev. Biol. 178, 498–513 (1996).
Leibl, M.A. et al. Expression of endothelin 3 by mesenchymal cells of embryonic mouse caecum. Gut 44, 246–252 (1999).
Natarajan, D., Marcos-Gutierrez, C., Pachnis, V. & de Graaff, E. Requirement of signalling by receptor tyrosine kinase RET for the directed migration of enteric nervous system progenitor cells during mammalian embryogenesis. Development 129, 5151–5160 (2002).
de Graaff, E. et al. Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev. 15, 2433–2444 (2001).
Carrasquillo, M.M. et al. Genome-wide association study and mouse model identify interaction between RET and EDNRB pathways in Hirschsprung disease. Nat. Genet. 32, 237–244 (2002).
Barlow, A., de Graaff, E. & Pachnis, V. Enteric nervous system progenitors are coordinately controlled by the G protein-coupled receptor EDNRB and the receptor tyrosine kinase RET. Neuron 40, 905–916 (2003).
Iwashita, T., Kruger, G.M., Pardal, R., Kiel, M.J. & Morrison, S.J. Hirschsprung disease is linked to defects in neural crest stem cell function. Science 301, 972–976 (2003).
Hearn, C.J., Murphy, M. & Newgreen, D. GDNF and ET-3 differentially modulate the numbers of avian enteric neural crest cells and enteric neurons in vitro. Dev. Biol. 197, 93–105 (1998).
Wu, J.J., Chen, J.X., Rothman, T.P. & Gershon, M.D. Inhibition of in vitro enteric neuronal development by endothelin-3: mediation by endothelin B receptors. Development 126, 1161–1173 (1999).
Paratore, C., Eichenberger, C., Suter, U. & Sommer, L. Sox10 haploinsufficiency affects maintenance of progenitor cells in a mouse model of Hirschsprung disease. Hum. Mol. Genet. 11, 3075–3085 (2002).
Kim, J., Lo, L., Dormand, E. & Anderson, D.J. SOX10 Maintains Multipotency and Inhibits Neuronal Differentiation of Neural Crest Stem Cells. Neuron 38, 17–31 (2003).
Kruger, G.M. et al. Temporally distinct requirements for endothelin receptor B in the generation and migration of gut neural crest stem cells. Neuron 40, 917–929 (2003).
Seidman, J.G. & Seidman, C. Transcription factor haploinsufficiency: when half a loaf is not enough. J. Clin. Invest. 109, 451–455 (2002).
Acknowledgements
We thank J. Burch and K. Zaret for critical comments on the manuscript, the members of the laboratories of M.K.S. and K. Zaret for discussions and H. Duchow and J. Levorse for establishing the P1 transgenic founders. The Fox Chase Cancer Center Automated DNA Sequencing and Transgenic facilities, supported by US National Cancer Institute Core Grant, carried out the sequencing and transgene injections, respectively. E.M.S. is supported by grants from Foundation for Digestive Health and Nutrition Research Scholar Award, Howard Hughes Medical Institute Scholar award and US National Institutes of Health. H.O.L. is supported by the Institutional Research Training Grant from National Cancer Institute. M.K.S. is supported by grants from the PEW Scholars Program in Biomedical Sciences and US National Institute of Dental and Craniofacial Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Zhu, L., Lee, HO., Jordan, C. et al. Spatiotemporal regulation of endothelin receptor-B by SOX10 in neural crest–derived enteric neuron precursors. Nat Genet 36, 732–737 (2004). https://doi.org/10.1038/ng1371
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1371
This article is cited by
-
TGFβR-1/ALK5 inhibitor RepSox induces enteric glia-to-neuron transition and influences gastrointestinal mobility in adult mice
Acta Pharmacologica Sinica (2023)
-
Epilation induces hair and skin pigmentation through an EDN3/EDNRB-dependent regenerative response of melanocyte stem cells
Scientific Reports (2017)
-
Identification of different mechanisms leading to PAX6 down-regulation as potential events contributing to the onset of Hirschsprung disease
Scientific Reports (2016)
-
Down-regulation of miR-206 is associated with Hirschsprung disease and suppresses cell migration and proliferation in cell models
Scientific Reports (2015)
-
Development and developmental disorders of the enteric nervous system
Nature Reviews Gastroenterology & Hepatology (2013)
