
Abstract
Cells are controlled by the complex and dynamic actions of thousands of genes. With the sequencing of many genomes, the key problem has shifted from identifying genes to knowing what the genes do; we need a framework for expressing that knowledge. Even the most rigorous attempts to construct ontological frameworks describing gene function (e.g., the Gene Ontology project) ultimately rely on manual curation and are thus labor-intensive and subjective. But an alternative exists: the field of functional genomics is piecing together networks of gene interactions, and although these data are currently incomplete and error-prone, they provide a glimpse of a new, probabilistic view of gene function. We outline such a framework, which revolves around a statistical description of gene interactions derived from large, systematically compiled data sets. In this probabilistic view, pleiotropy is implicit, all data have errors and the definition of gene function is an iterative process that ultimately converges on the correct functions. The relationships between the genes are defined by the data, not by hand. Even this comprehensive view fails to capture key aspects of gene function, not least their dynamics in time and space, showing that there are limitations to the model that must ultimately be addressed.
Similar content being viewed by others
Main
The necessity for a logical framework
A primary issue in biology is describing the global organization of genes into systems and understanding the coordination of these systems in the cell. The sequencing of complete genomes has yielded the full parts lists of several organisms, and we now have powerful techniques to sample different aspects of gene function at a genome-wide level. Together, these advances hold great promise in leading us closer to a complete description of the molecular biology of the cell. For us to reach this goal, however, we need some coherent framework in which to express what we learn about gene function through the accumulation of data. What do we mean by 'gene function'? More importantly, what should we mean by this? What are the key properties of genes that we should measure to adequately describe their function in the cell? If we have all the measurements, how should we integrate them into a complete description of the molecular machineries that are the basis of a cell? Our framework for thinking about gene function inevitably colors the questions we ask and the conclusions we draw; therefore, defining a rational framework is not merely an exercise in abstraction but a key tool in understanding how an organism works.
Great effort (e.g., refs. 1–8) has already gone into defining precisely what we mean by the functions of genes, as well as how we should record this information. These summaries help us by identifying common features of genes with similar functions and giving clues to the organization and control of processes in the cell. They may also help us to see whether this organization can explain observed biological properties and guide us toward new processes. The functional framework itself, perhaps even more than the raw data on which it was based, becomes useful for comparing how processes are organized in different organisms, uncovering common themes and giving insight into the biological evolution of complexity.
The framework we use for describing gene function is of such importance that it is worth considering the possible forms that such a framework might take and the possible ways to construct it. In particular, it is now routine to generate vast data sets rapidly; developing the conceptual tools to integrate and examine these data is thus of paramount importance. Here, we argue that functional genomics has created an opportunity for substantially refining our existing frameworks of gene function, specifically the ontologies capturing systems, pathways and interactions, and that there is much to be gained by considering an alternate, probabilistic view of gene function that has only recently become feasible.
What features should a functional framework possess?
Although we may not yet have a rigorous definition of gene function, we already understand a great deal about how genes act together in the cell. Any reasonable framework seeking to describe gene function should include these features. We know that many (if not most) genes carry out not a single function in the cell, but several. For example, some TAFs have a role both in transcriptional initiation9 and in DNA repair10,11; ras regulates both mitogenesis and cytoskeletal rearrangements12; and p53 (if we believe all the literature) can run an entire cell almost single-handedly. Whatever framework we choose must therefore allow for pleiotropy. We also know that cellular processes seem to be organized in a hierarchical manner. Consider the example of the protein ORC2, which binds the origin of DNA replication to initiate the process of DNA replication13,14,15. The specific function of ORC2 is to bind DNA and, along with the proteins ORC1–ORC6, it forms the origin recognition complex16,17,18. The origin recognition complex is a single component of the prereplicative complex, which in turn is one component required for initiation of DNA replication, and so on up the hierarchy of functions, from specific to general, detailed to global.
In addition to encapsulating both pleiotropy and a hierarchical organization of functions, we also need a framework that can rapidly accommodate new data. As the rate at which new data are being produced is ever increasing, the nearer one can get to an automated system, the better. This is a key feature for any workable framework. Finally, as well as being able to encapsulate our current state of knowledge regarding gene function, a useful framework should serve as a guide for future analysis, capable of highlighting unexplored processes and connections that arise from new data. Some of these may correctly overturn previous models, and a good framework must have the flexibility to allow this kind of change.
All in all, this is not a trivial problem. We want a framework that describes gene function in a hierarchical structure that can accommodate pleiotropy, that not only encapsulates all our current knowledge but can also rapidly pull in new data and direct our research towards new, uncharacterized phenomena. Where to even start? In the next section we contrast two possible approaches to this problem.
Two methods for constructing a hierarchical ontology
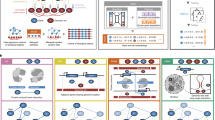
Two approaches seem feasible for constructing a rational hierarchical ontology for analyzing gene function: the 'top-down' and 'bottom-up' strategies (Fig. 1). The top-down approach, exemplified by the Gene Ontology project1,8, involves first defining an almost comprehensive list of gene function categories and organizing them into a hierarchy and, second, fitting individual genes into those categories. In this approach, 'what a gene does' is to be associated with a series of attributes covering its molecular and cellular functions (for example). The choice of categories, their hierarchical organization and the assignment of genes into these categories are all done through meticulous manual curation; this is ultimately a subjective process. In contrast, the bottom-up approach uses statistical methods to integrate multiple data sets, generating networks of gene linkages. These networks have features that can be extracted and used as the framework for describing and categorizing gene function. In the bottom-up view of gene function, 'what a gene does' is to interact with other genes, no more no less. In this admittedly simple view, the way in which a gene interacts and the topological features of the network in which a gene sits define its function. A key difference between these two approaches is that in a top-down system, the functional hierarchy is fixed by the subjective manual curation of the underlying data, whereas in a bottom-up approach, the data directly reveal the hierarchy. In the former case, what a gene does is defined by the processes it carries out; in the latter, it is the way a gene links to other genes that defines both its function and the processes themselves.

In a top-down approach, expert curators draw up a hierarchical ontology for describing different aspects of gene function (boxes 1–6) from their knowledge of biology; they then manually assign genes (circles A–C) to categories according to evidence from literature. Although we depict here a method in which genes are placed into a single category, genes can be associated with multiple categories, and these associations can be weighted. In the alternative, bottom-up approach, gene linkages are imported from diverse data sets and integrated into a complex network of genes (small circles). The features of this network define both the processes (dashed regions 1–4) and the associations of each gene with each of the processes.
The top-down approach has many merits: it consolidates the observations of uncountable researchers into a reasonable, though subjective, view of the current state of knowledge of gene function. This strategy creates an ontology that agrees, by design, with the current opinions of how genes mesh together functionally, with a nice side effect of having easy-to-read gene functions. In practice (e.g., as in the Gene Ontology project), curators begin with the sum of knowledge in the field, manually organize the observations into hierarchical categories of increasing precision and then place genes into these categories by examining the scientific literature, perhaps aided by computational approaches19,20,21. As new literature becomes available, the categories and gene assignments are manually modified. However powerful this approach—for example, curators may filter noise and suppress errors during this process—it suffers from certain shortcomings: there is no objective way to define the categories or to place the genes into the categories. It can be difficult to assess the quality of those assignments, as the evidence placing genes into a category may differ, and thus the quality of assignments varies in each category. Also, improvements to the hierarchy are difficult to automate and must usually be manual. Given the incredible speed with which data are now being gathered along with bewildering complexity of the data sets generated by many functional genomics approaches, this is a key limitation. Some of these limitations can perhaps be addressed by bottom-up approaches, which rely entirely on the integration of multiple data sets to map out a framework for describing gene function.
Constructing a probabilistic network model
Rather than impose a subjective functional hierarchy on the genes, as is done in the top-down approaches illustrated above, we would prefer to let the data construct the network and the implicit hierarchies directly. How might we integrate the many available data sets to give such a bottom-up network? Such a process of integration requires us first to deal with diverse data sets (e.g., physical interaction data, microarray coexpression data and genetic interaction studies) and, second, to be able to separate signal from noise. Merely summing the connections seen in all the hundreds of available error-strewn data sets would clearly result in a tangled network of little biological utility. Any researcher familiar with the large data sets generated by functional genomics approaches is acutely aware of the noise and error in such sets, and dealing with these errors is an essential part of any integrative approach.
To deal with noisy data sets, one must use statistical methods to ensure that each data set is quality-controlled and given only the weight that corresponds to its quality. For each incorporated data set, we can measure the error associated with the data by establishing objective, quantitative tests ('benchmarks') to distinguish accurate from inaccurate data. We expect that correct data sets should in general perform better than incorrect datasets on these benchmarks. For example, we might ask how often known protein interactions are recovered by a high-throughput interaction screen22,23, how often proteins observed to interact in a high-throughput screen actually localize to the same subcellular compartment24 or to what extent the interaction partners' mRNAs tend to be coexpressed across DNA microarray experiments25. We might even measure properties of the resultant properties interaction network26,27. This testing of the noisy data sets in the bottom-up approach explicitly relies on independent high quality data or manual annotations, such as those created in the top-down descriptions of gene function. Benchmarks provide a numerical estimate of the average error rate in a set of experiments and thereby make it feasible to combine interactions from different experiments (Fig. 2). Because the error is quantified, interactions from each data set can be weighted according to their measured performances on the benchmarks, and interactions can be assigned a joint confidence based on the combined weight of evidence. This measurement of error and weighted association between genes is the essence of the bottom-up approach. There are many different schemes for establishing the weights of each data set based on its underlying errors; these include Bayesian statistics, used recently for scoring physical interactions23,28, or heuristic or other probabilistic approaches29,30,31. Similarly, there are many approaches for ensuring the quality of the integrated data, such as using independent benchmarks or even saving aside portions of benchmarks for final tests of accuracy, akin to the cross-validation procedures used commonly in computer sciences. In outline, the methods give a similar output: they result in networks in which the links between the genes are no longer binary (i.e., YES-NO, linked-unlinked) but are probabilistic weights. The weight attached to each gene-gene linkage derives both from the 'tightness' of the association and the extent of error in measuring the linkage.

Data sets from diverse experiments are individually tested for their quality against a benchmark set and are weighted accordingly. Various statistical approaches can then be used to integrate these weighted matrices to yield a final matrix that contains all the probabilistic linkages between genes. Graphical networks can be derived from this matrix by extracting all links that exceed a specified confidence threshold. Thus, the integrated matrix can yield multiple networks of differing complexity and confidence.
In addition to being able to deal with error in the data sets that underlie the construction of the network, this integrative approach also has the excellent qualities that it improves iteratively as more data are added and, furthermore, that it can be used for the integration of diverse data sets. We can illustrate this by showing how one might begin to construct a complex network model of gene function in yeast (Fig. 3). The conceptually simplest network, and the one that many people recognize most easily, is a map of the physical interactions between proteins, and we take this as our starting point. Using the methods outlined above, we can integrate evidence from different data sets including both small-scale and large-scale (e.g., high-throughput yeast two-hybrid32,33,34,35 or affinity purification of complexes36,37) data sets and obtain a tolerably accurate view of the total possible network of interactions that might occur between cellular proteins. Links seen in multiple data sets (even if each of these is very noisy) reinforce and greatly improve the quality of the network; thus, even links observed in weak or noisy data sets are of value as they may sum with other such links to form statistically significant informational interactions. Because each linkage is a weight rather than a binary interaction, it is possible to view the network at differing degrees of confidence (Fig. 3).

Three networks are shown, each with a different level of confidence. The links derive from refs. 32,33,35–37 and are compared with benchmarks as described in ref. 25, with high confidence (3,344 interactions, estimated accuracy 78%) corresponding to interactions found in two or more screens combined with interactions from ref. 36, medium confidence (7,328 interactions, estimated accuracy 45%) corresponding to high-confidence interactions plus those from ref. 35 and low confidence (10,435 interactions, estimated accuracy 31%) including all interactions from the above screens.
Physical interaction data sets do not sample all the possible biologically significant interactions between genes. To do this, we need to add other data sets to the physical interaction map. A key aspect of this integrative approach is that it can deal easily with very diverse data sets: genetic interaction data sets can be imported as easily as physical interaction data, computational predictions of linkages as easily as microarray correlations. We can (for example) add links deriving from genetic interactions38 to the physical interaction map (Fig. 3) and obtain a hybrid network. In this network, links between genes no longer represent any obvious physical reality but are informational constructs: a link between two genes is not a single defined connection but a sum of different linkages that exceed a statistical threshold. Because of this generality, we anticipate that many distinct types of functional genomics data, including genetic or physical interactions, colocalization and expression patterns, could be combined in this framework.
The network obtained is not a uniform landscape of gene linkages, but has 'features', areas of high connectivity separated by more sparsely connected genes. Computational clustering of genes according to their connectivities39,40,41,42,43 shows that these features may correspond to known cellular machines (e.g., the ribosome, the proteasome, etc.)44,45 but may also be totally unexplored46,47,48,49. The network as a stand-alone description of gene function is not directly interpretable by us. What do those highly connected clusters of genes represent? How do they relate to the processes that we already know? The key is that whatever the features are, they arise directly out of the data in an unbiased manner. Adding subjective 'labels' to these features (e.g., the splicing machinery) is vital for us to be able to extract their meaning as useful, but the labels do not dictate the form and hierarchical organization of the network. A reasonable strategy would therefore be to construct the network and hierarchy by the bottom-up approach, then to interpret it with labels from the top-down approach, looking especially to rationalize areas of disagreement, which have the potential of being new connections between systems, unappreciated in the top-down approach but uncovered by the bottom-up synthesis of functional genomics data.
The bottom-up approach described here naturally leads to an entirely probabilistic description of gene function. Not only are the links between genes actually weighted, but also the descriptions of a 'process' and the relative involvement of a gene in that process are mathematical, probabilistic constructs that depend on the network features. These probabilistic connections arise in part from our experimental uncertainty in connecting genes together but also may reflect the stochastic nature of protein function, with finite copies of proteins forced to partition between alternate tasks in a cell. 'What a gene does' is defined by where it resides in the network and the probabilistic paths that link it to features. The organization of the features, their interconnections and the locations of each gene in this landscape arise purely from the data and form a pleasingly simple source of a hierarchical framework for describing gene function. Like many deceptively simple solutions to complex problems, however, probabilistic networks are not without their quirks and pitfalls. We consider some of these in the next section.
Properties and limitations of networks
Certain points are immediately obvious from looking at the kind of networks shown in Figure 3. First, the links are not distributed uniformly, but there are regions of high connectivity interspersed with barer, less connected areas; the highly connected areas probably correspond to biological processes or machineries such as the splicing machinery or DNA polymerase. Second, defining exactly where one highly connected region starts and another stops or the amount of connectedness required to say 'this is a connected region' is not rigid. Thus, biological processes (i.e., the highly connected regions) are not fixed entities but can be defined according to different criteria. One might consider as few as three interlinked genes to be a process or define a fully linked arrangement of ten genes as a process. Processes can thus be defined at different levels of complexity, which results directly in a hierarchical organization of subnetworks, networks, supernetworks. The networks thus implicitly capture the hierarchical organization of biological processes in the cell, and this hierarchy can be explored by clustering the genes according to their network connections, the genes' precise functions being reflected in their memberships in different clusters39,40,41,42,43,44,45,46,47,48,49. Third, however one defines a process in the full network, these processes are highly interlinked. Thus, these networks encapsulate the linked nature of molecular biology as we know it. For example, DNA repair is closely linked to DNA replication, the anaphase promoting complex is linked to cell-cycle regulation and so forth. Last, any gene is linked to multiple processes by a relatively short path. Pleiotropy is therefore implicit in these probabilistic networks. Because most biological networks examined so far are 'small-world' networks27,50,51,52, the path length from any gene to any process, or between any processes, is remarkably short, reinforcing both pleiotropy and the linked nature of processes.
Despite the strengths of the bottom-up, integrative approach, the current models for networks that result have several key shortcomings that should be addressed. First, they do not show either time or space (not insignificant omissions!), describing all linkages as equivalent static entities. Second, the networks represent 'genotypic' averages across cells, tissues, mutants and conditions. Thus, although each 'node' in the network represents a single gene, links attached to any node may in fact be a sum of data relating not to the 'normal', wild-type gene but to other forms (e.g., genetic interactions between point mutants or knockouts of the genes, or physical interactions between activated forms or single domains of the encoded proteins). Many of the different data sets used to construct the network may even derive from mutually excusive conditions, such as wild-type and mutant. Each node is thus an 'average gene', and the links to that average gene are the sums of data. Ultimately this problem of how to interpolate wild-type function by integrating data from perturbed states is a crucial one in much of biology, not only in these networks, but it is nonetheless one that we need to be aware of and attempt to address in the future.
Regarding time and space, network models are currently static summations of all available data. Genes have dynamic expression patterns in time and space, and the associations between genes and proteins are extensively regulated, rarely occurring constitutively. Thus, we might imagine the vertices of the network winking in and out of existence as the genes are expressed, and the edges only truly connecting the genes when given conditions are met. The network should thus be regarded as containing all possible linkages, regardless of which may occur in a given circumstance.
One conceptual solution to these problems is to consider the networks we have described so far as representing an organism's 'master' network, of which only pieces (subnetworks) will be active at any one time or in any one condition or cell type. The master network and subnetworks bear the same relationship to each other as do the genome and transcriptome—the former represents the organism's potential, the latter the actual genes observed under given conditions. Likewise, the master network represents the union of all possible gene linkages, and the conditional subnetworks reflect the state of the cell. For example, vertices might only be present conditional on the genes' expression, whereas edges might be conditional on the genes' activities. The conditional subnetworks would thus capture some aspects of the time dependency of cellular activities. The subcellular location might be implicit in the links themselves; links between molecules in different subcellular compartments are presumably less likely. Thus, we might imagine capturing the activity of the system as subnetworks dynamically evolving over time and conditions.
These are issues that must ultimately be resolved to represent the true state of relationships between genes in a cell. But they do not diminish the probabilistic network as a useful framework for considering gene function.
The universal network
We might consider the entire set of statistically significant linkages between genes in any one organism as the master informational network of the organism. Such networks are most robust in model organisms for which many large data sets have been accumulated; Saccharomyces cerevisiae is the best example. As model organisms have been essential for the generation of testable hypotheses in higher eukaryotes including humans, one might imagine constructing a network in one model organism, basing it entirely on data gathered in that system, and then using orthology to transport all the information in the network into another genome. The start network is the sum of all experimental data derived in one organism, and the resulting network is the sum of all testable hypotheses in the second organism. Doing this requires robust methods for the identification of orthologous sequences in the two genomes and makes the large assumption that the network underlying the basic biology of the cell in one organism is similar in form to that in a second. In Figure 4, we illustrate how even with these caveats, a network constructed in yeast can form a tremendously useful scaffold for understanding the function of Caenorhabditis elegans genes. The networks may thus provide an objective view of gene function that can be transported between organisms, and perhaps iteratively improved with data from across organisms47,53,54. Thus, one might consider constructing a universal network, the union of individual organisms' networks, perhaps with linkages and vertices flagged by the organisms from which they derive (such as in the Genome Knowledgebase; http://www.genomeknowledge.org/), that would represent the cumulative body of knowledge of gene function, even across evolutionary divergence of the systems. Even if such a grand goal may be currently unattainable, transporting entire networks from model organisms into higher genomes may already provide a valuable framework for generating testable hypotheses in these higher organisms.

A network of S. cerevisiae genes (the medium-confidence network from ref. 22) was 'transported' into C. elegans by mapping each yeast gene in the network to its worm counterpart using the Inparanoid algorithm55. (a) In the network, each node is a worm gene (or in some cases a number of worm paralogs), and each link between genes is hypothetical, based uniquely on links between yeast genes. (b) To test whether the features seen in the worm networks have any functional relevance, we examined whether genes with related loss-of-function phenotypes (as determined by RNAi56) are found in similar areas of the network. Genes with nonviable RNAi phenotypes are shown as red nodes, and links between nonviable genes are shown as red edges; genes with growth-defective RNAi phenotypes are shown in green. (c) Clusters of linkages in the networks correspond to known molecular processes. (d) Close-up of the proteasome. Large red circles mark nodes that were not found to have nonviable RNAi phenotypes in ref. 56 but were found to have nonviable phenotypes in other RNAi analyses57,58.
Conclusion
Having a rational framework for describing gene function is essential for us to be able to understand what every gene does and how gene functions are coordinated in the cell. Here, we illustrate how such a framework can come out of the integration of diverse large data sets into a probabilistic network of gene interactions. Both the framework itself and its hierarchical organization arise directly from raw data without any requirement for manual curation or assumptions about underlying cellular processes—the networks and framework also improve iteratively and automatically as new data are added. In this view of gene function, 'what a gene does' is defined simply by the way in which it interacts with other genes. Because the association between any two genes in this model is probabilistic rather than binary, the function of a gene is also probabilistic rather than concretely defined. The organization of the interactions of all the genes in these network is not uniform but ranges from areas of high connectivity to far emptier spaces; the areas of high connectivity may define individual biological processes. Defining a biological process in this way is, like the definition of gene function, a probabilistic approach. Both gene functions and biological processes are thus probabilistic constructs in this view. Furthermore, in this model of function, both pleiotropy and the hierarchical organization of biological processes are implicit.
This way to view gene function, and the underlying methods used to evaluate and integrate large data sets, may provide us with a practical way to use the enormous quantity of functional data being generated by genomics analyses to inform our understanding of the cell and of gene function in general. Although there are still key limitations to this probabilistic approach, we believe that it provides an important and productive view of gene function. Furthermore, as new data sets are incorporated into probabilistic networks in the future, they will iteratively converge on a more complete description of the molecular networks governing the fundamental biology of the cell.
References
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Bateman, A. et al. The Pfam protein families database. Nucleic Acids Res. 30, 276–280 (2002).
Kanehisa, M. et al. The KEGG databases at GenomeNet. Nucleic Acids Res. 30, 42–46 (2002).
Karp, P.D. et al. The EcoCyc Database. Nucleic Acids Res. 30, 56–58 (2002).
Karp, P.D. et al. The MetaCyc Database. Nucleic Acids Res. 30, 59–61 (2002).
Mewes, H.W. et al. MIPS: a database for genomes and protein sequences. Nucleic Acids Res. 30, 31–34 (2002).
Mulder, N.J. et al. The InterPro Database, 2003 brings increased coverage and new features. Nucleic Acids Res. 31, 315–318 (2003).
The Gene Ontology Consortium. Creating the gene ontology resource: design and implementation. Genome Res. 11, 1425–1433 (2001).
Tjian, R. The biochemistry of transcription in eukaryotes: a paradigm for multisubunit regulatory complexes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 351, 491–499 (1996).
Brand, M. et al. UV-damaged DNA-binding protein in the TFTC complex links DNA damage recognition to nucleosome acetylation. EMBO J. 20, 3187–3196 (2001).
Martinez, E. et al. Human STAGA complex is a chromatin-acetylating transcription coactivator that interacts with pre-mRNA splicing and DNA damage-binding factors in vivo. Mol. Cell. Biol. 21, 6782–6795 (2001).
Hall, A. The cellular functions of small GTP-binding proteins. Science 249, 635–640 (1990).
Ritzi, M. et al. Human minichromosome maintenance proteins and human origin recognition complex 2 protein on chromatin. J. Biol. Chem. 273, 24543–24549 (1998).
Rowles, A. et al. Interaction between the origin recognition complex and the replication licensing system in Xenopus. Cell 87, 287–296 (1996).
Coleman, T.R., Carpenter, P.B. & Dunphy, W.G. The Xenopus Cdc6 protein is essential for the initiation of a single round of DNA replication in cell-free extracts. Cell 87, 53–63 (1996).
Bell, S.P. & Stillman, B. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 357, 128–134 (1992).
Vashee, S. et al. Assembly of the human origin recognition complex. J. Biol. Chem. 276, 26666–26673 (2001).
Dhar, S.K., Delmolino, L. & Dutta, A. Architecture of the human origin recognition complex. J. Biol. Chem. 276, 29067–29071 (2001).
Raychaudhuri, S. et al. Associating genes with gene ontology codes using a maximum entropy analysis of biomedical literature. Genome Res. 12, 203–214 (2002).
Troyanskaya, O.G. et al. A Bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae). Proc. Natl. Acad. Sci. USA 100, 8348–8353 (2003).
Clare, A. & King, R.D. Machine learning of functional class from phenotype data. Bioinformatics 18, 160–166 (2002).
Von Mering, C. et al. Comparative assessment of large-scale data sets of protein-protein interactions. Nature 417, 399–403 (2002).
Jansen, R. et al. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science 302, 449–453 (2003).
Schwikowski, B., Uetz, P. & Fields, S. A network of protein-protein interactions in yeast. Nat. Biotechnol. 18, 1257–1261 (2000).
Deane, C.M. et al. Protein interactions: two methods for assessment of the reliability of high throughput observations. Mol. Cell. Proteomics 1, 349–356 (2002).
Saito, R., Suzuki, H. & Hayashizaki, Y. Interaction generality, a measurement to assess the reliability of a protein-protein interaction. Nucleic Acids Res. 30, 1163–1168 (2002).
Goldberg, D.S. & Roth, F.P. Assessing experimentally derived interactions in a small world. Proc. Natl. Acad. Sci. USA 100, 4372–4376 (2003).
Jansen, R. et al. Integration of genomic datasets to predict protein complexes in yeast. J. Struct. Funct. Genomics 2, 71–81 (2002).
Huynen, M. et al. Predicting protein function by genomic context: quantitative evaluation and qualitative inferences. Genome Res. 10, 1204–1210 (2000).
von Mering, C. et al. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261 (2003).
Marcotte, E.M. et al. A combined algorithm for genome-wide prediction of protein function. Nature 402, 83–86 (1999).
Ito, T. et al. Toward a protein-protein interaction map of the budding yeast: A comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc. Natl. Acad. Sci. USA 97, 1143–1147 (2000).
Ito, T. et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA 98, 4569–4574 (2001).
Tong, A.H. et al. A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science 295, 321–324 (2002).
Uetz, P. et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403, 623–627 (2000).
Gavin, A.C. et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147 (2002).
Ho, Y. et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415, 180–183 (2002).
Tong, A.H. et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294, 2364–2368 (2001).
Rives, A.W. & Galitski, T. Modular organization of cellular networks. Proc. Natl. Acad. Sci. USA 100, 1128–1133 (2003).
Spirin, V. & Mirny, L.A. Protein complexes and functional modules in molecular networks. Proc. Natl. Acad. Sci. USA 100, 12123–12128 (2003).
Tornow, S. & Mewes, H.W. Functional modules by relating protein interaction networks and gene expression. Nucleic Acids Res. 31, 6283–6289 (2003).
Ideker, T. & Lauffenburger, D. Building with a scaffold: emerging strategies for high- to low-level cellular modeling. Trends Biotechnol. 21, 255–262 (2003).
Segal, E. et al. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 34, 166–176 (2003).
Von Mering, C. et al. Genome evolution reveals biochemical networks and functional modules. Proc. Natl. Acad. Sci. USA 100, 15428–15433 (2003).
Krause, R., von Mering, C. & Bork, P. A comprehensive set of protein complexes in yeast: mining large scale protein-protein interaction screens. Bioinformatics 19, 1901–1908 (2003).
Manke, T., Bringas, R. & Vingron, M. Correlating protein-DNA and protein-protein interaction networks. J. Mol. Biol. 333, 75–85 (2003).
Stuart, J.M. et al. A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255 (2003).
Date, S.V. & Marcotte, E.M. Discovery of uncharacterized cellular systems by genome-wide analysis of functional linkages. Nat. Biotechnol. 21, 1055–1062 (2003).
Wu, L.F. et al. Large-scale prediction of Saccharomyces cerevisiae gene function using overlapping transcriptional clusters. Nat. Genet. 31, 255–265 (2002).
Snel, B., Bork, P. & Huynen, M.A. The identification of functional modules from the genomic association of genes. Proc. Natl. Acad. Sci. USA 99, 5890–5895 (2002).
Wagner, A. & Fell, D.A. The small world inside large metabolic networks. Proc. R. Soc. Lond. B Biol. Sci. 268, 1803–1810 (2001).
Watts, D.J. & Strogatz, S.H. Collective dynamics of 'small-world' networks. Nature 393, 440–442 (1998).
Matthews, L.R. et al. Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or “interologs”. Genome Res. 11, 2120–2126 (2001).
Marcotte, E. & Date, S. Exploiting big biology: integrating large-scale biological data for function inference. Brief. Bioinform. 2, 363–374 (2001).
Remm, M., Storm, C.E. & Sonnhammer, E.L. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 314, 1041–1052 (2001).
Kamath, R.S. et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237 (2003).
Gonczy, P. et al. Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature 408, 331–336 (2000).
Piano, F., Schetter, A.J., Mangone, M., Stein, L. & Kemphues, K.J. RNAi analysis of genes expressed in the ovary of Caenorhabditis elegans. Curr. Biol. 10, 1619–1622 (2000).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Fraser, A., Marcotte, E. A probabilistic view of gene function. Nat Genet 36, 559–564 (2004). https://doi.org/10.1038/ng1370
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1370
This article is cited by
-
BIONIC: biological network integration using convolutions
Nature Methods (2022)
-
BIONIC: discovering new biology through deep learning-based network integration
Nature Methods (2022)
-
Key mendelian variants
Nature Genetics (2015)
-
A gene ontology inferred from molecular networks
Nature Biotechnology (2013)
-
Quantitative utilization of prior biological knowledge in the Bayesian network modeling of gene expression data
BMC Bioinformatics (2011)
