
Abstract
Pineapple (Ananas comosus (L.) Merr.) is the most economically valuable crop possessing crassulacean acid metabolism (CAM), a photosynthetic carbon assimilation pathway with high water-use efficiency, and the second most important tropical fruit. We sequenced the genomes of pineapple varieties F153 and MD2 and a wild pineapple relative, Ananas bracteatus accession CB5. The pineapple genome has one fewer ancient whole-genome duplication event than sequenced grass genomes and a conserved karyotype with seven chromosomes from before the ρ duplication event. The pineapple lineage has transitioned from C3 photosynthesis to CAM, with CAM-related genes exhibiting a diel expression pattern in photosynthetic tissues. CAM pathway genes were enriched with cis-regulatory elements associated with the regulation of circadian clock genes, providing the first cis-regulatory link between CAM and circadian clock regulation. Pineapple CAM photosynthesis evolved by the reconfiguration of pathways in C3 plants, through the regulatory neofunctionalization of preexisting genes and not through the acquisition of neofunctionalized genes via whole-genome or tandem gene duplication.
Similar content being viewed by others

Main
Christopher Columbus arrived in Guadeloupe in the West Indies on 4 November 1493 during his second voyage to the New World. At a Carib village, he and his sailors encountered pineapple plants and fruit, with the astonishing flavor and fragrance delighting them then and us today. At that time, pineapple was already cultivated on a continent-wide scale following its initial domestication in northern South America, possibly more than 6,000 years before the present1. By the end of the sixteenth century, pineapple had become pantropical. Because of the success of industrial production in Hawaii in the last century, pineapple is now not only a routine part of our diet, but also has captured public imagination and become part of popular culture2,3. Today, pineapple is cultivated on 1.02 million hectares of land in over 80 countries worldwide, and 24.8 million metric tonnes of fruit are produced annually with a gross production value approaching $9 billion. Pineapple has outstanding nutritional and medicinal properties2 and is a model for studying the evolution of CAM photosynthesis, which has arisen convergently in many semiarid regions4. Cultivated pineapple, A. comosus (L.) Merr., is self-incompatible5, whereas wild species are self-compatible, providing an opportunity to dissect the molecular basis of self-incompatibility in monocots. As part of the Bromeliaceae family, the pineapple lineage diverged from the lineage leading to grasses (Poaceae) early in the history of the Poales, about 100 million years ago6,7, offering an outgroup and evolutionary reference for the investigation of cereal genome evolution.
Results
Genome assembly, scaffold anchoring and annotation
The genome of pineapple variety F153, cultivated by Del Monte for 80 years, was sequenced and assembled using 400× Illumina reads, 2× Moleculo synthetic long reads, 1× 454 reads, 5× PacBio single-molecule long reads and 9,400 BACs. Because of self-incompatibility, pineapple has high levels of heterozygosity and is cultivated through clonal propagation. To overcome the difficulties in assembling this highly heterozygous genome, we applied a genetic approach to reduce the complexity of the genome using a cross between F153 and the A. bracteatus (Lindl.) Schult. & Schult.f. CB5 accession from Brazil, generating 100× CB5 and 120× F1 genome sequences. Because the F1 plant has a haploid genome from F153 and CB5, its sequences were used for haplotype phasing to improve the assembly (Supplementary Table 1). The final assembly using this approach substantially improved the initial Illumina-only assembly and spanned 382 Mb, 72.6% of the estimated 526 Mb of the pineapple genome8. The contig N50 was 126.5 kb, and the scaffold N50 was 11.8 Mb (Supplementary Table 2). Transposable elements (TEs) accounted for 44% of the assembled genome and 69% of the raw reads, indicating that 25% of the unassembled genome consists of TEs. The remaining 2.4% are centromeres, telomeres, rDNAs and other highly repetitive sequences. The GC content was 38.3% across the genome and 51.4% in coding sequences.
We sequenced 93 F1 individuals from the cross between A. comosus F153 and A. bracteatus CB5 at 10× genome equivalents each and identified SNPs using the F153 genome as a reference, yielding 296,896 segregating SNPs from F153. We constructed a genetic map for F153, spanning 3,208.6 cM at an average of 98.4 kb/cM, resulting in 25 linkage groups corresponding to the haploid chromosome number. A total of 564 scaffolds were anchored to the genetic map, covering 316 Mb, or 82.7%, of the assembled genome (Supplementary Table 3). Scaffolds that mapped to multiple linkage groups were reassembled, with breakpoints approximated using information from individual SNPs, thereby correcting 119 chimeric scaffolds. Of the 18 telomeric tracks found, 16 were at the ends of linkage groups (Supplementary Table 4).
We used MAKER for gene annotation9 and obtained 27,024 gene models, 89% of which were categorized as complete (Online Methods and Supplementary Table 5). We identified 10,151 alternative splicing events, with intron retention accounting for 62.8% of these events (Supplementary Table 6). Analysis of small RNA libraries sequenced from leaves, flowers and fruits identified 32 microRNA families, including 11 that were specific to pineapple (Supplementary Table 7).
Transposable elements and expression of retrotransposons
Long terminal repeat (LTR) retrotransposons were identified using structural criteria10,11. About 44% of the assembly was accounted for by TEs (Supplementary Table 8). LTR retrotransposons were the most abundant of these elements, representing 33% of the assembly. We compared the abundance of LTR retrotransposons in the assembly and the raw reads. The most abundant elements in the raw reads were under-represented in the assembly because of an obligate masking step (Supplementary Table 9). The Pusofa family made up 28% of all LTR retrotransposon–related sequences in the raw reads but only accounted for 0.5% of all LTR retrotransposon–related sequences in the assembly. In contrast, Wufer elements, constituting the most abundant family in the assembly (7% of LTR retrotransposons), accounted for ∼1.7% of LTR retrotransposons in the raw reads. Screening of the raw reads showed that at least 52% of the nuclear genome is derived from LTR retrotransposons, indicating a total TE content of 69% in the pineapple genome. The abundance of Pusofa elements, accounting for 28% of LTR retrotransposons and 15% of the pineapple genome, is particularly interesting because this level of dominance by a single TE family is not generally observed. In addition, we identified 20 separate instances in which an LTR retrotransposon had incorporated fragments from one or two genes into the sequence of the TE. A recent wave of LTR retrotransposon insertion appears to have occurred in the pineapple lineage about 1.5–2 million years ago (Fig. 1).

The unrooted phylogenetic tree of Gypsy and Copia elements was constructed on the basis of 6,379 aligned sequences corresponding to the reverse-transcriptase domain.
About 0.26% of RNA sequencing (RNA-seq) reads from nine tissues originated from LTR retrotransposons, with the proportion ranging from 0.16% to 0.52% per tissue (Supplementary Table 10). High LTR expression levels correlated with relatively low copy number (Supplementary Fig. 1). Among reads that mapped to intact elements (0.05% of RNA-seq reads), the most abundantly expressed family was Sira, comprising Copia elements expressed in all nine tissues and accounting for 13% of all LTR retrotransposons expressed but only 0.2% of LTR retrotransposons in the raw reads, demonstrating an inverse correlation12 (Supplementary Fig. 1). Different TE families exhibited different expression biases, as the Sira family was most highly expressed in flowers or floral tissues, the Beka family was most highly expressed in mature fruit and the Ovalut family was most highly expressed in young fruit (Supplementary Fig. 2 and Supplementary Table 10). Individual elements within a family contributed differentially to the total RNA reads for the family. For instance, of the four subfamilies of Sira, subfamily sira_1 contributed 96% of the RNA-seq reads that mapped to this family. The tissue-specific expression patterns seemed to be largely the same for each subfamily of any given family (Supplementary Fig. 3).
Heterozygosity in F153, MD2 and CB5
MD2 has been the dominant pineapple variety for the global fresh-fruit market for the last 30 years and is a hybrid from the Pineapple Research Institute in Hawaii with a complex pedigree involving five generations of hybridization. We sequenced the genomes of MD2 and a wild accession of A. bracteatus, CB5, at 100× coverage using Illumina paired-end reads of libraries with different insert sizes. De novo assembly of these two genomes yielded short contigs owing to heterozygosity. The F153 genome was used as a reference for assembling these two genomes and for assessment of within-genome heterozygosity. F153 had a combined heterozygosity rate of 1.89%, with 1.54% SNPs and 0.35% indels, whereas MD2 had a heterozygosity rate of 1.98%, with 1.71% SNPs and 0.27% indels. The wild A. bracteatus CB5 accession had a higher heterozygosity rate of 2.93%, with 2.53% SNPs and 0.40% indels (Supplementary Table 11). Two homologous pairs of F153 BACs were identified by probes designed from coding genes and sequenced using Sanger methods to verify the heterozygosity rate. The resulting rate was 2.13%, with 1.21% SNPs and 0.92% indels, indicating an underestimate of the proportion of indels in the three genomes due to the use of a single reference genome. The vast majority of heterozygous sites were intergenic, but F153 and MD2 had 100,743 and 91,876 synonymous and 195,488 and 323,836 nonsynonymous sites, respectively (Supplementary Table 11). CB5 had 186,520 synonymous and 351,908 nonsynonymous sites.
Pineapple karyotype evolution
Intragenomic syntenic analyses of pineapple show clear evidence of at least two ancient whole-genome duplication (WGD) events. Structural comparison of pineapple with itself identified 388 intragenomic blocks, including 4,891 pineapple gene pairs, derived from WGD events (Supplementary Figs. 4 and 5). Collectively, these collinear blocks spanned 64% of the annotated gene space and involved each of the 25 pineapple linkage groups, providing strong support for the occurrence of WGD events. Syntenic depth analyses13,14 indicated that 35% of the pineapple genome has more than one duplicated segment, as expected if more than one WGD occurred in the pineapple lineage.
The chromosomal organization of pineapple reflects its evolutionary trajectory following the σ and τ WGD events in monocots13,14, starting from a seven-chromosome ancestral monocot genome. We organized the 25 extant chromosomes into major groups corresponding to regions most clearly identifiable as originating from one of the seven chromosomes existing before the τ WGD, Anc1 to Anc7 (Fig. 2). We inferred the 14 chromosomes present after the τ WGD, with the two chromosomes derived from each ancestral chromosome referred to as, for example, Anc11 and Anc12. Disrupting this general one-to-one pairing, a translocation of Anc51 into Anc31 can be inferred, as well as translocations of Anc52 into Anc42 and part of Anc42 into Anc32. These events reduced the karyotype to 12 chromosomes before the σ WGD.

Shown are the 25 pineapple chromosomes organized into the pairs of paired chromosomes that arose after two WGD events. Each color represents one of the seven ancestral chromosomes. The left and right pairs represent the two subgenomes produced by WGD τ, and within each pair are the two subgenomes produced by WGD σ.
Immediately following the σ event, there were 24 chromosomes, which merged into the 16 extant chromosomes—3, 4, 8, 10, 11, 12, 13, 14, 16, 17, 18, 19, 21, 22, 23 and 25. One Anc22 chromosome appears to have inserted into one Anc11 chromosome to produce extant chromosome 5, whereas the other Anc22 chromosome appears to have fused with one Anc32 chromosome to produce chromosome 1. Two Anc1 chromosome fissions and one Anc7 chromosome fission produced chromosomes 12, 20 and 24, respectively (Fig. 2).
The high level of retention of most chromosomal identities from the two ancestral monocot WGD events makes pineapple a conservative reference genome for monocots. Pineapple has few chromosomal rearrangements and has kept 25 of the 28 potential chromosomes expected from two doublings starting from seven ancestral chromosomes (7 × 2 × 2 = 28). Similarly, the grapevine genome has had a crucial role in clarifying eudicot genome evolution15, with 17 chromosomes intact out of the 21 predicted from the γ whole-genome triplication event that gave rise to much of the eudicot clade, also originating from seven ancestral chromosomes (7 × 3 = 21). The pineapple genome could serve the same comparative role for the monocots because it has conserved most of its karyotype structure during evolution of its genome.
Revised dating of monocot whole-genome duplication events
Syntenic analysis of the pineapple genome clarified the genome duplication history of the monocot lineage. We validated and refined the phylogenetic dating of three WGD events inferred by previous studies13,14,16 (Fig. 3a). Although the pan-cereal genome duplication event (ρ) has been relatively well studied14, the exact timing of more ancient WGD events (σ and τ) has remained controversial because of the high level of degeneration of phylogenetic signals and lack of proper outgroups for each duplication event13,17. Because of the pivotal phylogenetic position of pineapple at the base of the Poales, we circumscribed the placement of these ancient events on the basis of an integrated syntenic and phylogenetic approach16,18,19.

(a) Dating of WGD events on the monocot tree of life. Circles represent known WGD events identified previously. The pineapple genome sequence clarified the dating of the three WGD events in the grass lineage: ρ, σ and τ. Taxon labels are colored according to photosynthetic metabolism: C3, C4 or CAM. (b) Genomic alignment for Amborella trichopoda, A. comosus (pineapple) and Oryza sativa (rice), tracking gene positions through multiple species and copy numbers arising from multiple genome duplication events. Macrosynteny patterns show that a typical ancestral region in the basal angiosperm Amborella can be tracked to up to four regions in pineapple owing to the two genome duplication events, σ and τ, and to up to eight regions in rice. Gray wedges in the background highlight major syntenic blocks spanning more than 30 genes between the genomes (highlighted by one syntenic set shown in red). (c) Microcollinearity patterns between genomic regions from A. trichopoda, A. comosus (pineapple) and O. sativa (rice). Rectangles represent predicted gene models, with blue and green showing relative gene orientation. Gray wedges connect matching gene pairs, with one set highlighted in red.
Up to four pineapple regions could be aligned to each genomic region in the basal angiosperm Amborella trichopoda, which has not undergone WGD19 (Fig. 3b). Both the Amborella to pineapple comparison and the pineapple self-comparison supported two genome doublings in pineapple (Fig. 3c and Supplementary Fig. 6).
An extensive level of synteny conservation was found between the pineapple and grass genomes, with some large blocks containing over 300 gene pairs (Supplementary Table 12). Alignment of the rice genome to pineapple showed predominantly 4:2 patterns of syntenic depth (Supplementary Fig. 5). Microsynteny analyses (Fig. 3c and Supplementary Fig. 6) showed that each pineapple region had up to two highly syntenic rice regions, suggesting a shared duplication (σ) followed by one independent WGD (ρ) in rice. Higher degrees of microsynteny were observed between rice and pineapple orthologs than between rice and pineapple out-paralogs (Supplementary Fig. 7). In addition, the 4:2 syntenic relationship matched the expected patterns of fractionated gene content in rice following an independent WGD in its lineage. Retained duplicate genes in rice identified in syntenic blocks in the pineapple genome were sorted into gene families, and the timing of duplication events relative to speciation events was inferred through analyses of the gene family phylogenies (Supplementary Fig. 8). The gene family phylogenetic trees and all grass-pineapple syntenic block relationships suggest that the most recent WGD evident in the pineapple genome is the σ WGD, an event shared with all Poales members (Fig. 3).
Comparisons of the grass and pineapple genomes have refined previously published time brackets for both the pan-cereal ρ event and the shared σ event13,16. The ρ duplication is inferred to have occurred before radiation of the lineages leading to rice, wheat and maize but after divergence of the lineages leading to the grasses and pineapple within the Poales 95–115 million years ago6,7. The earlier σ WGD occurred after the lineage leading to the Poales diverged from the lineages leading to banana and the palms 100–120 million years ago18. Pineapple represents the closest sequenced lineage to the grasses lacking the pan-grass ρ WGD event, which makes it an excellent outgroup for comparative grass genomic studies (Fig. 3).
Comparative genomics across the monocots
Genome comparisons of pineapple with other non-cereal monocot clades unambiguously identify previously elusive lineage-specific WGD events. Synteny and phylogenomic analyses of banana, palm and grass genomes had indicated the existence of shared and lineage-specific WGD events6,16,18. However, precision in dating these events has been limited by sparse sampling of non-cereal monocot genomes.
Genome comparisons of non-cereal genomes to pineapple have much simpler synteny patterns than those using cereals, facilitating easier interpretation. Oil palm had one round of independent WGD, giving rise to mostly 2:2 syntenic depth in comparison with pineapple. Although banana had three independent WGD events in its lineage, giving rise to intricate patterns of mostly 8:2 syntenic depth in comparison to pineapple (Supplementary Fig. 8), our reconstructions of Zingiberales events were considerably less complicated than previous grass to banana comparisons13,18. Comparisons of pineapple to orchid in the Asparagales lineage were less definitive, owing to limited contiguity in the current orchid genome assembly20. Phylogenomic analyses including genes from the orchid, Phalaenopsis equestris, and gene sequences from transcriptome data for agave and garden asparagus, also in the Asparagales lineage, indicate that an earlier WGD event, τ, occurred in a common ancestor of Asparagales and commelinids, with the latter including the Poales, Arecales and Zingiberales (Fig. 2a).
Analysis of the synteny between duckweed (Spirodela polyrhiza) and pineapple together with phylogenomic analyses narrowed estimates of the timing of the τ WGD. The duckweed genome in the Alismatales lineage represents one of the earliest diverging monocots17. A duckweed to pineapple comparison showed 4:4 syntenic depth, consistent with two known Alismatales-specific WGD events17 and also confirming the independence of the two pineapple WGDs (σ and τ; Fig. 2). This inference was further supported in gene tree analyses (Supplementary Fig. 8). Consequently, we placed the τ WGD after the divergence of Alismatales and commelinids but before the divergence of Asparagales and commelinid (Fig. 2), implying a date between 135 and 110 million years ago6.
Study of lineage-specific gene family mobility in grasses
Arabidopsis thaliana genes have moved around its genome over recent evolutionary time21, inserting into new places probably by some form of translocation or recombination22. To distinguish between gene insertion in a query genome and gene deletion in an outgroup genome, at least two outgroups are required for a confident inference23. Although Brassicales gene movements have been studied23, the analysis of mobile genes in grasses has been hindered by the lack of closely related non-grass genomes, a need now fulfilled by pineapple.
Using pineapple and rice as outgroups, we tested whether the same gene families inferred to be mobile in A. thaliana (using a papaya outgroup) were also mobile in Sorghum bicolor (sorghum) (using a pineapple outgroup). The most mobile larger gene families in A. thaliana were F-box genes, MADS-box genes, defensins and NBS-LRR disease resistance genes23. We queried the A. thaliana genome using Arabidopsis lyrata, peach and grape as outgroups to determine the mobility of genes in A. thaliana. We used the same methods to query sorghum against rice and pineapple to determine gene mobility. Our test was whether the number of mobile genes in a family was significantly higher than the number of non-mobile, that is, syntenic, genes; if so, a gene family was determined to be mobile. We found that the gene families that tended to be mobile in A. thaliana also tended to be mobile in sorghum (Supplementary Table 13), with a few exceptions. The MADS-box genes, although mobile in the Arabidopsis lineage, were not mobile in the Sorghum lineage.
Plant MADS-box genes are classified as type I or type II based on their specified protein sequences. Type II proteins are composed of the most conserved MADS (M) domain for DNA binding, a keratin (K) domain for protein-protein interaction, an intervening domain located between the M and K domains, and a C-terminal domain that is mainly responsible for transcription activation24. The structure of type I proteins is simpler because these lack the K domain. Type I MADS-box genes experienced a faster pace of birth and death than type II genes owing in part to a higher frequency of gene duplications25. Type II MADS-box genes tended to be syntenous in both A. thaliana and sorghum in comparisons with the respective outgroups (Supplementary Table 13). Type I MADS-box genes tended to be mobile in sorghum, but there were fewer of these genes, suggesting either loss in the grasses or expansion in the Arabidopsis lineage.
The GDSL-like lipase/acylhydrolase gene family was not mobile in the Brassicales (Arabidopsis lineage) but was mobile in the Poales (Sorghum lineage) (Supplementary Table 13). The GDSL esterases/lipases are mainly involved in regulation of plant development, morphogenesis, synthesis of secondary metabolites and defense responses. This gene family has expanded in the monocot lineage in comparison to eudicots26. Much of GDSL family expansion was via gene mobility and likely has a role specific to grasses. These results demonstrate that pineapple is a useful and, at present, unique outgroup to the grass genomes for evolutionary inference.
Evolution of CAM photosynthesis
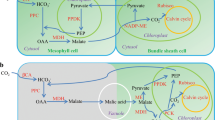
Drought is responsible for the majority of global crop loss, so understanding the mechanisms that plants have evolved to survive water stress is vital for engineering drought tolerance in crops. Plants use CAM to thrive in water-limited environments, potentially achieving greater net carbon dioxide uptake than their C3 and C4 counterparts27. By using an alternate carbon assimilation pathway that allows carbon dioxide to be fixed nocturnally by PEPC and stored transiently as malic acid in the vacuole (Fig. 4), CAM plants can keep their stomata closed during the daytime while the stored malic acid is decarboxylated and the carbon dioxide released is refixed through the Calvin-Benson cycle, greatly reducing water loss through evapotranspiration28. High water-use efficiency (WUE) and drought tolerance thus make CAM an attractive pathway by which to engineer crop plants for climate change29. The core CAM enzymatic steps are well characterized and are similar to those in C4 plants30, but the regulatory elements of CAM are largely unknown31. CAM photosynthesis is a recurrent adaptation, with numerous independent origins across 35 diverse families of vascular plants32.

(a) Pineapple leaf tissue used to survey the diurnal expression patterns of CAM pathway genes. The fully expanded D leaf of field-grown pineapple is shown. Green (photosynthetic) tissue at the leaf tip and white (non-photosynthetic) tissue at the leaf base were collected to distinguish CAM-related gene expression from non-CAM-related circadian oscillation. (b) Overview of the carboxylation (top) and decarboxylation (bottom) pathways of CAM. CAM enzymes are shown in blue. (c) Expression pattern and cis-regulatory elements of pineapple carbon fixation genes across the diurnal expression data. log2-transformed fragments mapped per kilobase of transcript length per million total mapped reads (FPKM) expression profiles are shown. Four known circadian clock–related binding motif sequences were searched in the 1-kb region upstream of each gene. (d) Summary table of the number of putative carbon fixation genes in pineapple, orchid, rice and maize. (e) Gene regulatory network of green leaf tissue. Only the largest module of the network was kept. Genes related to CAM and their interaction partners are highlighted in yellow.
We identified genes in the CAM pathway on the basis of homology to C3 and C4 pathway orthologs in maize, sorghum and rice. The pineapple genome contained 38 putative genes involved in the carbon fixation module of CAM, including for the key carbonic anhydrase (CA), phosphoenolpyruvate carboxylase (PEPC), phosphoenolpyruvate carboxylase kinase (PPCK), NAD- and NADP-linked malic enzymes (ME), malate dehydrogenase (MDH), phosphoenolpyruvate carboxykinase (PEPCK) and pyruvate, orthophosphate dikinase (PPDK) (Supplementary Table 14). Using PEPCK (rather than ME) as its principal decarboxylating enzyme during the daytime33, pineapple is distinctive among CAM plants in showing high activities of the alternative glycolytic enzyme inorganic pyrophosphate (PPi)-dependent phosphofructokinase (pyrophosphate:fructose-6-phosphate 1-phosphotransferase)34 and in possessing vacuolar transporters for soluble sugars35,36, which form the main pool of transitory carbohydrates supplying PEP for nocturnal carbon dioxide fixation and malic acid synthesis37,38 (Fig. 4b). Pineapple contains fewer of these core metabolic genes than other monocots (Fig. 4d).
To investigate the diel expression patterns of CAM pathway genes, we collected RNA-seq samples at 2-h intervals over a 24-h period from the photosynthetic (green tip) and non-photosynthetic (white base) leaf tissues of field-grown pineapple (Fig. 4a). On the basis of contrasting expression patterns in the two tissues, we were able to distinguish gene family members involved in carbon fixation from non-CAM-related members involved in other processes. Nine genes (encoding PEPC, PPCK, PEPCK, PPDK, three copies of CA and two MDH isoforms) had a diurnal expression pattern in the green leaf tissue with low or no expression in the white leaf tissue (Fig. 4c). CAM photosynthesis is divided into four temporal phases that are largely controlled by the circadian clock. Genes under circadian clock control were enriched in cis-regulatory elements, including the morning (CCACAC) and evening (AAAATATC) elements39. The diurnally expressed photosynthetic genes were enriched (P = 0.002) in known circadian clock cis-regulatory elements in comparison to the non-photosynthetic gene copies (Fig. 4c), suggesting that the carbon fixation pathway in pineapple is regulated by circadian clock components through cis-regulatory elements.
CA, by catalyzing the conversion of carbon dioxide into bicarbonate, is responsible for the first step in carbon dioxide fixation in C4 and CAM photosynthesis. Of the three CA families (α, β and γ) in pineapple, only βCA showed a nighttime and early-morning expression profile in green leaf tissue, as the major protein for carbon fixation. The promoter regions of all three βCA genes contained a CCA1-binding site that can bind to both circadian core oscillators, CIRCADIAN CLOCK ASSOCIATED 1 (CCA1) and LATE ELONGATED HYPOCOTYL (LHY). Of all the βCA genes in orchid, rice, maize and sorghum, only one gene in sorghum (Sobic.003G234500) contained a CCA1-binding site (Supplementary Table 15) in its promoter, but this gene has no known photosynthetic function40. These findings indicate that the βCA genes in pineapple are temporally regulated by the circadian clock to synchronize the expression of the enzyme with stomatal opening at night for maximum carbon dioxide fixation in pineapple.
We identified putative regulators of CAM by surveying gene interaction networks. CAM pathway genes were highly connected in the gene interaction network (Fig. 4e and Supplementary Fig. 9). CAM-related genes showed dramatic differences in their regulatory patterns based on their gene interactions (Supplementary Table 16). From the network, the increase in expression of βCA in green leaf cells was mainly contributed by the appearance of about 243 potential activators and also by the disappearance of two potential repressors. PPCK showed similar regulatory patterns, although the number of repression controllers identified was much higher than for βCA. In contrast, increased expression of PEPC was mainly related to the release from repression by potential repression controllers (35) and relatively less to the appearance of potential activators (1). Three isoforms of MDH (Aco006122.1, Aco010232.1 and Aco004996.1) showed similar regulatory patterns. Different enzymes involved in CAM photosynthesis used different regulatory mechanisms, as reflected in both their interaction and regulatory patterns (Supplementary Table 16). This result provides strong molecular evidence as to how the regulatory mechanisms controlling the expression of CAM-related genes could have evolved independently so often: the capacity was always present but was repressed at the trans-acting, cell-specific and individual gene level.
Discussion
Pineapple is self-incompatible, and all pre-Columbian and most post-Columbian varieties were selected from variants with somatic mutations, in comparison to the extensive breeding history of most crops. Sequencing the genomes of two leading commercial varieties of pineapple, F153 and MD2, identified heterozygosity within each genome at a rate of about 2%, which is much higher than for seed-propagated crops but similar to the rate for clonally propagated crops. Self-incompatibility combined with clonal propagation contributes to and maintains the high level of heterozygosity in pineapple. The inbreeding depression from a self-compatible pineapple mutant was so severe that most seedlings died after two generations of self41. The high frequency of nonsynonymous SNPs in F153 and MD2 may be the cause of such unusually severe inbreeding depression (Supplementary Table 11). The abundance of retrotransposons, such as the Pusofa family (28% of LTR retrotransposons and 15% of the pineapple genome), might have contributed to genome instability in pineapple. Any search for somatic mutations caused by LTR retrotransposons, including those potentially associated with pineapple cultivar improvement, would be best focused on the families that are most highly expressed.
The modified carbon assimilation pathways of CAM and C4 photosynthesis result in higher WUE, a highly desirable trait given the need to double food production by 2050 under a changing climate. CAM and C4 photosynthesis use many of the same enzymes for concentrating carbon dioxide but differ in the spatial (C4) versus temporal (CAM) separation of carbon fixation. Understanding the evolution of CAM and C4 photosynthesis may expedite projects to convert C3 into C4 rice42 and C3 into CAM poplar29. CAM plants have higher WUE than C3 and C4 plants and may be better suited for engineering crop drought tolerance. All plants contain the necessary genes for CAM photosynthesis, and the evolution of CAM simply requires the rerouting of preexisting pathways. CAM pathway genes are enriched in circadian clock–associated cis-regulatory elements, providing the first cis-regulatory link, to our knowledge, between CAM and the circadian clock. Consistent with this link, βCA genes in pineapple contain a CCA1-binding site, which is absent in the corresponding genes from C3 and C4 monocots. Regulation of CAM is complex, and CAM-related enzymes use different regulatory mechanisms, which explains how CAM evolved independently many times during evolution: in non-CAM plants, the gene content encoding the enzymatic machinery is present, but diel expression patterns are likely silenced or not activated sufficiently at the cis-acting, cell-specific individual gene level. This work provides the first detailed analysis of the expression and regulation patterns of genes associated with CAM and could ultimately be used to engineer better WUE and drought tolerance in crop plants.
Methods
Nuclear DNA preparation.
Fresh leaf tissues from pineapple varieties F153 and MD2 and wild species A. bracteatus accession CB5 were collected, and nuclear DNA was isolated following the procedure described previously43.
Flow cytometry analysis of CB5 genome size.
The procedure used to analyze the nuclear DNA content of A. bracteatus accession CB5 was described previously44. The nuclear DNA content estimated by flow cytometry reflects the diploid, or 2C, genome size, but for sequencing purposes the haploid, or 1C, value is used and converted to the number of nucleotides using the equation 1 pg = 978 Mb (ref. 45). The estimated genome size of A. bracteatus CB5 is 1C = 592 Mb (2C = 1.21 pg), close to the previously reported flow cytometry–based value of 1C = 526 Mb (for A. comosus)8.
Genome sequencing.
Raw sequences for F153 were generated primarily using Illumina sequencing, following a standard protocol, with the HiSeq 2000 platform. Four paired-end libraries were created with inserts of 180 bp, 500 bp, 3 kb and 8 kb, generating 33×, 150×, 41.8× and 25.5× coverage, respectively. A paired-end 20-kb insert library was generated for scaffolding using the Roche/454 circularization protocol with sequencing carried out on the 454 FLX+ platform. We generated 1.2 Gb of sequence (∼2× coverage) using Moleculo chemistry, with average read lengths of 5 kb, and 2.8 Gb (∼5× coverage) using PacBio P6-C4 chemistry. A total of 9,400 BACs were sequenced using a random pooling strategy with 48 BACs per pool. Each pool was sequenced to produce Illumina HiSeq 2500 paired-end 2 × 150 bp sequencing.
Raw sequences for MD2 and CB5 were generated using Illumina sequencing, following standard protocols, with the HiSeq 2000 platform (for 180-bp and 500-bp insert libraries) and the HiSeq 2500 platform (for 3-kb and 8-kb insert libraries). The four paired-end libraries with inserts of 180 bp, 500 bp, 3 kb and 8 kb were sequenced to produce 33×, 50×, 35× and 12× coverage, respectively.
Genome assembly.
Genome assembly overview. The pineapple reference genome assembly incorporated data from a mixture of sequencing technologies, including whole-genome shotgun sequencing with Illumina, 454, PacBio and Moleculo technologies, as well as BAC pools sequenced with Illumina sequencing. The assembly underwent three major rounds of improvement by applying the different technologies (Supplementary Fig. 10 and Supplementary Table 17). The original F153 pineapple assembly was based on ALLPATHS-LG using Illumina whole-genome sequencing and 454 sequencing data (v1 assembly), was subsequently improved by incorporating the assembled BAC contigs (v2 assembly) and was finally improved by incorporating the PacBio and Moleculo data (v3 assembly). See the Supplementary Note for a detailed description of the assembly methods. The k-mer coverage of the F153 fragment library is shown in Supplementary Figure 11.
Genetic maps and chromosomal assembly. Ninety-three F1 individuals from a CB5 × F153 cross were sequenced to an average depth of 10× by whole-genome sequencing. The raw reads were mapped onto the genome assembly using Bowtie2. Segregating polymorphic SNPs were called using the Genome Analysis Toolkit (GATK). Only SNPs that were homozygous for the reference genotype in one parent and heterozygous in the other parent were used. SNPs were assigned to either an F153 or CB5 map depending on which parental genotype was heterozygous. The SNPs segregating for each parent were further divided into two pools using genotyping calls for a single F1 individual that was sequenced to higher coverage (50×), to group the SNPs according to the phase of the SNP. Using the deep sequenced F1 individual allowed all of the SNPs that contained the non-reference SNP on the same chromosome to be grouped together.
Individual SNPs had a high rate of missing data, and many heterozygous SNPs also could be scored incorrectly because of limited depth of sequencing coverage; however, by looking at several adjacent SNPs, a consensus SNP genotype for each scaffold piece could be determined. Adjacent SNPs mapping to 100-kb bins on each scaffold were combined into a consensus genotype for each individual. Scaffold segments 100 kb long with ≥15 SNPs had consensus genotypes determined. These consensus genotypes were ordered into genetic maps. The F153 map consisted of 3,125 scaffold segments containing 928,659 segregating SNPs and was assembled into the 25 linkage groups corresponding to the haploid chromosome number of pineapple. A consensus order for the scaffolds in chromosomal pseudomolecules was determined, with breakpoints (represented as intervals) approximated using the information from individual SNPs. Chimeric scaffolds were split at the largest gap in the inferred breakpoints. When no gaps could be found within the inferred breakpoints, the closest gap was identified instead.
Genome annotation.
We used MAKER to generate a first-pass gene annotation. MAKER is a computational pipeline for genome annotation that can integrate multiple tiers of coding evidence, including ab initio gene predictions, transcript evidence and protein evidence9. Ab initio gene models were evaluated against matching transcript and protein evidence to select the model for each gene that was most consistent on the basis of an AED metric9.
Input data for MAKER were prepared as follows. First, ab initio gene predictors, including SNAP46, GENEMARK47 and AUGUSTUS48, were each trained with nearly 'full-length' pineapple transcripts. The pineapple transcripts were constructed using PASA49 and were evaluated against UniProt plant proteins to identify the set of nearly full-length candidates that covered at least 95% of any target protein. The pineapple transcripts were sampled from major tissues, including flower, fruit, leaf and root. Comprehensive transcriptome assembly was carried out using both de novo Trinity and reference-guided Trinity50, with the results combined and used together as mRNA evidence for MAKER. Plant proteins were downloaded from UniProt (last accessed on 21 September 2014) and used as plant-specific evidence for MAKER.
MAKER was run on the pineapple v3 scaffold assembly with the above evidence twice, once with and once without masking with a pineapple-specific repeat library, for the purpose of comparison. Putative proteins over 30 amino acids in length were kept. Additionally, we set up a JBrowse instance51 during structural annotation to visualize the gene models along with the aligned annotation evidence (see URLs).
For the final gene set, a MAKER run without repeat masking was selected, followed by extensive filtering of TE-related genes. The original MAKER run produced 31,893 genes, from which we removed 4,850 TE-related genes and 19 that were broken during linkage group construction. For the 27,024 remaining genes, we obtained 24,063 (89.0%) complete gene models, with 11% categorized as partial (Supplementary Table 5).
For functional annotation, we inferred the human readable protein description for each predicted pineapple protein using AHRD (see URLs), on the basis of names from three protein databases: SWISS-PROT, TrEMBL and TAIR10. The InterPro domains, Gene Ontology (GO) terms and KEGG pathway information associated with each protein were computed using InterProScan52.
Synteny analysis.
We performed synteny searches to compare the pineapple genome structure with that of other related plant genomes. To call syntenic blocks, we performed all-against-all LAST53 and chained the LAST hits with a distance cutoff of 20 genes, also requiring at least four gene pairs per syntenic block using QUOTA-ALIGN54. Synteny was searched for by performing comparisons of the pineapple genome with other selected genomes (Amborella19, banana18, date palm55, duckweed17, grape15, oil palm56, orchid20, rice57 and sorghum58). The resulting dot plots were inspected to confirm the paleoploidy level of pineapple in relation to the other genomes by counting the syntenic depth at each genomic region.
Phylogenetic dating of whole-genome duplication events.
We used an integrated pipeline of spatial and temporal evidence to circumscribe WGD events16. Briefly, we started with homologous gene detection and then used two parallel methodologies to provide separate lines of evidence to place the events on the tree of life. The 'spatial' signal relies on extracted syntenic blocks (macrosynteny) and gene order alignments (microsynteny). Analysis of synteny patterns was conducted using CoGe comparative genomics tools (see URLs)59. The inferred syntenic depth ratio of syntenic blocks allows determination of the paleopolyploidy level13,17,54. The 'temporal' signal relies on the construction of gene families using sequence similarity. A clustering of coalescence among gene duplicates was used to infer likely genome-wide events16. The latter method is supplemented by using only the syntenic gene pairs in the structural data set to offer higher precision during inference.
Ancestral chromosome reconstruction.
We identified syntenic blocks for duckweed and pineapple using the SynMap procedure (default settings) on the CoGe platform59. For the many duckweed regions showing fourfold matching blocks in pineapple, we identified the start and end points of the four matching regions in pineapple. We then examined all duplicates found in pineapple versus pineapple SynMap analysis, without restricting ourselves to duplicates satisfying the strict syntenic block conditions. From these data, we extracted all sets of gene quadruples consisting of two pairs of close matches (generally >73% identical) with the four interpair matches scoring lower (<74% identical). Almost all of these gene quadruples fell into the pattern consisting of seven sets of four chromosomes (or large chromosomal segments).
Gene family analysis.
Gene models were sorted into gene families circumscribed from 22 plant genomes using BLASTX (best BLAST hit; e-value cutoff of 1 × 10−10) (ref. 19). The taxa used to estimate gene family circumscriptions (orthogroups) are represented in Figure 3. Putative paralog pairs from both the σ (2,750 pairs) and τ (1,292 pairs) WGD events were used to collapse gene families that were improperly split. The peptide sequences for each corrected gene family were aligned using default settings for peptides in PASTA (v1.6.4)60. Peptide alignments were then converted to the corresponding codon alignments using pal2nal61. Gene trees for the nucleotide alignments were estimated using RAxML v7.3.0 (ref. 62), rooting to Amborella, Vitis vinifera or A. thaliana with a generalized time reversible (GTR) + Γ model over 500 bootstraps.
The relationships among species, as denoted in Figure 3, were used to generate hypotheses to test for the presence of a polyploid event along the phylogeny. For each node along the backbone of the monocot clade, a hypothesis was generated that consisted of taxa descendent of the node, which would share the polyploid event, and taxa in the sister group to the node, which would not share the polyploid event. Gene trees were queried using putative paralogs from both the σ and τ duplications. For each pair of genes, the last common ancestor (LCA) node was identified, and the taxa descendent of the node and those found in the sister group were compared to the expectation with the generated hypotheses. If a hypothesis matched what was found for the node, then the bootstrap value of that node was used to count support for the event. For each node, we report LCA nodes that had bootstrap values of 80 or greater and those that had bootstrap values of less than 80 but greater than 50 (Supplementary Fig. 8).
We identified 697 LCA nodes for putative σ paralogs across 986 gene trees that had bootstrap values of 50 or greater. Of these nodes, 359 were found to represent well-defined hypotheses with sampling of taxa from both the descendent and sister groups. We note that the reduction in LCA nodes is due to the conservative nature of the hypothesis-testing algorithm, which requires sampling of representative taxa for both descendent and sister groups. Of these LCA nodes, 258 (159 with bootstrap values ≥80 and 99 with bootstrap values ≥50) placed the σ WGD before the divergence of pineapple from the rest of the Poales, which represents the overwhelming majority (71.9%) of the LCA nodes (Supplementary Fig. 8). The next best-supported timing for the σ WGD was on the lineage leading to the commelinids, with estimated duplications in just 42 gene trees (11.7%).
A total of 192 LCA nodes for putative τ paralogs were found in 361 gene trees that had bootstrap values of 50 or greater. We identified 83 LCA nodes that represented well-defined hypotheses. The predominantly supported placement of the τ WGD was shown to be after the divergence of the Spirodela (Alismatales) lineage from the rest of the monocots and was shared by Asparagales, commelinids and Poales with a total of 45 LCA nodes (28 with bootstrap values ≥80 and 17 with bootstrap values ≥50), or 54.2% of the LCA nodes (Supplementary Fig. 10). The second best-supported timing for the τ WGD was on the lineage leading to the common ancestor of monocots and eudocots, with estimated duplications in just 23 gene trees (27.7%).
In summary, phylogenomic analyses support the results of the comparative synteny analyses, placing the σ WGD on the lineage leading to the Poales crown group and the τ WGD between the divergences of Alismatales and Asparagales from the lineage leading to the commelinids.
Plant materials for CAM photosynthesis analysis.
Leaves from A. comosus cultivar MD2 were collected from the field at Dole Plantation (Wahiawa, Hawaii) for RNA extraction and physiology studies. The D leaf (the youngest physiologically mature leaf, fourth from the apex) was collected from five individual plants as one replicate, and three biological replicates were collected every hour between 10 a.m. on 24 October 2013 and 9 a.m. on 25 October 2013, with the exception of two time points at 2 p.m. on 24 October and 1 a.m. on 25 October. The sunset time on 24 October was 6 p.m. HST, and the sunrise time on 25 October was 6:32 a.m. HST. For the time-course experiment, two regions of the D leaf were used for transcriptomic studies: the white base and green tip. Thirteen time points (6 p.m., 8 p.m., 10 p.m., midnight, 2 a.m., 4 a.m., 6 a.m., 8 a.m., 10 a.m., noon, 1 p.m., 3 p.m. and 4 p.m.) were chosen for RNA-seq library construction. All leaf segments were frozen in liquid nitrogen immediately after they were collected in the field and were stored at −80 °C until ground into powder in liquid nitrogen.
RNA extraction and library construction.
Total RNA was extracted from ground leaf using the Qiagen RNeasy Plant Mini kit (74904), following the manufacturer's protocol. DNA was then removed using the DNA-free DNA Removal kit (Life Technologies, AM1906M). A single indexed RNA-seq library was constructed using the Illumina TruSeq Stranded RNA Sample Preparation kit (RS-122-2001) and then sequenced using the Illumina HiSeq 2500 platform in paired-end 100-nt mode. Three biological replicates were studied for each time point.
Sequencing read processing and estimation of gene expression.
The trimmed paired-end reads for each sample were aligned to repeat-masked pineapple assembly v3 using TopHat v2.0.9 with default settings63. The normalized FPKM value for each sample was estimated by Cufflinks v2.2.1, followed by Cuffnorm v2.2.1, using the default setting with pineapple gene model annotation provided (-g option).
Gene network construction using diurnal expression profiles.
Gene regulatory networks for white leaf base and green leaf tip tissues, designated as the 'base network' and 'tip network', respectively, were constructed using the PCA-CMI algorithm on the basis of gene expression data, and 15,483 genes (201,537 interactions) and 13,543 genes (188,391 interactions) were included in the base and tip networks, respectively. Isolated interactions were trimmed, and only the largest module was retained in which 11,079 genes (183,168 interactions, or 90.9% of the 201,537 interactions) and 7,506 genes (177,308 interactions, or 94.1% of the 188,391 interactions) were present in the base and tip networks, respectively. Topology analysis was then conducted for the two networks using the MCL algorithm.
Cis element annotation and enrichment analysis.
Five known circadian clock–related motif sequences were searched for in the 1-kb promoter sequences upstream of pineapple genes involved in carbon fixation. These five motifs included the morning element (CCACAC), the evening element (AAAATATCT), the CCA1-binding site (AAAAATCT), the G-box element (G-box; CACGTG) and the TCP15-binding motif (TCP15; NGGNCCCAC)39,64,65,66. Enrichment of cis-regulatory elements in the promoters of photosynthetic genes in comparison to non-photosynthetic genes was tested using Fisher's exact test. The βCA genes from the orchid, rice, maize and sorghum genomes were annotated on the basis of sequence homology using BLASTP, and their promoter regions were searched for cis-regulatory element motif sequences. The sequences of all genomes used in the comparisons were downloaded from Phytozome v10.1, except for that of orchid, which was downloaded separately (see URLs).
URLs.
JBrowse instance to visualize the gene models and aligned annotation evidence, http://peach.fafu.edu.cn/html/jbrowse/JBrowse-1.11.5/?data=Pineapple_genome_project; Phytozome v10.1, http://phytozome.jgi.doe.gov/; orchid genome download, ftp://ftp.genomics.org.cn/from_BGISZ/20130120/; Automated Assignment of Human Readable Descriptions (AHRD), https://github.com/groupschoof/AHRD/; CoGe, http://genomevolution.org/.
Accession codes.
The pineapple genome sequence, annotation and RNA-seq data have been deposited at the iPlant CoGe database and can be downloaded from https://genomevolution.org/CoGe/NotebookView.pl?nid=937. Pineapple tissue RNA-seq data and pineapple time-course RNA-seq data are available from https://de.iplantcollaborative.org/de/?type=data&folder=/iplant/home/cmwai/coge_data/Pineapple_tissue_RNAseq. Pineapple genome resources are also available in Phytozome.
References
Clement, C.R., de Cristo-Araújo, M., Coppens D'Eeckenbrugge, G., Alves Pereira, A. & Picanço-Rodrigues, D. Origin and domestication of native Amazonian crops. Diversity 2, 72–106 (2010).
Bartholomew, D.P., Paull, R.E. & Rohrbach, K.G. The Pineapple: Botany, Production, and Uses (CABI, 2002).
Beauman, F. The Pineapple: King of Fruits (Random House, 2006).
Yang, X. et al. A roadmap for research on crassulacean acid metabolism (CAM) to enhance. New Phytol. 207, 491–504 (2015).
Brewbaker, J.L. & Gorrez, D.D. Genetics of self-incompatibility in the monocot genera, Ananas (pineapple) and Gasteria. Am. J. Bot. 54, 611–616 (1967).
Magallón, S., Gómez-Acevedo, S., Sánchez-Reyes, L.L. & Hernández-Hernández, T. A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity. New Phytol. 207, 437–453 (2015).
Givnish, T.J. et al. Adaptive radiation, correlated and contingent evolution, and net species diversification in Bromeliaceae. Mol. Phylogenet. Evol. 71, 55–78 (2014).
Arumuganathan, K. & Earle, E. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9, 208–218 (1991).
Cantarel, B.L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
McCarthy, E.M. & McDonald, J.F. LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics 19, 362–367 (2003).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Meyers, B.C., Tingey, S.V. & Morgante, M. Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res. 11, 1660–1676 (2001).
Tang, H., Bowers, J.E., Wang, X. & Paterson, A.H. Angiosperm genome comparisons reveal early polyploidy in the monocot lineage. Proc. Natl. Acad. Sci. USA 107, 472–477 (2010).
Paterson, A.H., Bowers, J.E. & Chapman, B.A. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. USA 101, 9903–9908 (2004).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Jiao, Y., Li, J., Tang, H. & Paterson, A.H. Integrated syntenic and phylogenomic analyses reveal an ancient genome duplication in monocots. Plant Cell Online 26, 2792–2802 (2014).
Wang, W. et al. The Spirodela polyrhiza genome reveals insights into its neotenous reduction fast growth and aquatic lifestyle. Nat. Commun. 5, 3311 (2014).
D'Hont, A. et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213–217 (2012).
Amborella Genome Project. The Amborella genome and the evolution of flowering plants. Science 342, 1241089 (2013).
Cai, J. et al. The genome sequence of the orchid Phalaenopsis equestris. Nat. Genet. 47, 65–72 (2015).
Freeling, M. et al. Many or most genes in Arabidopsis transposed after the origin of the order Brassicales. Genome Res. 18, 1924–1937 (2008).
Woodhouse, M.R. et al. Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homeologs. PLoS Biol. 8, e1000409 (2010).
Woodhouse, M.R., Tang, H. & Freeling, M. Different gene families in Arabidopsis thaliana transposed in different epochs and at different frequencies throughout the rosids. Plant Cell Online 23, 4241–4253 (2011).
Kramer, E.M., Dorit, R.L. & Irish, V.F. Molecular evolution of genes controlling petal and stamen development: duplication and divergence within the APETALA3 and PISTILLATA MADS-box gene lineages. Genetics 149, 765–783 (1998).
Nam, J. et al. Type I MADS-box genes have experienced faster birth-and-death evolution than type II MADS-box genes in angiosperms. Proc. Natl. Acad. Sci. USA 101, 1910–1915 (2004).
Chepyshko, H., Lai, C.-P., Huang, L.-M., Liu, J.-H. & Shaw, J.-F. Multifunctionality and diversity of GDSL esterase/lipase gene family in rice (Oryza sativa L. japonica) genome: new insights from bioinformatics analysis. BMC Genomics 13, 309 (2012).
Nobel, P.S. Achievable productivities of certain CAM plants: basis for high values compared with C3 and C4 plants. New Phytol. 119, 183–205 (1991).
Osmond, C. Crassulacean acid metabolism: a curiosity in context. Annu. Rev. Plant Physiol. 29, 379–414 (1978).
Borland, A.M. et al. Engineering crassulacean acid metabolism to improve water-use efficiency. Trends Plant Sci. 19, 327–338 (2014).
Christin, P.-A. et al. Shared origins of a key enzyme during the evolution of C4 and CAM metabolism. J. Exp. Bot. 65, 3609–3621 (2014).
Edwards, E.J. & Ogburn, R.M. Angiosperm responses to a low-CO2 world: CAM and C4 photosynthesis as parallel evolutionary trajectories. Int. J. Plant Sci. 173, 724–733 (2012).
Silvera, K. et al. Evolution along the crassulacean acid metabolism continuum. Funct. Plant Biol. 37, 995–1010 (2010).
Dittrich, P., Campbell, W.H. & Black, C. Phosphoenolpyruvate carboxykinase in plants exhibiting crassulacean acid metabolism. Plant Physiol. 52, 357–361 (1973).
Carnal, N.W. & Black, C.C. Phosphofructokinase activities in photosynthetic organisms: the occurrence of pyrophosphate-dependent 6-phosphofructokinase in plants and algae. Plant Physiol. 71, 150–155 (1983).
McRae, S.R., Christopher, J.T., Smith, J.A.C. & Holtum, J.A. Sucrose transport across the vacuolar membrane of Ananas comosus. Funct. Plant Biol. 29, 717–724 (2002).
Antony, E. et al. Cloning, localization and expression analysis of vacuolar sugar transporters in the CAM plant Ananas comosus (pineapple). J. Exp. Bot. 59, 1895–1908 (2008).
Holtum, J.A., Smith, J.A.C. & Neuhaus, H.E. Intracellular transport and pathways of carbon flow in plants with crassulacean acid metabolism. Funct. Plant Biol. 32, 429–449 (2005).
Kenyon, W.H., Severson, R.F. & Black, C.C. Maintenance carbon cycle in crassulacean acid metabolism plant leaves: source and compartmentation of carbon for nocturnal malate synthesis. Plant Physiol. 77, 183–189 (1985).
Michael, T.P. et al. Network discovery pipeline elucidates conserved time-of-day–specific cis-regulatory modules. PLoS Genet. 4, e14 (2008).
Wang, X. et al. Comparative genomic analysis of C4 photosynthetic pathway evolution in grasses. Genome Biol. 10, R68 (2009).
Collins, J.L. The Pineapple: Botany, Cultivation and Utilization (Interscience Publishers, 1960).
von Caemmerer, S., Quick, W.P. & Furbank, R.T. The development of C4 rice: current progress and future challenges. Science 336, 1671–1672 (2012).
Ming, R. et al. Genome of the long-living sacred lotus (Nelumbo nucifera Gaertn.). Genome Biol. 14, R41 (2013).
VanBuren, R. et al. Longli is not a hybrid of Longan and Lychee as revealed by genome size analysis and trichome morphology. Trop. Plant Biol. 4, 228–236 (2011).
Dolezel, J., Bartos, J., Voglmayr, H. & Greilhuber, J. Nuclear DNA content and genome size of trout and human. Cytometry A 51, 127–128, author reply 129 (2003).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y.O. & Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506 (2005).
Stanke, M., Schoffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62 (2006).
Haas, B.J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Grabherr, M.G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Skinner, M.E., Uzilov, A.V., Stein, L.D., Mungall, C.J. & Holmes, I.H. JBrowse: a next-generation genome browser. Genome Res. 19, 1630–1638 (2009).
Zdobnov, E.M. & Apweiler, R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Kielbasa, S.M., Wan, R., Sato, K., Horton, P. & Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493 (2011).
Tang, H. et al. Screening synteny blocks in pairwise genome comparisons through integer programming. BMC Bioinformatics 12, 102 (2011).
Al-Dous, E.K. et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat. Biotechnol. 29, 521–527 (2011).
Singh, R. et al. Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature 500, 335–339 (2013).
International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 436, 793–800 (2005).
Paterson, A.H. et al. The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556 (2009).
Lyons, E. et al. Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiol. 148, 1772–1781 (2008).
Mirarab, S., Nguyen, N. & Warnow, T. in Research in Computational Molecular Biology 177–191 (Springer, 2015).
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612 (2006).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood–based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Hudson, M.E. & Quail, P.H. Identification of promoter motifs involved in the network of phytochrome A–regulated gene expression by combined analysis of genomic sequence and microarray data. Plant Physiol. 133, 1605–1616 (2003).
Franco-Zorrilla, J.M. et al. DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc. Natl. Acad. Sci. USA 111, 2367–2372 (2014).
Michael, T.P. & McClung, C.R. Phase-specific circadian clock regulatory elements in Arabidopsis. Plant Physiol. 130, 627–638 (2002).
Acknowledgements
We thank R. Kai and C. Mayo Riley for maintaining the pineapple plants and the collection of leaf tissues; M. Conway at Dole Plantation for assistance in time-course leaf sample collection; G. Sanewski for providing the MD2 pedigree; and M. Cushman for providing clarifying comments on the manuscript. This project is supported by funding from the Fujian Agriculture and Forestry University to R.M.; a USDA T-START grant through the University of Hawaii to Q.Y., R.M., P.H.M. and R.E.P.; and funding from the University of Illinois at Urbana-Champaign to R.M. H.T. is supported by the 100 Talent Plan award from the Fujian provincial government. Analyses of the pineapple genome are supported by the following funding sources: US National Science Foundation (NSF) Plant Genome Program grant 0922545 to R.M., P.H.M. and Q.Y. and NSF grant DBI-1401572 to R.V.; NSF grant IOS-1444567 to J.H.L.-M.; and US National Institutes of Health award R01-HG006677 and US NSF awards DBI-1350041 and DBI-1265383 to M.C.S. W.C.Y., H.-B.G., H.G., G.A.T., X.Y. and J.C.C. acknowledge support from the US Department of Energy, Office of Science, Genomic Science Program, under award DE-SC0008834.
Author information
Authors and Affiliations
Contributions
R.M., Q.Y., R.E.P., P.H.M., R.V. and C.M.W. conceived the experiments. L.H., L.Z., W.M., A.G.H. and C.L.W. sequenced the genomes. M.C.S., E.B., H.L., J.G. and F.J.S. assembled the genome. H.T., C.M. and Z.Y. annotated the genome. R.M., R.V., C.M.W., J.E.B., E.L., M.-L.W., J.C., Jisen Zhang, Z. Lin, Jian Zhang, H.W., H.Z., W.C.Y., H.D.P., C.Z., M.W., P.P.E., R.G., H.-B.G., H.G., G.Z., R. Singh, A.S., X.M., Y.Z., A.H., M.R.M., Z. Liao, J.F., J.L., X. Zhang, Q.Z., W.H., Y.Q., K.W., L.-Y.C., N.S., Y.-R.L., L.-Y.L., V.B., G.A.T., K.H., F.Z., R. Sunkar, J.H.L.-M., T.M., J.L.B., M.F., D.S., A.H.P., X. Zhu, X.Y., J.A.C.S., J.C.C., R.E.P. and Q.Y. analyzed the genomes. R.M., R.V., C.M.W., H.T., M.C.S., D.S., M.W., M.F., X. Zhu, X.Y., J.A.C.S. and J.C.C. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Correlation between family copy number and expression level of LTR elements.
The data indicate that high expression levels of LTR elements are correlated with a relatively low copy number of their family.
Supplementary Figure 2 Expression of intact LTR retrotransposons in nine pineapple tissue samples.
This heat map shows the number of RNA-seq reads mapped to the top 40 most highly expressed LTR retrotransposon families. Family names are shown as row labels, and tissue names are given as column labels. From top to bottom, the rows are sorted by total counts of mapped reads in families.
Supplementary Figure 3 Expression of subfamilies of LTR retrotransposons in nine pineapple tissue samples.
The heat map shows the number of RNA-seq reads mapped to the top ten most highly expressed LTR retrotransposon families. Each row represents a subfamily, and each column represents a tissue. The numbers following family names give subfamily IDs. From top to bottom, the rows are sorted by total counts of mapped reads in families. Within each family, the rows are further sorted by total counts of mapped reads in subfamilies.
Supplementary Figure 4 Synonymous substitutions per site (Ks) values between inferred whole-genome duplicates in pineapple.
(a) Syntenic dot plot in pineapple versus pineapple comparison, with Ks values color coded; only the gene pairs with a Ks value between 0 and 2 are plotted. (b) Histogram of Ks values for pineapple-rice orthologs, rice whole-genome duplicates and pineapple whole-genome duplicates.
Supplementary Figure 5 Pairwise genome comparisons between pineapple and ten related plant species.
Pairwise comparisons (dot plots) between pineapple (y axis) and a total of ten related plant genomes (x axis), including (a–j) Amborella, banana, date palm, duckweed, grape, oil palm, orchid, pineapple (i.e., self-comparison), rice and sorghum. For clarity, only gene pairs within synteny blocks of at least size 4 are shown.
Supplementary Figure 6 Microsynteny fractionation for 4:1 pineapple to Amborella, providing evidence that pineapple has undergone two WGDs in its lineage since their divergence.
Five exemplar regions are shown. Each panel contains multiple parallel tracks representing syntenic regions in rice and pineapple. Connecting lines show sequence similarities between the regions. CoGe, https://genomevolution.org/r/e426, https://genomevolution.org/r/e428, https://genomevolution.org/r/e427, https://genomevolution.org/r/e448 and https://genomevolution.org/r/e446.
Supplementary Figure 7 Microsynteny fractionation for 1:2 pineapple to rice, providing evidence that rice has undergone one WGD in its lineage (ρ) since its divergence from pineapple.
Three exemplar regions are shown. Each panel contains multiple parallel tracks representing syntenic regions in Amborella and pineapple. Connecting lines show sequence similarities between the regions. CoGe, https://genomevolution.org/r/e3kg, https://genomevolution.org/r/e3kw, https://genomevolution.org/r/e3k4.
Supplementary Figure 8 Dating of whole-genome duplication (WGD) events on the flowering plant tree.
Letters represent previously identified WGDs. Estimated gene family phylogenies including genes on syntenic blocks corresponding to the σ and τ WGDs were queried to identify the timing of implied gene duplications relative to speciation events. The numbers below each lineage in the monocot clade represent gene duplication events corresponding to the σ (green) and t (purple) synteny blocks. Trees with inferred duplication events supported by greater than 80% (left) and between 80% and 50% (right) bootstrap support values are shown for each node. Taxon names are color coded as in Figure 2.
Supplementary Figure 9 Property of leaf green tip gene interaction network.
(a,c,d) Distributions of the node degree, diameter and betweenness attribute. (b) Relationship between node degree and frequency in logarithmic coordinates.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–11, Supplementary Tables 1–4 and 6–17, and Supplementary Note. (PDF 2174 kb)
Supplementary Table 5
Summary of gene model annotations. (XLSX 2068 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article

Cite this article
Ming, R., VanBuren, R., Wai, C. et al. The pineapple genome and the evolution of CAM photosynthesis. Nat Genet 47, 1435–1442 (2015). https://doi.org/10.1038/ng.3435
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3435
This article is cited by
-
TIP aquaporins in Cyperus esculentus: genome-wide identification, expression profiles, subcellular localizations, and interaction patterns
BMC Plant Biology (2024)
-
Genomic and transcriptomic analysis of sacred fig (Ficus religiosa)
BMC Genomics (2023)
-
Characterization and expression profiles of the B-box gene family during plant growth and under low-nitrogen stress in Saccharum
BMC Genomics (2023)
-
Comparative transcriptome analysis reveals candidate genes for cold stress response and early flowering in pineapple
Scientific Reports (2023)
-
Selection signatures and population dynamics of transposable elements in lima bean
Communications Biology (2023)
