
Abstract
The unique anatomical features of turtles have raised unanswered questions about the origin of their unique body plan. We generated and analyzed draft genomes of the soft-shell turtle (Pelodiscus sinensis) and the green sea turtle (Chelonia mydas); our results indicated the close relationship of the turtles to the bird-crocodilian lineage, from which they split ∼267.9–248.3 million years ago (Upper Permian to Triassic). We also found extensive expansion of olfactory receptor genes in these turtles. Embryonic gene expression analysis identified an hourglass-like divergence of turtle and chicken embryogenesis, with maximal conservation around the vertebrate phylotypic period, rather than at later stages that show the amniote-common pattern. Wnt5a expression was found in the growth zone of the dorsal shell, supporting the possible co-option of limb-associated Wnt signaling in the acquisition of this turtle-specific novelty. Our results suggest that turtle evolution was accompanied by an unexpectedly conservative vertebrate phylotypic period, followed by turtle-specific repatterning of development to yield the novel structure of the shell.
Similar content being viewed by others

Main
The unique anatomy of turtles has raised questions about their evolution1. Their armor, even compared to other armored tetrapods (for example, the armadillo and Indian rhinoceros), is distinct in that the dorsal part of the shell (carapace) represents transformed vertebrae and ribs. In addition, their shoulder blades or scapulae1 display an inside-out topology against the rib cage (Supplementary Fig. 1 and Supplementary Note), and the lack of a temporal fenestra further complicates the reconstruction of their phylogenetic position1,2.
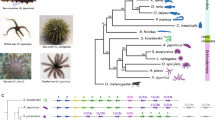
Three major hypotheses have been proposed for the evolutionary origin of turtles, including that they (i) constitute early-diverged reptiles, called anapsids3, (ii) are a sister group of the lizard-snake-tuatara (Lepidosauria) clade4 or (iii) are closely related to a lineage that includes crocodilians and birds (Archosauria)5,6,7,8. Even using molecular approaches, inconsistency still remains6,7,8,9. To clarify the evolution of the turtle-specific body plan, we first addressed the question of evolutionary origin of the turtle by performing the first genome-wide phylogenetic analysis with two turtle genomes sequenced in this project (the green sea turtle, C. mydas, and the Chinese soft-shell turtle, P. sinensis; Fig. 1a). In brief, the fragmented genomic DNA libraries of the two turtles were independently shotgun sequenced using the HiSeq 2000 sequencer and assembled using the SOAPdenovo assembler (Online Methods). The generated turtle genomes were both around 2.2 Gb in size, with the N50 lengths of scaffolds longer than 3.3 Mb (Table 1, Supplementary Figs. 2–5 and Supplementary Tables 1–9).

(a) Two genome-sequenced turtles, the soft-shell turtle (P. sinensis) and the green sea turtle (C. mydas). (b) Estimated divergence times of 12 vertebrate species calculated using the first and second codon positions of 1,113 single-copy coding genes (Supplementary Tables 9 and 10). Tree topology is supported by 100% bootstrap values and further statistical assessment (Supplementary Fig. 5 and Supplementary Tables 11–13). The black ellipses on the nodes indicate the 95% credibility intervals of the estimated posterior distributions of the divergence times. The red circles indicate the fossil calibration times used for setting the upper and lower bounds of the estimates. MYA, million years ago.
On the basis of the largest turtle data set so far, our phylogenetic analysis, with an orthologous set of 1,113 single-copy coding genes, robustly indicated that turtles are likely to be a sister group of crocodilians and birds (Fig. 1b, Supplementary Figs. 6 and 7 and Supplementary Tables 10–13), implying that the temporal fenestrae in the turtle skull were most likely secondarily lost in the turtle lineage1. A molecular evolutionary clock analysis with time constraints based on the fossil records estimated that turtles diverged from archosaurians approximately 257.4 million years ago, with a 95% credibility interval between 267.9 and 248.3 million years ago (Fig. 1b, Supplementary Fig. 7 and Supplementary Table 12). These results are consistent with the oldest turtle fossil (from 220 million years ago), named Odontochelys10. The estimated time range corresponds to the Upper Permian to Triassic period (Fig. 1b), overlapping or following shortly after the Permian extinction event11; this raises the question of whether the emergence of the turtle group was related to this severe extinction event, which especially involved the extinction of marine species.
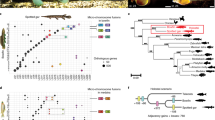
Taking into consideration the phylogenetic position of turtles, we next searched for genes that could potentially explain turtle-specific characteristics (Supplementary Fig. 8 and Supplementary Tables 14–23). Unexpectedly, we found that the olfactory receptor family was highly expanded in both turtle species (Fig. 2 and Supplementary Tables 14–20). In particular, the soft-shell turtle contained 1,137 intact, possibly functional olfactory receptor genes, a number comparable to or even greater than the number of olfactory receptor genes found in most mammals12. Olfactory receptor gene expansion was observed mainly in the α subtype of the class I olfactory receptor genes, suggesting that turtles have superior olfaction ability against a wide variety of hydrophilic substances12 (Fig. 2a and Supplementary Tables 19 and 20). Detailed analyses with genomic sequences further clarified that the majority of the expansion occurred after the split of the two turtle species (Fig. 2b) and that the expansion was most likely facilitated by a gene duplication process, as inferred by the clustered distribution of the olfactory receptor genes in the genome (Fig. 2c,d). These results call into question the general proposition based on mammalian studies13,14 that vertebrates that expand their niche back into aquatic environments tend to reduce the number of olfactory receptor genes. Other than olfactory receptor gene expansion, we found that many genes involved in taste perception (Supplementary Tables 21–23) were lost in the two turtle species. Further investigation of the lost genes in the two turtles identified the loss of many orthologs that are known to be important for normal development in different species, including the genes encoding UNC homeobox, FGF-binding protein 3, CXCL10 and Agouti signaling protein (Supplementary Table 23). These results, together with the identification of many other genes that show accelerated evolutionary rate in turtles (Supplementary Table 24; for example, Bmp receptor 1b, Kit, Jak1 and Eya4), suggest that turtle evolution has included many alterations of the signaling cascade that is presumably involved in morphogenesis. Finally, a possible connection to longevity in turtles was also found (Supplementary Table 24); the most accelerated gene in turtles, showing evidence of positive selection (with the rate of nonsynonymous nucleotide substitutions exceeding the rate of neutral mutations, dN/dS ratio > 1), was microsomal glutathione S-transferase 3 (Mgst3; dN/dS = 5.68), which is reported to function in antioxidative stress, and disrupting the homolog Mgst3-like in Drosophila melanogaster reduces lifespan15.

(a) A neighbor-joining tree constructed with all the intact group α olfactory receptors from eight vertebrate species (soft-shell turtle, green sea turtle, chicken, zebra finch, anole lizard, human, dog and Western clawed frog), with the group β olfactory receptors as the outgroup. Bootstrap values (from 500 resamplings) are shown on the branches. The scale bar represents the number of amino-acid substitutions per site. (b) Expansion of group α olfactory receptor genes in the evolution of tetrapods. Numbers in boxes indicate the current number of intact group α olfactory receptor genes in each species. The number of group α olfactory receptor genes in an ancestral species is shown in an ellipse at each node, and the numbers of gene gains and losses are shown on each branch with plus and minus signs, respectively. For divergence times, we used the median values obtained from TimeTree30. Note that the majority of the expansion of the group α olfactory receptor genes occurred independently in each turtle lineage. The same color code for species is used in a,b. (c,d) Genomic clusters of olfactory receptor genes in scaffolds 55 and 145 of the soft-shell turtle genome. Vertical red bars represent class I (c) and class II (d) olfactory receptor genes. Bars above and below the horizontal line indicate opposite directions of transcription. Long bars depict intact olfactory receptor genes, whereas short bars depict olfactory receptor pseudogenes or gene fragments.
In addition to changes in genomic sequences, we also investigated alterations in embryonic gene regulation that occurred after the split from the bird-crocodilian lineage. According to the recently supported developmental hourglass model16,17,18,19,20,21, the evolutionary changes underlying major adult morphological evolution occurred primarily in the developmental stages after the period of the vertebrate common plan or the period that serves as the source of the vertebrate basic body plan, namely, the vertebrate phylotypic period22. However, the hourglass model has not been tested in non-model organisms, particularly in those with the atypical anatomical features of turtles; therefore, we tested whether the model held true in turtle-chicken comparison. Taking advantage of RNA sequencing (RNA-seq) technology and our previously established method21 based on hierarchical Bayes statistics, our cross-species approach comparing whole-embryo gene expression profiles (GXPs) clearly demonstrated an hourglass-like GXP divergence in the embryogenesis of the soft-shell turtle and the chicken (Fig. 3a and Supplementary Figs. 9–12). However, the result was not robust enough to suggest that the most conserved developmental stage in turtles and birds corresponds to the vertebrate phylotype. The conserved stage could be one occurring later than the vertebrate phylotype, as a previous developmental study23 demonstrated that turtles have a typical amniote-common plan during embryogenesis and develop turtle-specific characteristics thereafter (for example, the scapula primordium first arises outside the rib cage and only later comes to lie inside the rib cage), as if the embryo is recapitulating its own evolutionary history23. If the most conserved developmental stage between the two species indeed corresponds to the stage of the amniote-common plan (approximately Tokita-Kuratani24 stage (TK) 13–14), this would indicate that the conserved stage may change depending on how distantly related the species are that are being compared, similar to the idea from the nested hourglasses model18 (Fig. 3b), justifying, in part, the hierarchical relationship between ontogeny and phylogeny once proposed by Karl von Baer25. Further investigation using a statistically robust cross-species comparative analysis indicated that the soft-shell turtle TK11 and the chicken HH16 developmental stages showed the most similar GXPs (Fig. 3c, Supplementary Figs. 13 and 14 and Supplementary Table 25). Considering that the chicken stage corresponds to the previously identified phylotypic period21, turtle stage TK11 would be an attractive candidate for the vertebrate phylotypic period. In addition to the conservation between turtle and chicken at the level of gene regulation, the identified stages showed notable similarity in morphology (Fig. 3d and Supplementary Table 26), despite the large differences in their final anatomy, the size of their eggs and the actual time scale required for embryogenesis as well as the geological time scale passed since their split (approximately 230 million years ago; Fig. 1), indicating that the morphological and molecular patterns are not uncoupled, in contrast to recent implications from plant development26. Taken together, these results suggest that turtles indeed conform to the developmental hourglass model (Supplementary Fig. 15) by first establishing an ancient vertebrate body plan and by developing turtle-specific characteristics thereafter.

(a) Distances of whole-embryo GXPs (from depth-controlled, TMM (trimmed mean of M values)-normalized data) for 11,602 orthologs in selected developmental stages of the soft-shell turtle and chicken (Supplementary Fig. 16). Error bars, s.d. ANOVA P value under heteroscedasticity = 7 × 10−7. (b) The hypothetical model (nested hourglass)18 in which both an hourglass-like divergence and a recapitulation-like relationship between ontogeny and phylogeny can be justified. The model infers that the most conserved developmental stage changes depending on how distantly related the species are that are being compared. Comparisons within vertebrate embryos gives a vertebrate phylotype (blue arrow), and comparisons within amniotes gives an amniote-type stage (red arrow) that emerges later than the vertebrate phylotype stage. (c) An all-to-all comparison of turtle and chicken GXP distances (total Manhattan) indicates that the highest similarity occurs between turtle stage TK11 and chicken stage HH16 embryos (see Supplementary Fig. 18 for statistical assessment). HH16 is the stage previously identified as the vertebrate phylotypic period21, which does not coincide with the model in b. Error bars, s.d. (d) Morphological appearance of the soft-shell turtle (stage TK11) and chicken (stage HH16) embryos that showed the highest GXP similarity (Supplementary Table 25). Scale bars, 1 mm.
The above results suggest that turtle-specific global repatterning of gene regulation begins after TK11 or the phylotypic period. Although turtle and chicken express many shared developmental genes in the embryo during the putative phylotypic period (Fig. 4a and Supplementary Tables 27 and 28) and have the fewest expanded or contracted gene family members expressed (Supplementary Fig. 16) at this stage, later stages showed increasing differences in their molecular patterns. We found 233 genes that showed turtle-specific increasing expression patterns after the phylotype (Fig. 4b). Considering that the chicken orthologs did not show this type of increasing expression (Supplementary Figs. 17 and 18), these 233 genes represent attractive candidates for clarifying the genomic nature of turtle-specific morphological oddities. Furthermore, our Gene Ontology (GO)-based statistical analysis identified many genes that are potentially involved in ossification and extracellular matrix regulation (Fig. 4c), suggesting the involvement of morphological characteristics appearing in turtle embryogenesis, such as extensive ossification in the shell and folding of the body wall23,24. The morphological specifications of turtle embryogenesis after the identified phylotypic period include the formation of the novel turtle structure called the carapacial ridge27, which is considered to be responsible for the flabellate expansion of the turtle ribs in late development27. Previous molecular studies27,28 have identified many carapacial ridge–specific coding genes, whereas no study so far has investigated carapacial ridge–specific microRNA (miRNA) expression, despite the increasing number of reports claiming the crucial roles of miRNA in various developmental processes. We therefore performed a small RNA-seq analysis of three tissues from soft-shell turtle embryos—limb, body wall and carapacial ridge (Fig. 4d and Supplementary Figs. 19–21)—and further predicted possible miRNAs by referring to the genome sequence (Supplementary Table 29). Unexpectedly, we found expression of a large number of specific miRNAs in all of the tissues (Fig. 4d and Supplementary Table 30), including the carapacial ridge (212 miRNAs). Although no definitive conclusion can be made regarding the functions of these miRNAs, our preliminary prediction-based analysis implied the possible involvement of Wnt signaling (Supplementary Fig. 21 and Supplementary Tables 31–33).

(a) Shared expression of developmental genes (Supplementary Table 27) in the phylotypic stages of turtle and chicken embryos. The log10-transformed relative expression levels of 11,602 orthologous genes from mapped-10M reads (data set based on randomly selected 10M tags mapped to the genome; Online Methods), with TMM-normalized data, were graphed on a scatterplot. Essentially the same results were obtained from other data sets (all-read-data, RPKM (reads per kilobase per million mapped reads) and TMM normalizations). The results of a statistical test to determine the groups of genes that have more similar expression can be found in the Supplementary Note. (b) Genes that showed a statistically significant increase in their expression level after the phylotypic period (IAP; Online Methods). Each line represents the mean expression level of each IAP (increased expression after the phylotype) gene calculated, with two biological replications, for each stage. The names of the genes with the top three highest expression levels in TK23 are shown. Consequently, 233 turtle IAP genes were found. See Supplementary Figure 18 for the expression pattern of the chicken orthologs of the turtle IAP genes. (c) Over-represented GO annotations for 233 turtle IAP genes with read depth–controlled, TMM-normalized data. Only the results corroborated by all of the data sets (mapped-10M reads (Online Methods), all reads, RPKM normalization and TMM normalization) are shown. Shown are P values calculated by Fisher's exact test. (d) High numbers of tissue-specific miRNAs were identified (also in the carapacial ridge) in the embryo after the phylotypic period.
Ann Burke29 was the first to point out the similarity of the apical ectodermal ridge of limbs and the carapacial ridge of the turtle shell. Later, increasing molecular evidence supported this hypothesis. Previous studies27,28 have shown the carapacial ridge–specific activation of Wnt downstream genes (for example, Lef1 expression and nuclear localization of β-catenin) and the essential role of LEF1 in carapacial ridge formation27; however, no Wnt ligand expression has been identified. We therefore annotated all the Wnt genes in the soft-shell turtle and green sea turtle genomes, finding a total of 20 (Supplementary Table 31), and studied their expression patterns in soft-shell turtle embryos at stage TK14, the stage when the carapacial ridge begins to be apparent (Fig. 5a). Notably, we found that Wnt5a was the only Wnt gene expressed in the turtle carapacial ridge region (Fig. 5b,c and Supplementary Fig. 22). With respect to the evolutionary scenario of the carapacial ridge, Wnt5a expression was also found in both the forelimbs and the hindlimbs, as in other amniotes, implying that part of the gene regulatory network involved in carapacial ridge development has been co-opted, most likely from the limb buds28,29. However, this hypothesis has to be considered with caution, particularly because we still lack functional evidence of Wnt5a involvement in carapacial ridge formation. Taking the findings together, the exact roles of the carapacial ridge–expressed Wnt and miRNAs remain to be elucidated; however, our series of genome-scale results indicate the co-option of the Wnt signaling pathway in turtles and provide a basis for understanding shell evolution.

(a) Whole-mount in situ hybridization (ISH) was performed for all Wnt genes in the genome. Wnt5a (red outline) is specifically expressed in the carapacial ridge (red arrowheads), whereas most of the other genes show similar expression patterns to their known mouse and chicken counterparts. Scale bars, 0.5 mm. (b) Soft-shell turtle embryo at stage TK14. (c) Carapacial ridge expression of Wnt5a confirmed by ISH on a 6-μm paraffin transverse section (the sectioned level for ISH is indicated by the dashed line in b). The arrowhead indicates the carapacial ridge; the arrow indicates the body wall. NT, neural tube; NC, notochord. Scale bars in b and c, 0.5 mm.
In summary, our study both highlights the evolution of the turtle body plan and offers a model to explain, at the genomic level, how the vertebrate developmental program can change to produce major evolutionary novelties in morphological phenotypes.
Methods
Source and sequencing of genomic DNA and error correction.
The soft-shell turtle was purchased from a local farmer in Japan, and the green sea turtle was provided by the Genome 10K Project (originally collected in Ocean Park, Hong Kong). Genomic DNA was extracted from the whole blood of a female individual in each species, and we constructed a total of 18 (for the soft-shell turtle) and 17 (for the green sea turtle) libraries consisting of short-insert (170-bp, 500-bp and 800-bp) and long-insert (2-kb, 5-kb, 10-kb, 20-kb and 40-kb) libraries. Sequencing was performed using the Illumina HiSeq 2000 system, and read error correction was performed for the short-insert libraries (on the basis of the K-mer frequency distribution curve; Supplementary Note). Data accession numbers are given in Supplementary Table 34.
Genome assembly.
Filtered and corrected data were assembled using SOAPdenovo31,32. We first generated contigs by constructing a de Bruijn graph with the reads from the K-mer–split short-insert library data. The graph was then simplified to generate the contigs by removing tips, merging bubbles and solving repeats. All sequenced reads were then realigned onto the contig sequences, and scaffolds were constructed by weighting the rates of consistent and conflicting paired-end relationships. Finally, we retrieved the read pairs with one end that uniquely mapped to the contig and the other end located in the gap region, and performed a local assembly for these collected reads to fill the gaps.
Repeat annotation and whole-genome alignment.
Repeat detection was performed using the program RepeatMasker and the Genetic Information Research Institute (GIRI) repeat library. For homology-based prediction of repeats, we used the library of known repeats in the Repbase33 database (v2008-08-01, Repbase-16.02) with RepeatMasker (v3.2.6) and RepeatProteinMask to identify transposable elements at the DNA and protein levels, respectively. The de novo prediction of repeats involved building a de novo repeat library with RepeatModeler34 and subsequently employing RepeatMasker. Tandem repeats were searched with the Tandem Repeats Finder (TRF)35. Whole-genome pairwise alignments were generated by LASTZ36.
Gene prediction for the two turtles and crocodilians.
Gene prediction for the two turtle genomes employed both the ab initio approach (GENSCAN37 (v2.5.5) and AUGUSTUS38 (v1.0)) and a homolog-based approach against the repeat-masked genome, and gene sets predicted by these two approaches were further consolidated with the GLEAN39 program. For the soft-shell turtle, an additional 146.7 Gb of RNA-seq data was used. The proteins of other vertebrate species were mapped to the genome using TBLASTN (Legacy Blast40 v2.2.23). Aligned sequences were then filtered and passed to GeneWise41 (v2.2.0) along with the query sequences. The resulting data sets were integrated by GLEAN39 into a consensus gene set. The best BLASTP match to the SwissProt and TrEMBL databases was used to assign function. The motifs and domains of the gene products were annotated with InterProScan42 against the protein databases ProDom, PRINTS, Pfam, SMART, PANTHER and PROSITE. Gene Ontology43 IDs for each gene were obtained from the corresponding InterPro entries. The above prediction pipeline was applied to the saltwater crocodile and American alligator genomes (from the Crocodile Genome Consortium), except for the integration step in the latter case. Gene family identification was performed using TreeFam32.
Gene prediction for the soft-shell turtle by the Ensembl prediction pipeline.
For gene expression comparison analyses between soft-shell turtle and chicken embryos, we generated and used another soft-shell turtle gene set that was created by the same Ensembl pipeline as the chicken gene set (see URLs).
GO analysis.
Over-represented GO terms were investigated by testing (Fisher's exact test) the bias in frequency toward other GO terms among certain gene sets, using the total set of defined GO terms as a control distribution. Developmental genes (5,659 in total) were defined as genes with developmental GO terms, and developmental GO terms were defined as those with GO:0032502 (developmental process) as an ancestor.
Animal care and use.
Experimental procedures and animal care were conducted in strict accordance with guidelines approved by the RIKEN Animal Experiments Committee (Approval IDs H14-23 and H16-10).
Phylogenetic tree reconstruction and divergence time estimation.
The coding sequences of single-copy gene families conserved among the soft-shell turtle, green sea turtle, anole lizard, saltwater crocodile, chicken, zebra finch, dog, human, platypus and Xenopus tropicalis were extracted and aligned with guidance from amino-acid alignments created by the MUSCLE program44. Sequences were then concatenated to one supergene sequence for each species. PhyML45,46 was applied to construct the phylogenetic tree under an HKY85+gamma or GTR+gamma model for nucleotide sequences and the JTT+gamma model for protein sequences. aLRT values were taken to assess the branch reliability in PhyML. RAxML47 was also applied for the same set of sequences to build a phylogenetic tree under a GTR+gamma or JTT+gamma model for nucleotide and protein sequences, respectively, with 1,000 rapid bootstraps employed to assess the branch reliability in RAxML. The same set of codon sequences at positions 1 and 2 was used for phylogenetic tree construction and estimation of the divergence time. The PAML mcmctree program (PAML version 4.5)48,49,50 was used to determine divergence times with the approximate likelihood calculation method and the 'correlated molecular clock' and 'REV' substitution model. Two independent runs were performed to confirm convergence.
Gene loss analysis and gene family expansion and contraction analysis.
Protein sequences of the two turtles and related species (chicken, anole lizard, X. tropicalis and zebra finch) were used in BLAST searches against human protein sequences (Ensembl Gene v.68), identifying homologs. Subsequently, human proteins that lacked homologs in both the turtle species but had homologs in the related species were identified as lost genes in turtle. For the statistical analysis of gene family expansion and contractions, we generated pairwise whole-genome alignments for anole lizard and soft-shell turtle and for anole lizard and green sea turtle using LASTZ51 and created three-way alignments using MULTIZ52. When an anole lizard gene fell in an area of conserved sequence and there was no homologous gene in the corresponding aligned sequences of the two turtle species, we hypothesized that gene loss potentially occurred at that locus in turtle (Supplementary Note). Frameshift mutations and those introducing premature stop codons in the coding sequences were also considered to represent gene loss.
Prediction of olfactory receptor genes.
Olfactory receptor genes were identified by previously described methods53, with the exception of a first-round TBLASTN54 search, in which 119 functional olfactory receptor genes from human, mouse and zebrafish were used as queries (Supplementary Note). To construct phylogenetic trees, the amino-acid sequences encoded by olfactory receptor genes were first aligned using the program E-INS-i in MAFFT55. We then constructed a phylogenetic tree using the neighbor-joining method56 with Poisson correction distances using the program LINTREE57. The numbers of olfactory receptor genes in ancestral species and those of gene gains or losses in evolution were calculated by the reconciled tree method53 with 70% bootstrap value cutoff.
Genes with accelerated evolutionary rate in the turtle lineage.
Homologous genes in soft-shell turtle, green sea turtle and other vertebrate species (chicken, zebra finch, anole lizard, X. tropicalis and platypus) were first identified with the all-against-all BLASTP program. Orthologs were defined by reciprocal best BLAST hits (RBBHs) in humans and the other species. The full orthologous gene sets were aligned using the program MUSCLE. We then compared a series of evolutionary models within the likelihood framework using the phylogenetic tree obtained by our analysis. A branch model50 was used to detect the average length (ω) across the tree (ω0), the ω value of the ancestor of all soft-shell turtles, the ω value for the green sea turtle branch (ω2) and the ω value for all of the other branches (ω1).
Embryo sampling and mRNA extraction.
Fertilized soft-shell turtle and chicken eggs purchased from local farms in Japan were incubated and staged according to previous descriptions24,58. Amniotic membranes were removed before mRNA extraction, and more than two individual embryos were pooled for each sample. The RNeasy Lipid Tissue kit (Qiagen) and the Ambion MicroPoly(A) Purist kit (Life Technologies) were used for mRNA extraction.
RNA-seq for transcriptome identification.
Three different types of sequencing were performed for transcriptome identification in the soft-shell turtle: (i) Titanium sequencing (about 2 Gb of clean sequence data), (ii) HiSeq strand-specific paired-end RNA-seq (two libraries were prepared by methods that retain strand-specific information, including a dUTP-based method59,60 (19 Gb of clean data) that was modified to comply with the Illumina TruSeq RNA sample prep kit and an original method developed at BGI Sequencing that was performed with Illumina HiSeq 2000 (26 Gb of clean data) and (iii) HiSeq non-stranded RNA-seq (deep-sequencing data for gene expression analysis was also used for transcriptome identification). Further details are given in the Supplementary Note.
RNA-seq for gene expression analysis and expression comparison.
Biological replicates for each developmental stage were created from an independent sample pool. Extracted mRNA samples were then sequenced with an Illumina HiSeq 2000 instrument. We identified 11,602 one-to-one orthologous genes in the soft-shell turtle and chicken using RBBH information from BLAST+ (v2.2.25)61. Gene expression scores were obtained from RNA-seq data by mapping clean reads to the genome using Burrows-Wheeler Aligner (BWA)62 software (v0.5.9-r16). SAMtools63, BEDtools64 and the DEGseq package65 for R (v2.14.2) were used to calculate the tag count data that were mapped to the coding regions. Normalization of the orthologous gene expression scores was performed with all samples at once by either RPKM or TMM normalization66. Pearson's correlation coefficients, Spearman correlation coefficients, total Euclidean distances (t-Euclidean) or total Manhattan distances (t-Manhattan) were used to estimate similarities in the gene expression profiles of the two samples being compared. Two independent random selections from all reads were performed to make the mapped-10M reads (sequencing depth–controlled data set based on randomly selected 10M tags mapped to the genome) data. The Welch two-sample t test or the Wilcoxon signed-rank test was used to detect the most conserved stages. The Holm-corrected α level was applied for these multiple comparisons. Only results reproduced by the data set from two different normalizations (RPKM67 and TMM66) were considered to be significant.
Genes with a significant increase in expression levels after the phylotypic period.
Turtle IAP genes were selected using the following criteria: (i) the mean expression level after the phylotypic period (TK15–TK23) was more than five times higher (Wilcoxon test) than during earlier stages (gastrula, neurula, TK7 and TK9) and (ii) the chicken orthologs of the turtle IAP genes (if any) did not show such increases in chicken (the average expression levels in HH28 and HH38 did not show more than five times higher expression than in the Prim–HH14 stages).
Wnt gene identification and cloning and whole-mount ISH.
In addition to constructing the predicted gene sets, we manually searched for Wnt genes using TBLASTN. Cloning of the probes and whole-mount ISH were performed using standard methods28 (Supplementary Note).
miRNA extraction, prediction and expression analysis.
Small RNA was extracted from dissected tissues using the mirVana microRNA Isolation kit (Life Technologies). Small RNA libraries were prepared and sequenced using an Illumina HiSeq 2000 (>24 million reads per sample). These small RNA reads, together with the miRNA sequences from chicken, zebra finch and Anolis carolinensis from miRBase (v.18), were used to predict miRNA sequences in the genome. The program miRDeep2 (v2.0.0.3)68 was used to predict miRNAs for this prediction. Only miRNA predictions that had P value lower than a significant Randfold α level (P < 0.05 mononucleotide shuffling and 999 permutations; see ref. 68 for details) were taken into account for subsequent comparisons. miRNA target prediction was performed with miRanda69 (v3.3a) using the annotated 3′ UTRs of soft-shell turtle genes.
Statistical tests.
To avoid an inflated type I error rate, an α level of 0.01 (further Bonferronni correction in case of multiple comparisons) was accepted for statistical significance throughout the analyses unless otherwise specified. Statistical methods were carefully chosen to properly reflect the population of interest. The Welch two-sample t test was used for two-sample comparisons when the data passed the Kolmogorov-Smirnov test for normal distribution; otherwise, the Wilcoxon signed-rank test was used.
URLs.
International Crocodilian Genomes Working Group, http://www.crocgenomes.org/; Genome 10K Project, http://genome10k.soe.ucsc.edu/; Genetic Information Research Institute (GIRI), http://www.girinst.org/; creation of the Ensembl chicken embryo gene set, http://www.ensembl.org/info/docs/genebuild/genome_annotation.html, LINTREE, http://www.personal.psu.edu/nxm2/software.htm; reconciled tree method, http://bioinfo.tmd.ac.jp/~niimura/software.html; RepeatMasker, http://www.repeatmasker.org/; LASTZ, http://www.bx.psu.edu/miller_lab/dist/README.lastz-1.02.00/README.lastz-1.02.00a.html.
Accession codes.
The Chinese soft-shell turtle and the green sea turtle draft genomes have been deposited in NCBI GenBank under accessions AGCU00000000 and AJIM00000000, respectively. The Chinese soft-shell turtle genome can also be accessed at the Ensembl database. Wnt gene sequences cloned for whole-mount ISH have been deposited in NCBI GenBank under accessions JQ968433, JQ968434, JQ968435, JQ968436, JQ968437, JQ968438, JQ968439, JQ968440, JQ968441, JQ968442, JQ968443, JQ968444, JQ968445, JQ968446, JQ968447, JQ968448, JQ968449, JQ968450, JQ968451, JQ968452. Soft-shell turtle and chicken RNA-seq data have been deposited in the DNA Data Bank of Japan (DDBJ) Sequence Read Archive under accession DRA000567. Soft-shell turtle RNA-seq data for small RNA are available under DDBJ Sequence Read Archive accession DRA000639. Additional information is provided in Supplementary Table 34.
Change history
28 May 2014
In the version of this article initially published, the claim that the ghrelin gene was lost specifically in the two turtle species was incorrect, and ghrelin sequences can be found in the genome sequences originally published. The error has been corrected in the HTML and PDF versions of the article.
References
Kuratani, S., Kuraku, S. & Nagashima, H. Evolutionary developmental perspective for the origin of turtles: the folding theory for the shell based on the developmental nature of the carapacial ridge. Evol. Dev. 13, 1–14 (2011).
Rieppel, O. Turtles as hopeful monsters. Bioessays 23, 987–991 (2001).
Romer, A.S. Vertebrate Paleontology 3rd edn (University of Chicago Press, Chicago, 1966).
Rieppel, O. & deBraga, M. Turtles as diapsid reptiles. Nature 384, 453–455 (1996).
Hedges, S.B., Moberg, K.D. & Maxson, L.R. Tetrapod phylogeny inferred from 18S and 28S ribosomal RNA sequences and a review of the evidence for amniote relationships. Mol. Biol. Evol. 7, 607–633 (1990).
Crawford, N.G. et al. More than 1000 ultraconserved elements provide evidence that turtles are the sister group of archosaurs. Biol. Lett. 8, 783–786 (2012).
Chiari, Y., Cahais, V., Galtier, N. & Delsuc, F. Phylogenomic analyses support the position of turtles as the sister group of birds and crocodiles (Archosauria). BMC Biol. 10, 65 (2012).
Tzika, A.C., Helaers, R., Schramm, G. & Milinkovitch, M.C. Reptilian-transcriptome v1.0, a glimpse in the brain transcriptome of five divergent Sauropsida lineages and the phylogenetic position of turtles. Evodevo. 2, 19 (2011).
Lyson, T.R. et al. MicroRNAs support a turtle + lizard clade. Biol. Lett. 8, 104–107 (2012).
Li, C., Wu, X.C., Rieppel, O., Wang, L.T. & Zhao, L.J. An ancestral turtle from the Late Triassic of southwestern China. Nature 456, 497–501 (2008).
Zhong-Qiang, C. & Benton, M.J. The timing and pattern of biotic recovery following the end-Permian mass extinction. Nat. Geosci. 5, 375–383 (2012).
Niimura, Y. Olfactory receptor multigene family in vertebrates: from the viewpoint of evolutionary genomics. Curr. Genomics 13, 103–114 (2012).
Hayden, S. et al. Ecological adaptation determines functional mammalian olfactory subgenomes. Genome Res. 20, 1–9 (2010).
Kishida, T., Kubota, S., Shirayama, Y. & Fukami, H. The olfactory receptor gene repertoires in secondary-adapted marine vertebrates: evidence for reduction of the functional proportions in cetaceans. Biol. Lett. 3, 428–430 (2007).
Toba, G. & Aigaki, T. Disruption of the microsomal glutathione S-transferase–like gene reduces life span of Drosophila melanogaster. Gene 253, 179–187 (2000).
Duboule, D. Temporal colinearity and the phylotypic progression: a basis for the stability of a vertebrate bauplan and the evolution of morphologies through heterochrony. Dev. Suppl. 135–142 (1994).
Raff, A. The Shape of Life: Genes, Development, and The Evolution of Animal Form (University of Chicago Press, Chicago, 1996).
Irie, N. & Sehara-Fujisawa, A. The vertebrate phylotypic stage and an early bilaterian-related stage in mouse embryogenesis defined by genomic information. BMC Biol. 5, 1 (2007).
Kalinka, A.T. et al. Gene expression divergence recapitulates the developmental hourglass model. Nature 468, 811–814 (2010).
Domazet-Lošo, T. & Tautz, D. A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns. Nature 468, 815–818 (2010).
Irie, N. & Kuratani, S. Comparative transcriptome analysis reveals vertebrate phylotypic period during organogenesis. Nature Commun. 2, 248 (2011).
Richardson, M.K., Minelli, A., Coates, M. & Hanken, J. Phylotypic stage theory. Trends Ecol. Evol. 13, 158 (1998).
Nagashima, H. et al. Evolution of the turtle body plan by the folding and creation of new muscle connections. Science 325, 193–196 (2009).
Tokita, M. & Kuratani, S. Normal embryonic stages of the Chinese softshelled turtle Pelodiscus sinensis (Trionychidae). Zoolog. Sci. 18, 705–715 (2001).
von Baer, K.E. Uber Entwickelungsgeschichte der Thiere: Beobachtung und Reflektion (Gebrüder Borntraeger, Koenigsberg, 1828).
Quint, M. et al. A transcriptomic hourglass in plant embryogenesis. Nature 490, 98–101 (2012).
Nagashima, H. et al. On the carapacial ridge in turtle embryos: its developmental origin, function and the chelonian body plan. Development 134, 2219–2226 (2007).
Kuraku, S., Usuda, R. & Kuratani, S. Comprehensive survey of carapacial ridge–specific genes in turtle implies co-option of some regulatory genes in carapace evolution. Evol. Dev. 7, 3–17 (2005).
Burke, A. Development of the turtle carapace: implications for the evolution of a novel bauplan. J. Morphol. 199, 363–378 (1989).
Hedges, S.B., Dudley, J. & Kumar, S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22, 2971–2972 (2006).
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010).
Li, R. et al. The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317 (2010).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Price, A.L., Jones, N.C. & Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 21 (suppl. 1), i351–i358 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Brudno, M. et al. Automated whole-genome multiple alignment of rat, mouse, and human. Genome Res. 14, 685–692 (2004).
Burge, C.B. & Karlin, S. Finding the genes in genomic DNA. Curr. Opin. Struct. Biol. 8, 346–354 (1998).
Keller, O., Kollmar, M., Stanke, M. & Waack, S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763 (2011).
Elsik, C.G. et al. Creating a honey bee consensus gene set. Genome Biol. 8, R13 (2007).
Kent, W.J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Zdobnov, E.M. & Apweiler, R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003).
Endres, C.S., Putman, N.F. & Lohmann, K.J. Perception of airborne odors by loggerhead sea turtles. J. Exp. Biol. 212, 3823–3827 (2009).
Rannala, B. & Yang, Z. Inferring speciation times under an episodic molecular clock. Syst. Biol. 56, 453–466 (2007).
Yang, Z. & Rannala, B. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Mol. Biol. Evol. 23, 212–226 (2006).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Harris, R. Improved Pairwise Alignment of Genomic DNA PhD Thesis, Penn. State Univ. (2007).
Blanchette, M. et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 14, 708–715 (2004).
Niimura, Y. & Nei, M. Extensive gains and losses of olfactory receptor genes in mammalian evolution. PLoS ONE 2, e708 (2007).
Altschul, S.F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Katoh, K., Kuma, K., Toh, H. & Miyata, T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518 (2005).
Saitou, N. & Nei, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 (1987).
Takezaki, N., Rzhetsky, A. & Nei, M. Phylogenetic test of the molecular clock and linearized trees. Mol. Biol. Evol. 12, 823–833 (1995).
Hamburger, V. & Hamilton, H.L. A series of normal stages in the development of the chick embryo. 1951. Dev. Dyn. 195, 231–272 (1992).
Levin, J.Z. et al. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 7, 709–715 (2010).
Parkhomchuk, D. et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 37, e123 (2009).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A.R. & Hall, I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Wang, L., Feng, Z., Wang, X. & Zhang, X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 (2010).
Robinson, M.D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Mortazavi, A., Williams, B.A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Friedländer, M.R., Mackowiak, S.D., Li, N., Chen, W. & Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37–52 (2012).
Enright, A.J. et al. MicroRNA targets in Drosophila. Genome Biol. 5, R1 (2003).
Acknowledgements
We would like to acknowledge the International Crocodilian Genomes Working Group, in particular, D.A. Ray of Mississippi State University, R.E. Green of the University of California at Santa Cruz and E.L. Braun of the University of Florida, for making their early drafts of the Crocodylus porosus (saltwater crocodile) and Alligator mississippiensis (American alligator) genomes available for the phylogenetic analysis. We acknowledge P. Martelli at Ocean Park Corporation Hong Kong for providing the green sea turtle samples and Daiwa-yoshoku Ltd. For providing the soft-shell turtle samples. We thank M. Noro, K. Yamanaka, K. Itomi and T. Kitazume for help collecting and sequencing the soft-shell turtle mRNA. The soft-shell turtle genome project was supported in part by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science & Technology, Japan (22128003 and 22128001), the NIBB Cooperative Research Program (Next-generation DNA Sequencing Initiative, 11-730) and the European Molecular Biology Laboratory and the Wellcome Trust (grant numbers WT095908 and WT098051). The green sea turtle genome project was supported by the China National GeneBank at BGI-Shenzhen.
Author information
Authors and Affiliations
Contributions
Z.W. coordinated genome assembly, annotation and bioinformatics analysis. J.P.-A. performed embryo collection, miRNA analysis and ISH experiments. W.L. coordinated RNA-seq and the project. Z.H., Z.X., D.F., B.W., Y.C., Y.Z., Juan Wang, H.Z., Junyi Wang and J.L. performed genome sequencing and assembly. C.L., Y.M. and L.Y. produced soft-shell turtle, green sea turtle and saltwater crocodile gene sets. Y.N., S. Kuraku, J.H., K.B., Q.L. and M.P. performed bioinformatics analysis. M.N. and S. Shigenobu prepared RNA-seq libraries. B.A. supervised the soft-shell turtle genome annotation. P.F. and S. Searle supervised the Ensembl project. A.Z. produced the soft-shell turtle gene set. S.W. developed the RNA-seq pipeline. Y.Y., S. Kuratani and Jun Wang supervised the project. G.Z. (headed the green sea turtle genome project) and N.I. (headed the soft-shell turtle genome project), coordinated and supervised the project, collected samples, performed analysis and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Note, Supplementary Figures 1–22 and Supplementary Tables 1–14, 19–22, 24–29, 31, 33 and 34 (PDF 5305 kb)
Supplementary Table 15
Nucleotide sequences of genes that are predicted to be expanded in the turtle lineage (XLS 4436 kb)
Supplementary Table 16
Table of gene family IDs and genes that are predicted to be contracted / expanded in the turtle lineage. (XLS 10661 kb)
Supplementary Table 17
Amino acid sequences of intact OR genes identified from genome sequences. (XLS 603 kb)
Supplementary Table 18
Nucleotide sequences of intact OR genes identified from genome sequences. (XLS 1466 kb)
Supplementary Table 23
List of genes that were predicted to be lost in two turtles, but exists in either of chicken, zebra finch, anole lizard, or X. tropicalis. (XLS 79 kb)
Supplementary Table 30
Predictions of miRNAs from P. sinensis TK14 carapacial ridge, limb and body wall. (XLS 802 kb)
Supplementary Table 32
miRNA target prediction statistics (XLS 3243 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article
Cite this article
Wang, Z., Pascual-Anaya, J., Zadissa, A. et al. The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat Genet 45, 701–706 (2013). https://doi.org/10.1038/ng.2615
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.2615
This article is cited by
-
Stability in gene expression and body-plan development leads to evolutionary conservation
EvoDevo (2023)
-
The first high-quality chromosome-level genome of Eretmochelys imbricata using HiFi and Hi-C data
Scientific Data (2023)
-
In situ hybridization analysis of olfactory receptor expression in the sea turtle olfactory organ
Cell and Tissue Research (2023)
-
Chromosome-level genome assembly of Asian yellow pond turtle (Mauremys mutica) with temperature-dependent sex determination system
Scientific Reports (2022)
-
Development of 105 SNP markers in endangered turtle species Pelodiscus sinensis using RAD-seq
Conservation Genetics Resources (2022)
