
Abstract
As top predators, falcons possess unique morphological, physiological and behavioral adaptations that allow them to be successful hunters: for example, the peregrine is renowned as the world's fastest animal. To examine the evolutionary basis of predatory adaptations, we sequenced the genomes of both the peregrine (Falco peregrinus) and saker falcon (Falco cherrug), and we present parallel, genome-wide evidence for evolutionary innovation and selection for a predatory lifestyle. The genomes, assembled using Illumina deep sequencing with greater than 100-fold coverage, are both approximately 1.2 Gb in length, with transcriptome-assisted prediction of approximately 16,200 genes for both species. Analysis of 8,424 orthologs in both falcons, chicken, zebra finch and turkey identified consistent evidence for genome-wide rapid evolution in these raptors. SNP-based inference showed contrasting recent demographic trajectories for the two falcons, and gene-based analysis highlighted falcon-specific evolutionary novelties for beak development and olfaction and specifically for homeostasis-related genes in the arid environment–adapted saker.
Similar content being viewed by others

Main
We carried out next-generation genome sequencing for the peregrine and saker falcon, generating 128.07 Gb and 136.21 Gb of sequence for F. peregrinus and F. cherrug, respectively (Supplementary Tables 1 and 2). Genome size was estimated at 1.2 Gb for both species (Supplementary Fig. 1 and Supplementary Table 3), suggesting genome coverage of 106.72× for F. peregrinus and 113.51× for F. cherrug (Supplementary Fig. 2). Assembly using SOAPdenovo1,2 resulted in contig and scaffold N50 values, respectively, of 28.6 kb and 3.89 Mb for F. peregrinus and 31.2 kb and 4.15 Mb for F. cherrug (Supplementary Table 4). Fosmid-based Sanger sequencing confirmed >99% (peregrine) and >97% (saker) coverage of euchromatic DNA (Supplementary Table 5). With repeats masked, protein-coding genes were predicted using homology and de novo methods and also with RNA sequencing (RNA-seq) data, employed to refine gene structure and identify novel genes (Supplementary Fig. 3). As a result, 16,263 genes were predicted for F. peregrines, and 16,204 were predicted for F. cherrug (Supplementary Table 6 and Supplementary Note). Approximately 92% of these genes were functionally annotated using homology-based methods (Supplementary Table 7).
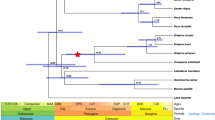
Comparative genome analysis was carried out to assess evolution and innovation within falcons using related genomes with comparable assembly quality (Supplementary Table 8). Orthologous genes were identified in the chicken, zebra finch, turkey, peregrine and saker using the program TreeFam3. For the five genome-enabled avian species, a maximum-likelihood phylogeny using 861,014 4-fold degenerate sites from 6,267 single-copy orthologs confirmed that chicken and turkey comprise one evolutionary branch and that zebra finch and the falcons form a second (Fig. 1a), supporting previous analysis using 19 nuclear sequences4. Analysis of the peregrine and saker using the same data set (Online Methods) dated the most recent common ancestor of the two falcon species to 2.1 (0.9–4.2) million years ago (Fig. 1a). More than 99.6% of the peregrine genome was syntenic to the saker genome (Supplementary Table 9).

(a) Phylogenetic tree constructed using fourfold degenerate sites from single-copy orthologs, with the branch lengths scaled to estimated divergence time. Branch-specific ω values calculated from concatenated and SATé-aligned single-copy orthologs are shown on the left of each branch, and gene gain (+) and loss (−) per million years are shown on the right. (b) Venn diagram showing shared and unique gene families in five avian species: red, peregrine; pink, saker; light blue, chicken; orange, turkey; green, zebra finch. (c) Neighbor-joining tree based on the amino-acid sequences encoded by olfactory receptor genes in the peregrine, saker, chicken and zebra finch. Bootstrap values are shown (clockwise) for the major olfactory receptor clades, namely γ-c, other γ, α, θ and TAAR. Note that both chicken and zebra finch show expansions in the olfactory receptor γ-c clade. Birds are represented by the same colors as in b.
Falcons were found to have less repetitive DNA (Table 1 and Supplementary Table 10), and their transposable element composition was most similar to that of zebra finch (with fewer DNA transposable elements and long interspersed nucleotide elements (LINEs) but not short interspersed nucleotide elements (SINES)) (Supplementary Tables 11 and 12). We found fewer large (>1-kb) segmental duplications in falcons than in either chicken or zebra finch (Table 1 and Supplementary Table 13), with these elements comprising less than 1% of both falcon genomes. We also found that fewer branch-specific insertions and/or deletions (indels) have accumulated in falcon genomes over evolutionary time (Table 1 and Supplementary Tables 14 and 15). TreeFam results showed, however, that falcons feature fewer lineage-specific genes than other birds (Fig. 1b and Table 1). It should be noted that our comparative genome analyses are based on alignments from the local to the scaffold level and are not based on whole-chromosome alignments.
The olfactory receptor gene repertoire, annotated on the basis of putative functionality and 7-TM (transmembrane) structure, showed the fewest intact olfactory receptor genes in falcons (n = 28; Supplementary Table 16), even though they have a larger olfactory bulb ratio than the zebra finch and a similar ratio to chicken5. These two traits have previously been thought to be positively correlated6 and linked to olfactory function7. Furthermore, a gene expansion in the olfactory receptor γ-c clade8 in chicken and zebra finch is not present in falcons (Fig. 1c and Supplementary Table 17), possibly reflecting their reliance on vision for locating prey5.
To compare selection at the gene level, two orthologous gene sets were compiled: the single-copy orthologs for the five avian species and a representative gene set from multiple-copy orthologs (n = 2,157). Analysis of branch-specific Ka/Ks (nonsynonymous-synonymous) substitution ratios (ω) showed that both falcons have higher branch ω values than other birds, independent of the gene set used (Fig. 1a and Supplementary Fig. 4), implying accelerated functional evolution in the falcon lineage. Supporting evidence for this comes from a recent work showing rapid phenotypic evolution and speciation in the falcon family9. We also calculated the ω value for each orthologous gene and found that the ω values in falcons with mean value of 0.39 were considerably larger than for the other five pairwise avian combinations (see distribution in Fig. 2a). However, these results need to be interpreted in the light of the relative paucity of avian genomes currently sequenced. We further examined both rapidly and slowly evolving gene categories by comparing the two falcons against the galliformes, zebra finch–chicken and zebra finch–turkey pairs, respectively. To account for rapid genome evolution in falcons, each ω value within these categories was normalized using the genome median ω of each species pair. Functional GO (Gene Ontology)10 categories involved in circulation (for example, heme synthesis), the nervous system, olfaction and sodium ion transport were found to have evolved rapidly in falcons (Supplementary Fig. 5 and Supplementary Table 18). Notable rapid evolution was also observed in the mitochondrial respiration chain when comparing falcons and galliformes birds but not in the comparisons of falcons with other species (Supplementary Table 18).

(a) Distribution of ω values among avian species. The estimates of ω for 8,424 orthologs show a highly significant shift toward larger values in falcons compared with other pairs (P = 1.354 × 10−189, 2.837 × 10−297, 1.621 × 10−284, 7.264 × 10−233 and 4.617 × 10−215 relative to the other indicated pairs, Mann Whitney test). Genes with Ka = 0 are included in relative frequencies but are not shown. The x axis shows log2 Ka/Ks, using 0.01 as the unit interval, and the y axis shows the number of orthologs in each interval. (b) Gene number differences in contracted and expanded protein families in the falcons compared with zebra finch. Zebra finch genes of unplaced sequence that had more than 97% sequence identity with their closest paralog were excluded to correct for the possible overestimation of zebra finch gene expansion14.
The gene families defined by TreeFam (Supplementary Table 19) were input into CAFE11 to examine changes in ortholog cluster sizes between putative ancestors and each species across our phylogeny (Fig. 1a). We found that net gene loss occurred in all avian genomes, but the rate of loss per unit of time was most rapid in falcons (Fig. 1a). We then used protein clustering12 to explore gene number variation in the same protein families between each falcon species and the zebra finch. Loss of protein families within falcons (extinction plus contraction) exceeded gain (innovation plus expansion) (Supplementary Tables 20–24), and contraction was greater than expansion for both the number of protein families and gene number (Fig. 2b).
We identified 879,812 and 761,748 heterozygous SNPs in the peregrine and saker genomes, respectively. Despite its lower number of heterozygous SNPs, the saker had a higher heterozygous SNP rate than the peregrine (Table 1), although rates for both falcons were lower than for either the chicken13 or zebra finch14. Lower genetic diversity might originate from recent population contractions (Fig. 3). A narrow distribution in genome-wide SNP density for the peregrine, unlike the saker, suggests a more heterogeneous SNP distribution, indicating that mutations in the peregrine genome are more evenly distributed (Fig. 3a). On the basis of local SNP densities, we used the pairwise sequentially Markovian coalescent (PSMC)15 to model the demographic history of both species (Fig. 3b). For the peregrine, we inferred demographic history from 2 million years ago to 10,000 years ago, whereas, for the saker, the analysis included both the saker and its ancestral hierofalcon16 because the fossil record indicates that the saker originated less than 34,000 years ago16. PSMC showed that both falcon species have experienced at least one bottleneck; however, while this lasted until 100,000 years ago for the peregrine, hierofalcon populations expanded around 300,000 years ago and started to expand again from 60,000 years ago, eventually giving rise to the four species in the hierofalcon group16. The peregrine, however, underwent a second bottleneck approximately 20,000 years ago, possibly owing to climate-driven habitat diminution (Supplementary Fig. 6).

(a) Distribution of SNP density across each falcon genome. Heterozygous SNPs between the two sets of falcon chromosomes were annotated, and heterozygosity density was observed in non-overlapping 50-kb windows. (b) PSMC inference of falcon population history based on autosomal data. The central bold lines represent inferred population sizes, and the 100 thin curves surrounding each line are the PSMC estimates generated using 100 sequences randomly resampled from the original sequence. The mutation rate on autosomes, which is used in time scaling, was estimated using zebra finch autosome data.
The word falcon comes from the Latin falco, meaning hook shaped, referring to its beak. The falcon beak is more robust, being longer, wider and deeper, than those of the chicken and zebra finch. We examined the evolution of the genes in two KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways (TGF-β and WNT)17, those in FGF signaling pathways (see URLs) and 98 other genes involved in the development of avian beaks (Online Methods). Bmp4 is a well-studied major-effect gene in beak development18, and exonization provides a major source of accelerated, lineage-specific evolution19. We found that falcons have gained two novel exons owing to splice-site mutations in Bmp4 (Supplementary Fig. 7) and have also gained a second copy of TGFB2 (a gene upregulated in curved beaks) and Dkk 2 (a gene related to longer and wider beaks in ducks)20. The two copies originated in different parts of a single ancestral gene (Supplementary Figs. 8 and 9). These changes can both influence gene expression21,22 and contribute to the regulation of development in falcon beaks (Table 2). Relative to chicken and zebra finch, eight genes in these pathways show evidence of positive selection in falcons (Supplementary Fig. 10), among which TGFβIIr has been shown to be functional in shaping avian beak morphology (Table 2). Although it has been shown that possible errors in multiple-sequence alignment have only a limited impact on estimates of Ka/Ks ratios at the genome level (Supplementary Fig. 11), we caution that the analysis of positive selection remains sensitive to the quality of the multiple-sequence alignments of separate orthologous genes and can be further complicated by variance in sequence divergence among them. Analysis of microRNA target sites provided further support for the presence of novel regulatory mechanisms in falcons (Table 2, Supplementary Fig. 10 and Supplementary Table 25).
Peregrines and sakers are parapatric breeding species, with the latter confined to the Palearctic and primarily inhabiting arid environments (Supplementary Fig. 6)16,23. Consequently, it is probable that sakers require greater maintenance of osmotic equilibrium and suffer heat stress more than peregrines. We compared the genes from two kidney-expressed KEGG pathways and other related genes involved in homeostasis in the two species. Rab11a and GNAS have major contributions to the V2R water conservation pathway17,24, and both have two more copies in saker than in peregrine (Supplementary Table 26).
The aldosterone-regulated sodium reabsorption pathway has a major role in determining sodium levels, and the activation of protein kinase C (PKC) inhibits sodium uptake25. We found that the saker has three more functional copies of PKCβ than peregrine. In saker, we also found the exonization of trpv1, a gene involved in promoting thermoregulatory cooling by stimulating sweat production and preemptive renal water reabsorption through the release of vasopressin (antidiuretic hormone)26 (Supplementary Fig. 12). Supporting evidence for water conservation in sakers comes from the observation that sakers secrete more sodium and chloride in their urine than do peregrines27. These results, when taken together, suggest a genetic basis by which sakers cope with desert and steppe habitats and heat stress. The functional expression of these genes related to water conservation and sodium secretion warrants future research.
The genome sequencing data presented here provide a resource for the future examination of evolution and adaptation in birds and in raptors in particular. Both falcon species are widely distributed across the globe and show a wide variety of local phenotypes and behaviors, and their conservation status varies substantially across their habitat ranges, with the saker falcon being globally classified as vulnerable. Both species have migratory as well as sedentary populations, and understanding the genetic basis of this wide diversity could provide a valuable tool to aid their long-term conservation.
Methods
Sampling and DNA extraction.
Approximately 2 ml of blood each was taken from one male saker and one male peregrine that were caught in the wild at the Abu Dhabi Falcon Hospital. Genomic DNA was extracted using gravity-flow, anion-exchange tips and buffers (Qiagen Blood & Cell Culture DNA Maxi Kit). Sex was determined using morphology and molecular assays (Supplementary Note).
Genome sequencing and assembly.
Sequencing was carried out using an Illumina HiSeq 2000. Paired-end libraries with insert sizes of 170, 500 and 800 bp (short inserts) and 5, 10 and 20 kb (long inserts) were constructed. A detailed description of library construction, sequencing and assembly, genome size estimation, annotation, gene prediction (homology, de novo and RNA-seq) and functional annotation, and RNA (mRNA and microRNA) sequencing is included in the Supplementary Note. Genome assembly quality was assessed using GC content (Supplementary Figs. 13 and 14) and Sanger sequencing of fosmid libraries (Supplementary Note). The lengths of the repetitive DNA sequences identified are listed in Supplementary Table 27.
Comparative genome analysis.
We employed the widely used program TreeFam3 to define orthologous genes (Supplementary Note) because it has been inferred that phylogeny-based software is more robust in inferring orthologs3. The analysis using LASTZ (see URLs) and MCscan30 in chicken, peregrine and saker showed that approximately 88.9% of TreeFam orthologs were supported by at least one synteny analysis, implying high quality for the orthologs defined (Supplementary Table 28 and Supplementary Note). On the basis of concatenated 4-fold degenerate sites from 6,267 single-copy orthologs, a maximum-likelihood phylogeny for the falcons, zebra finch, chicken and turkey genomes was constructed using PhyML31. Ortholog sequences were aligned using MUSCLE (see URLs) with the protein alignment as a guide. The optimal evolutionary model was GTR + I + G, selected using MODELTEST3.7 (ref. 32). The Bayesian relaxed molecular clock approach was used to estimate species divergence time using MCMCTREE in PAML33, based on the degenerate sites data set used above.
Whole-genome synteny analysis was performed for the five bird species. Pairwise alignments were first produced using LASTZ. ChainNet was then applied to merge traditional alignments into larger structures. Scaffolds of <5 kb in size were filtered out to avoid multiple hits for small scaffolds.
To detect segmental duplications, an alignment was generated using LASTZ with the parameters T = 2 (no transition), C (chain) = 2, H (inner) = 2,000, Y (ydrop) = 3,400, L (gappedthresh) = 6,000 and K (hspthresh) = 2,200. Before aligning, with repeat sequences masked, the genome assembly was split into 100 subfiles. The maximum simultaneous gap allowed was 100 bp. After aligning, the blocks obtained were conjoined to obtain larger blocks (chains). We unmasked the repeat sequences in the chains and then selected those with length of >500 bp and identity of >85% and aligned the chains using LASTZ again. Finally, we extracted the aligned chains with length of >1 kb and identity of >90%, considering these predictions to be segmental duplications. After removing the overlapping fragments, we obtained a nonredundant set. Because previous research34 has shown that highly similar alignments may represent allelic overlaps missed during the assembly process, we removed segmental duplications that were ≥98% identical. Furthermore, to examine the quality of the detected segmental duplications, the sequencing depth distributions of segmental duplications and regions without segmental duplications were compared for peregrine and saker, respectively (Supplementary Fig. 15).
For the assessment of lineage-specific indels, MULTIZ (see URLs) was used to integrate all the LASTZ alignments from the synteny analysis to obtain conserved elements among the genomes from the two falcons, zebra finch and chicken. For blocks longer than 1 kb, we identified species-specific short indels using the aligned data. Indels located within 50 bp of the end of the block and pairs of indels with a distance less than 50 bp were filtered out.
Avian olfactory receptor genes.
The olfactory receptor genes of the five avian species and of green anole, human, cow and dog were annotated using GenBank, Ensembl and published data8 (Supplementary Note). Intact avian olfactory receptor genes were aligned using SATé-II (ref. 35), from which a neighbor-joining tree was constructed using MEGA5.03 (ref. 36), with the Poisson model chosen as the substitution model as previously analyzed8. The reliability of the phylogenetic tree was evaluated with 1,000 bootstrap replicates.
Genome evolution analysis.
TreeFam was used to help define both single-copy and multiple-copy orthologs for the five avian species (Supplementary Fig. 16). For multiple-copy orthologs, we used single-copy genes in our outgroup (green anole) as a query and selected the best hits in the five birds as the representatives of the ortholog. Similar sequence depth was found for multiple-copy and single-copy genes (Supplementary Fig. 17), suggesting that gene duplications are unlikely to have been misidentified as alleles of the same locus. The two orthologous gene sets were then used for ω ratio calculations as follows.
First, on the basis of the concatenated ortholgs, branch-specific ω values for each avian species were calculated using codeml in PAML (Supplementary Note). Alignment quality is of major importance for ω estimation because errors can lead to the misidentification of synonymous sites as nonsynonymous sites37. To minimize the effect of alignment errors, two algorithms, SATé-II and DIALIGN-TX38, were chosen to align each ortholog, as they have been reported to be robust in dealing with global and local alignments, respectively (Supplementary Note). To further examine the influence of alignment quality on the ω analysis, we randomly selected 421 SATé-aligned orthologs and manually corrected them. All automatic alignment methods and manual corrections produced similar results (Supplementary Fig. 4). Therefore, for the following analysis, we used SATé, except for alignments involving too few taxa (≤3), where we used DIALIGN instead.
Second, PAML was used to calculate ω values for each ortholog, and comparisons were made of the ω distributions of all orthologs between the falcons and other species pairs (chicken and turkey, zebra finch and chicken, zebra finch and turkey, peregrine and zebra finch, and saker and zebra finch).
Third, we examined both rapidly and slowly evolving gene categories by comparing the two falcon genomes with the two galliformes genomes, zebra finch and chicken, and zebra finch and turkey. We calculated the median ω values for each gene ontology10 functional category containing at least ten genes found in the saker-peregrine lineage.
Assessment of the possible influence of sequencing errors indicated that they can have little influence on our ω calculations (Supplementary Fig. 11). Detailed information for the above analyses is provided in the Supplementary Note.
Gene gain and loss.
CAFE based on a random gene birth and death model11 was used to study gene gain and loss in gene families across our reconstructed phylogenetic tree with six species (Supplementary Fig. 18). The robustness of CAFE was tested by removing one species each time. The results showed that, independent of the species removed, falcons always featured the most rapid gene loss of the studied avian species (Supplementary Fig. 19), although we note that the avian species studied here are more divergent than mammals11. To further explore gain and loss between each falcon and its closest genome-enabled relative, zebra finch, we used protein clustering12 to explore gene number variation for the same protein families (Supplementary Note). Filtering was applied to clean up possible overestimated gene expansion in the zebra finch gene set. However, the patterns of gene turnover in the falcon species were similar with (Fig. 2b) and without (Supplementary Fig. 20) this filtering.
SNP calling and demographic history reconstruction.
We use SOAPsnp39 to detect SNPs between diploid chromosomes of both falcons (Supplementary Note). The SNP distribution was observed across each genome. To assess whether SNP quality scores or sequencing depth influenced SNP distribution, we compared the results achieved with varied quality scores (20 or 40) and different depth ranges (Supplementary Fig. 21). By comparison with the zebra finch genome, the autosomal mutation rate was estimated to be 1.65 × 10−9 mutations per year for both falcons. Generation time was inferred to be 6.0 and 6.6 years for peregrine and saker, respectively (Supplementary Note). The demographic history of each falcon was reconstructed using the PSMC model15 (Supplementary Note).
Beak development.
Genes in three signaling pathways (TGF-β, WNT and FGF) and 98 others (Supplementary Table 29) were analyzed in chicken, zebra finch, peregrine and saker. We examined (i) gene copy number variation (CNV); (ii) structural variations between orthologous genes; (iii) positively selected genes in falcons; and (iv) predicted microRNA target sites, identified on the basis of chicken embryonic microRNAs. Detailed methods are presented in the Supplementary Note.
Water conservation in the saker and peregrine.
Because the kidney is the most important organ to reabsorb water and secrete sodium, we focused on analyzing genes in the two kidney-related pathways in KEGG (V2R and aldosterone) and other genes involved in this process (Supplementary Note). Adopting the same method described for beak analysis, we examined CNVs and structural variations between the two falcon species.
Statistical analysis.
Differences in average ω values between falcons and other birds were tested using the Mann-Whitney test implemented in PAST40. In the LRT tests, P values were tested using the χ2 statistic adjusted by the FDR41 (false discovery rate) method (q < 0.05) to allow for multiple testing.
URLs.
LASTZ and MULTIZ, http://www.bx.psu.edu/miller_lab/; MUSCLE, http://www.drive5.com/muscle; FGF signaling pathways, http://www.sabiosciences.com/pathway.php?sn=FGF_Signaling.
Accession codes.
Assemblies and gene annotations of the peregrine and saker falcon genomes have been deposited at GenBank (BioProjects PRJNA159791 and PRJNA168071). Raw DNA and RNA sequencing reads have been submitted to the NCBI Short Read Archive database (SRA054256, SRX225198, SRX225199).
Accession codes
Primary accessions
NCBI Reference Sequence
Sequence Read Archive
References
Kim, E.B. et al. Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 479, 223–227 (2011).
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010).
Li, H. et al. TreeFam: a curated database of phylogenetic trees of animal gene families. Nucleic Acids Res. 34, D572–D580 (2006).
Hackett, S.J. et al. A phylogenomic study of birds reveals their evolutionary history. Science 320, 1763–1768 (2008).
Roper, T.J. Olfaction in birds. Adv. Stud. Behav. 28, 247–332 (1999).
Steiger, S.S., Fidler, A.E., Valcu, M. & Kempenaers, B. Avian olfactory receptor gene repertoires: evidence for a well-developed sense of smell in birds? Proc. R. Soc. Lond. B 275, 2309–2317 (2008).
Bang, B.G. Anatomical evidence for olfactory function in some species of birds. Nature 188, 547–549 (1960).
Steiger, S.S., Kuryshev, V.Y., Stensmyr, M.C., Kempenaers, B. & Mueller, J.C. A comparison of reptilian and avian olfactory receptor gene repertoires: species-specific expansion of group γ genes in birds. BMC Genomics 10, 446 (2009).
Hugall, A.F. & Stuart-Fox, D. Accelerated speciation in colour-polymorphic birds. Nature 485, 631–634 (2012).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Hahn, M.W., Demuth, J.P. & Han, S.G. Accelerated rate of gene gain and loss in primates. Genetics 177, 1941–1949 (2007).
Dalloul, R.A. et al. Multi-platform next-generation sequencing of the domestic turkey (Meleagris gallopavo): genome assembly and analysis. PLoS Biol. 8, e1000475 (2010).
Wong, G.K. et al. A genetic variation map for chicken with 2.8 million single-nucleotide polymorphisms. Nature 432, 717–722 (2004).
Warren, W.C. et al. The genome of a songbird. Nature 464, 757–762 (2010).
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011).
Nittinger, F., Haring, E., Pinsker, W., Wink, M. & Gamauf, A. Out of Africa? Phylogenetic relationships between Falco biarmicus and the other hierofalcons (Aves: Falconidae). J. Zoological Syst. Evol. Res. 43, 321–331 (2005).
Kanehisa, M., Goto, S., Furumichi, M., Tanabe, M. & Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 38, D355–D360 (2010).
Abzhanov, A., Protas, M., Grant, B.R., Grant, P.R. & Tabin, C.J. Bmp4 and morphological variation of beaks in Darwin's finches. Science 305, 1462–1465 (2004).
Sorek, R. The birth of new exons: mechanisms and evolutionary consequences. RNA 13, 1603–1608 (2007).
Brugmann, S.A. et al. Comparative gene expression analysis of avian embryonic facial structures reveals new candidates for human craniofacial disorders. Hum. Mol. Genet. 19, 920–930 (2010).
Menezes, R.X., Boetzer, M., Sieswerda, M., van Ommen, G.B. & Boer, J.M. Integrated analysis of DNA copy number and gene expression microarray data using gene sets. BMC Bioinformatics 10, 203 (2009).
Perry, G.H. et al. Diet and evolution of human amylase gene copy number variation. Nat. Genet. 39, 1256–1260 (2007).
del Hoyo, J., Elliot, A. & Sargatal, J. Handbook of the Birds of the World, Vol. 2 (Lynx Edicions, Barcelona, Spain, 1994).
Tajika, Y. et al. Differential regulation of AQP2 trafficking in endosomes by microtubules and actin filaments. Histochem. Cell Biol. 124, 1–12 (2005).
Hays, S.R., Baum, M. & Kokko, J.P. Effects of protein kinase C activation on sodium, potassium, chloride, and total CO2 transport in the rabbit cortical collecting tubule. J. Clin. Invest. 80, 1561–1570 (1987).
Sharif -Naeini, R., Ciura, S. & Bourque, C.W. TRPV1 gene required for thermosensory transduction and anticipatory secretion from vasopressin neurons during hyperthermia. Neuron 58, 179–185 (2008).
Cade, T.J. & Greenwald, L. Nasal salt secretion in falconiform birds. Condor 68, 338–350 (1966).
Abzhanov, A. et al. The calmodulin pathway and evolution of elongated beak morphology in Darwin's finches. Nature 442, 563–567 (2006).
Mallarino, R. et al. Two developmental modules establish 3D beak-shape variation in Darwin's finches. Proc. Natl. Acad. Sci. USA 108, 4057–4062 (2011).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488 (2008).
Guindon, S. & Gascuel, O. A simple, fast and accurate method to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003).
Posada, D. & Crandall, K.A. Modeltest: testing the model of DNA substitution. Bioinformatics 14, 817–818 (1998).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Bailey, J.A., Yavor, A.M., Massa, H.F., Trask, B.J. & Eichler, E.E. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 11, 1005–1017 (2001).
Liu, K. et al. SATé-II: very fast and accurate simultaneous estimation of multiple sequence alignments and phylogenetic trees. Syst. Biol. 61, 90–106 (2012).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Schneider, A. et al. Estimates of positive Darwinian selection are inflated by errors in sequencing, annotation, and alignment. Genome Biol. Evol. 1, 114–118 (2009).
Subramanian, A.R., Kaufmann, M. & Morgenstern, B. DIALIGN-TX: greedy and progressive approaches for segment-based multiple sequence alignment. Algorithms Mol. Biol. 3, 6 (2008).
Li, R. et al. SNP detection for massively parallel whole-genome resequencing. Genome Res. 19, 1124–1132 (2009).
Hammer, Ø., Harper, D.A.T. & Ryan, P.D. PAST: paleontological statistics software package for education and data analysis. Palaeontol. Electronica 4, 9 (2001).
Benjamini, Y. & Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29, 1165–1188 (2001).
Acknowledgements
This work was supported by the EAAD (Environment Agency of Abu Dhabi). We thank H.E. Mohammed Al Bowardi for his support. We also thank P. Orozco-terWengel and X. Fang for their critical comments on the manuscript, A. Subramanian for suggestions on analysis with DIALIGN-TX, S. Mirarab for suggestions on SATé-II alignment and E. Alm and B.J. Shapiro for advice on Ka/Ks normalization. We acknowledge staff at International Wildlife Consultants, EAAD and BGI-Shenzhen who helped conduct this study.
Author information
Authors and Affiliations
Contributions
M.W.B. led the UK team. Jun Wang and Junyi Wang led the BGI team. M.W.B., X.Z., A.D. and N.C.F. designed the study. M.G.M., A.D. and X.Z. sampled the falcons. Q.L., S.G., C.C., P.N., Y.L., S.L., Z.Y., G.L., Z.W. and Y.Y. performed genome sequencing and assembly. X.Z. and Yuanping Chen extracted the DNA and RNA. X.Z., S.P., J.H., L.H., H.H., J.X., P.X. and Ying Chen analyzed the data. X.Z. and M.W.B. interpreted the results and wrote the manuscript. All authors read and provided input for the manuscript and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–21, Supplementary Tables 1–29 and Supplementary Note (PDF 3185 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article
Cite this article
Zhan, X., Pan, S., Wang, J. et al. Peregrine and saker falcon genome sequences provide insights into evolution of a predatory lifestyle. Nat Genet 45, 563–566 (2013). https://doi.org/10.1038/ng.2588
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.2588
This article is cited by
-
Phylogenomic insights into the polyphyletic nature of Altai falcons within eastern sakers (Falco cherrug) and the origins of gyrfalcons (Falco rusticolus)
Scientific Reports (2023)
-
Genomic bases underlying the adaptive radiation of core landbirds
BMC Ecology and Evolution (2021)
-
Climate-driven flyway changes and memory-based long-distance migration
Nature (2021)
-
Status of the saker falcon in China
Science China Life Sciences (2021)
-
Signature of adaptive evolution in olfactory receptor genes in Cory’s Shearwater supports molecular basis for smell in procellariiform seabirds
Scientific Reports (2020)
