
Abstract
How an insect evolves to become a successful herbivore is of profound biological and practical importance. Herbivores are often adapted to feed on a specific group of evolutionarily and biochemically related host plants1, but the genetic and molecular bases for adaptation to plant defense compounds remain poorly understood2. We report the first whole-genome sequence of a basal lepidopteran species, Plutella xylostella, which contains 18,071 protein-coding and 1,412 unique genes with an expansion of gene families associated with perception and the detoxification of plant defense compounds. A recent expansion of retrotransposons near detoxification-related genes and a wider system used in the metabolism of plant defense compounds are shown to also be involved in the development of insecticide resistance. This work shows the genetic and molecular bases for the evolutionary success of this worldwide herbivore and offers wider insights into insect adaptation to plant feeding, as well as opening avenues for more sustainable pest management.
Similar content being viewed by others


Main
The global pest P. xylostella (Lepidoptera: Yponomeutidae) is thought to have coevolved with the crucifer plant family3 (Supplementary Fig. 1) and has become the most destructive pest of economically important food crops, including rapeseed, cauliflower and cabbage4. Recently, the total cost of damage and management worldwide was estimated at $4–5 billion per annum5,6. This insect is the first species to have evolved resistance to dichlorodiphenyltrichloroethane (DDT) in the 1950s7 and to Bacillus thuringiensis (Bt) toxins in the 1990s8 and has developed resistance to all classes of insecticide, making it increasingly difficult to control9,10. P. xylostella provides an exceptional system for understanding the genetic and molecular bases of how insect herbivores cope with the broad range of plant defenses and chemicals encountered in the environment (Supplementary Fig. 2).
We used a P. xylostella strain (Fuzhou-S) collected from a field in Fuzhou in southeastern China (26.08 °N, 119.28 °E) for sequencing (Supplementary Fig. 1). Whole-genome shotgun–based Illumina sequencing of single individuals (Supplementary Table 1), even after ten generations of laboratory inbreeding, resulted in a poor initial assembly (N50 = 2.4 kb), owing to high levels of heterozygosity (Supplementary Figs. 3 and 4 and Supplementary Table 2). Subsequently, we sequenced 100,800 fosmid clones (comprising ∼10× the genome length) to a depth of 200× (Supplementary Fig. 5 and Supplementary Tables 3–5), assembling the resulting sequence data into 1,819 scaffolds, with an N50 of 737 kb, spanning ∼394 Mb of the genome sequence (version 1; Supplementary Fig. 6 and Supplementary Table 6). The assembly covered 85.5% of a set of protein-coding ESTs (Supplementary Tables 7 and 8) generated by transcriptome sequencing11. Alignment of a subject scaffold against a 126-kb BAC (GenBank GU058050) from an alternative strain (Geneva 88) showed extensive structural variations between haplotypes. However, the coding sequence of the nicotinic acetylcholine receptor α6 gene (spanning >75 kb)12 on the BAC and the subject scaffold was relatively conserved (Supplementary Fig. 7). Whole-genome shotgun reads from three libraries (500 bp, 5 kb and 10 kb) were mapped to the BAC and corresponding scaffold, covering 86.7% and 98.1% of sites, respectively (Supplementary Fig. 7), indicating high polymorphism levels between the alleles. Genome-wide exploration of variation identified abundant SNPs, insertions and/or deletions (indels), structural variations and complex segmental duplication patterns within the sequenced population of the Fuzhou-S strain (Fig. 1, Supplementary Figs. 8 and 9, Supplementary Tables 9–13 and Supplementary Note). Thus, we generated a genome of ∼343 Mb (version 2) for annotation and analysis by masking ∼50 Mb of possible allelic redundancy in the version 1 assembly (Supplementary Fig. 10, Supplementary Table 14 and Supplementary Note).

The outermost circle shows the reference genome assembly with a 100-kb unit scale. Scaffolds that could be assigned to linkage groups are joined in arbitrary order to generate the partial sequences of 28 chromosomes (detailed in the Supplementary Note). The green segment represents the scaffolds that were unable to be assigned (Un). The innermost circle denotes segmental duplications (of ≥8 kb), with connections shown between segment origins and duplication locations. Segmental duplication pairs with 100% similarity are shown in red, and those with ≥90% similarity are shown in blue. Histograms indicate the number of SNPs (red, outer circle) and indels (light green, inner circle) in 30-kb and 50-kb windows, respectively.
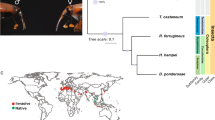
The P. xylostella genome is predicted to contain 18,071 protein-coding genes (Supplementary Fig. 11 and Supplementary Tables 15–18) and 781 non-coding RNAs (Supplementary Table 19), with 33.97% of the genome made up of repetitive sequences (Supplementary Fig. 12, Supplementary Table 20 and Supplementary Note). Compared with the genomes of other sequenced insect species, the P. xylostella genome possesses a relatively larger set of genes and a moderate number of gene families (Supplementary Table 21), suggesting the expansion of certain gene families. In addition to 1,683 Lepidoptera-specific genes (Supplementary Table 22 and Supplementary Note), we found 1,412 P. xylostella–specific genes (Supplementary Fig. 13), exceeding in number the 463 Bombyx mori–specific genes13 and the 1,184 Danaus plexippus–specific genes14 (Fig. 2). The P. xylostella–specific genes were largely involved in biological pathways essential for environmental information processing, chromosomal replication and/or repair, transcriptional regulation and carbohydrate and protein metabolism (Supplementary Fig. 14 and Supplementary Table 23). These findings suggest that P. xylostella has an intrinsic capacity to swiftly respond to environmental stress and genetic damage.

The red dots (for calibration) represent the divergence time (295.4–238.5 million years ago) of Drosophila melanogaster and Culicidae and the divergence time (307.2–238.5 million years ago) of D. melanogaster and Apis mellifera, which are based on fossil evidence. The Arachnida, Tetranychus urticae, was used as an outgroup, and a bootstrap value was set as 1,000. 1:1:1 orthologs include the common orthologs with the same number of copies in different species, N:N:N orthologs include the common orthologs with different copy numbers in the different species, patchy orthologs include the orthologs existing in at least one species of vertebrates and insects, other orthologs include the unclassified orthologs, and unclustered gene include the genes that cannot be clustered into known gene families.
Phylogenetic analysis indicated that the estimated divergence time of insect orders was approximately 265–332 million years ago (Fig. 2). This is around the time of the divergence of mono- and dicotyledonous plants (∼304 million years ago)15, consistent with the coevolution and concurrent diversification of insect herbivores and their host plants. It can be predicted that P. xylostella became a cruciferous specialist when Cruciferae diverged from Caricaceae (∼54–90 million years ago)16. This estimated time provides additional evidence to support our estimation of the divergence time (∼124 million years ago) of P. xylostella from two other Lepidoptera, B. mori and D. plexippus (Fig. 2). The genome-based phylogeny showed that P. xylostella is a basal lepidopteran species (Fig. 2), and this idea is well supported by its modal karyotype of n = 31 (refs. 17,18) and the molecular phylogeny of Lepidoptera19,20, indicating the importance of P. xylostella in the history of lepidopteran evolution.
On the basis of P. xylostella transcriptome data11, we identified 354 preferentially expressed genes in larvae (Supplementary Fig. 15), and a set of these genes is involved in sulfate metabolism, some of which were validated using quantitative RT-PCR for gene expression analysis (Supplementary Figs. 16–18, Supplementary Table 24 and Supplementary Note). Glucosinolate sulfatase (GSSs) enables P. xylostella to feed on a broad range of cruciferous plants by catalyzing the conversion of glucosinolate defense compounds into desulfoglucosinolates, thus preventing the formation of toxic hydrolysis products3 (Supplementary Fig. 2). In order to function, all sulfatases require post-translational modification by sulfatase-modifying factor 1 (encoded by SUMF1)21, which regulates the sulfatase whose higher activities depend on greater amounts of sulfatase and SUMF1 transcripts22. We found that high expression of P. xylostella SUMF1 in third-instar larvae was coupled with significantly higher expression of the two GSS genes relative to other members of the P. xylostella sulfatase gene family (Fig. 3). We propose that the coevolution of SUMF1 and GSS genes was key in P. xylostella becoming such a successful herbivore of cruciferous plants (Supplementary Fig. 2). Furthermore, a new gene, predicted to be a sodium-independent sulfate anion transporter, was highly expressed in all larval stages and in the midgut (Fig. 4) and is likely associated with the excretion of toxic sulfates23.

Phylogenetic tree and tandem duplication of the sulfatase gene families, including two GSSs, are shown. Gene expression levels are scaled using fragments per kilobase of transcript per million fragments mapped (FPKM) values, and circle sizes vary according to the levels of expression. The two GSSs and SUMF1 are highly coexpressed but are not expressed with the pseudo-SUMF1 (truncated) gene.

Expression values measured in reads per kilobase of exon per million mapped sequence reads (RPKM) are plotted, and the most significant BLASTX results against the NCBI nr database (E value ≤ 1 × 10−10) of the predicted protein-coding genes are shown. SNMP, sensory neuron membrane protein; UGT, UDP glucosyltransferase; AOX, aldehyde oxidase; CAT, catalase; CSP, chemosensory protein; POD, peroxidase; SUC, β-fructofiranosidase; β-GLU, β -glucosidase; SUT, sodium-independent sulfate anion transporter; SPT, sodium-dependent phosphate transporter; OBP, odorant binding protein; P450, cytochrome P450 monooxygenase; GST, glutathione S-transferase; COE, carboxylesterase; GOX, glucose oxidase; SUMF, sulfatase modifying factor; GSS, glucosinolate sulfatase.
In comparisons with the larval midgut proteome of the polyphagous lepidopteran Helicoverpa armigera24, we found similar digestive enzymes encoded by P. xylostella larval preferentially expressed genes that were expressed predominantly in the midgut (Supplementary Fig. 19 and Supplementary Table 25). The abundant larval midgut-specific serine proteinase genes in the P. xylostella genome may circumvent the action of insecticidal plant protease inhibitors through differential expression in response to different plant hosts25 (Supplementary Fig. 20). Among the P. xylostella larval preferentially expressed genes, we identified a set of genes, including GOX (encoding glucose oxidase), related to the host range of herbivores26 and involved in the perception of chemical signals from host plants and defense against secondary plant compounds (Fig. 4, Supplementary Table 25 and Supplementary Note), suggesting the presence of a complex chemoreception network and multiple detoxification mechanisms.
We identified five chemoreception gene families related to larval feeding preferences and adult searching for host plants: odorant receptors (ORs), odorant-binding proteins (OBPs), gustatory receptors (GRs), ionotropic receptors (IRs) and chemosensory proteins (CSPs) (Supplementary Fig. 21, Supplementary Table 26 and Supplementary Note). Notable among these genes is an expansion of ORs but not GRs, as reported in the B. mori genome27. Species-specific expansion of CSPs in moths is less than that observed in butterflies18. Lifecycle- and tissue-specific expression of ORs identified 30 variable, 23 constitutive and 9 adult-specific expression patterns (Supplementary Fig. 22), indicating that P. xylostella possesses a high potential for adaptation to chemical cues from host plants (Supplementary Fig. 2).
Detoxification pathways used by insect herbivores against plant defense compounds may be co-opted for insecticide tolerance28 or resistance (Supplementary Fig. 2). We found that P. xylostella possessed an overall larger set of insecticide resistance–related genes than B. mori, which is monophagous and has had little exposure to insecticide over 5,000 years of domestication13 (Supplementary Table 27). We identified in the P. xylostella genome apparent gene duplications of most ATP-binding cassette (ABC) transporter families and three classes of major metabolic enzymes, the P450 monooxygenases (P450s), glutathione S-transferases (GSTs) and carboxylesterase (COEs) (Supplementary Fig. 23 and Supplementary Table 26). These genes are known to have important roles in xenobiotic detoxification in insects29,30 (Supplementary Note). Among the four gene families, the ABC transporter gene family in P. xylostella is much more expanded compared to the corresponding family in B. mori (Fig. 5a). Larval transcriptomes were sequenced from the Fuzhou-S strain that was genotyped and from two substrains selected for resistance to chlorpyrifos or fipronil11. ABC transporter genes were upregulated more frequently than GSTs, COEs or P450s in insecticide-resistant larvae (Supplementary Fig. 24), highlighting the potential role of ABC transporters in detoxification.

(a) Neighbor-joining tree showing expansions of ABC transporter genes in P. xylostella (yellow) and B. mori (fuchsia). The arthropod-specific ABCH clade is highlighted by the red background. (b) Principal-component analysis for the average SNPs, indels, structural variations (SVs) and transposable elements (TEs) in the gene families of ABC transporters, P450s, GSTs and COEs. The first two components represent 82.2% of the accumulated information on variations, and the red circle encompasses closely associated variables. CDS, coding sequence. (c) Percentages of transposable element orders within or around the gene families in the genomes of P. xylostella and B. mori. The numbers of transposable elements per gene family are shown above. (d) Box plots of the estimated expansion times of LTR transposable elements for the two species. The dashed lines represent up to 1.5 times the interquartile range33. MYA, million years ago.
We then investigated the genomic variations and transposable elements in genes and their 2-kb upstream regions in these four families, some of which were validated using Sanger sequencing (Supplementary Tables 28–31 and Supplementary Note). On average, transposable elements (∼20 per gene) were abundant, followed in frequency by structural variations (∼16), SNPs (∼6) and indels (<1), near these gene families (Supplementary Fig. 25). The coding sequences of COEs were rich in SNPs (Supplementary Fig. 25a), which can be critical in determining COE substrate specificity and catalytic activity under xenobiotic stresses31. Principal-component analysis indicated that intronic regions consistently harbored all types of variations, whereas structural variations and transposable elements frequently occurred in coding sequences, which may largely affect gene functions (Fig. 5b). Transposable elements were abundant within or near the P450s involved in induced xenobiotic detoxification in insects, whereas those related to constitutive developmental metabolism were free of transposable element insertions32. Our findings show that numerous transposable elements accompany the gene families involved in metabolic detoxification sensitive to external stresses (Supplementary Table 32). These associations seem to be a consistent trend in Lepidoptera (Supplementary Fig. 25b). The transposable element orders of long terminal repeat (LTR) and long interspersed nuclear element (LINE) were predominant in P. xylostella and B. mori, respectively, and the proportional composition of various transposable element orders tended to be similar in different gene families for each of the species (Fig. 5c). A recent expansion of the LTR retrotransposons (>90%) in the P. xylostella genome has occurred over the past 2 million years, occurring much later than the expansion of B. mori LTRs (Fig. 5d) and possibly reflecting the timing of extensive adaptive evolutionary events in P. xylostella33. The polymorphism within the P. xylostella genome might support adaptation to host plant defenses and insecticides by providing a repertoire of alternative alleles or cis-regulatory elements29 and genetic variations34 for gene expression.
In this project, we developed a new approach for non-model insect genome sequencing using next-generation sequencing technology and de novo assembly of the highly polymorphic genome. Analyses identify complex patterns of heterozygosity, the expansion of gene families associated with perception and the detoxification of plant defense compounds and the recent expansion of retrotransposons near detoxification genes. These adaptations reflect the diversity and ubiquity of toxins in its host plants and underlie the capacity of P. xylostella to rapidly develop insecticide resistance. This study provides insights into the genetic plasticity of P. xylostella that underlies its success as a worldwide herbivore. The genomic resources described here will facilitate future studies on the adaptation and evolution of other arthropods and support the incorporation of molecular information into the development of strategies for more sustainable agriculture.
URLs.
FTP site, ftp://ftp.genomics.org.cn/pub/Plutellaxylostella/; LASTZ, http://www.bx.psu.edu/miller_lab/dist/README.lastz-1.02.00/README.lastz-1.02.00a.html; Infonet Biovision, http://www.infonet-biovision.org/; North American Moth Photographers Group, http://mothphotographersgroup.msstate.edu/MainMenu.shtml; Interactive Agricultural Ecological Atlas of Russia and Neighboring Countries, http://www.agroatlas.ru/; the diamondback moth (DBM) genome database, http://iae.fafu.edu.cn/DBM.
Methods
Strain for sequencing.
A strain of the diamondback moth (DBM) (Fuzhou-S), P. xylostella, was reared on radish seedlings without exposure to insecticides for 5 years, spanning at least 100 generations. An inbred line was developed by successive single-pair sibling matings. Male pupae were used for genome sequencing.
Whole-genome shotgun sequencing and assembly.
Individual DNA from the inbred F1, F4 and F10 insects was used for construction of paired-end libraries (Supplementary Table 1). Sequencing was performed using the Illumina Genome Analyzer IIx or HiSeq 2000 platform. Short reads were assembled using SOAPdenovo35.
Fosmid-to-fosmid sequencing and assembly.
DNA was extracted from a pool of ∼1,000 male pupae using a CATB-based method. A fosmid library with insert sizes ranging from 35 to 40 kb was constructed. We sequenced 100,800 single colonies to achieve 10× coverage of the genome. For each colony, two paired-end libraries with 250-bp and 500-bp fragments were constructed and sequenced. On average, each library was sequenced >200× with a total of 114 lanes and an output of 855 Gb. Vector or contaminated DNA and poor reads with >10% unknown nucleotides or >40 bases with quality value of ≤5 were filtered out36.
Genome assembly.
We developed custom software (Rabbit) for assembling sequences with large overlaps (>2 kb). Rabbit contains three modules: Relation Finder, Overlapper and Redundancy Remover.
We used the Poisson-based K-mer model to determine repeat sequences, segmental duplications or divergent haplotypes. Each K-mer was defined as either a 'repeat' or 'unique' K-mer, depending on whether its occurrence frequency was greater or less than twice the average frequency, respectively (Supplementary Fig. 10), using the Poisson model

where λ is the expected frequency for K-mers, y is the given frequency of a particular K-mer and P is the occurrence probability of a given K-mer frequency. Therefore, the probability of a unique K-mer being greater than twice the expected frequency is given by the following equation.

Few unique K-mers can occur with a frequency larger than twice the expected value, especially when the expected frequency is ≥20 (Supplementary Table 14). Rabbit is capable of connecting these unique regions and removing redundancy. We chose K = 17 bp36,37 and trimmed repeat sequence ends (Supplementary Fig. 4).
We used SSPACE38 to build scaffolds and SOAP-GapCloser35 to fill the gap with 131.2× whole-genome shotgun short reads (Supplementary Table 1). This resulted in a genome with 394 Mb (version 1), slightly larger than the estimated haploid genome size (339.4 Mb)17. We extracted all similar sequences with LAST39 and retained one copy of the sequences containing >40% unique K-mers and masked the others with 'n' to generate a revised genome of ∼343 Mb (version 2).
Digital gene expression (DGE).
Quantitative RNA-seq was conducted for newly laid eggs, fourth-instar larvae, the midguts of fourth-instar larvae, pupae (>2 d), virgin male and female adults, and the heads of fourth-instar larvae and male or female adults. Paired-end libraries (insert size of 200 bp) were sequenced with read length of 49 bp. The RPKM40 values were calculated for DGE profiling.
Larval preferentially expressed gene analysis.
On the basis of the DBM genome and the transcriptomes for newly laid eggs, third-instar larvae, pupae and virgin adults, we analyzed differential gene expressions in four developmental stages using the same statistical approach11. The larval preferentially expressed genes were defined as genes that were highly expressed in the larval stage compared to the other three developmental stages, with RPKM ratio ≥ 8 fold (upregulated) and false discovery rate (FDR) ≤ 0.001.
Gene prediction.
We used Augustus (v 2.5.5)41, Genscan42 and SNAP43 for de novo gene prediction, compared the candidate genes to the transposable element protein database using BLASTP (1 × 10−5) and removed genes that showed over 50% similarity to the transposable elements. The predicted proteomes of D. melanogaster, B. mori, Anopheles gambiae and Tribolium castaneum were aligned with the DBM genome using TBLASTN (1 × 10−5). High-scoring segment pairs (HSPs) were grouped using Solar (v. 0.9.6)36. We extracted target gene fragments and extended 500 bp at both ends. GeneWise (v. 2.2.0)44 was used for the alignment of fragments to a protein set. We clustered the predicted genes with an overlap cutoff of >50 bp. The results of de novo and homolog-based predictions were incorporated into a gene set using GLEAN45.
Integration of transcriptome data with the GLEAN set.
Transcriptome reads11 were mapped onto the genome using TopHat46. We then used Cufflinks47 (with default parameters) to assemble transcripts and integrated the transcripts with the GLEAN set by filtering out redundancy and the genes with ≥10% uncertain bases and coding region lengths of ≤150 bp.
Functional annotation.
The integrated gene set was translated into amino-acid sequences, which were used to search the InterPro database48 by Iprscan (v 4.7)49. We used BLAST to search the metabolic pathway database50 (release58) in KEGG and homologs in the SwissProt and TrEMBL databases in UniProt51 (release 2011-01).
Annotation of repetitive sequences.
We used RepeatProteinMask and RepeatMasker (version 3.2.9) from Repbase (version 16.03)52 to search for transposable elements. We constructed a de novo repeat library using RepeatScout (v 1.0.5)53, Piler (v 1.0)54 and LTR_FINDER (v 1.0.5)55 and annotated the transposable element regions with RepeatMasker. Simple tandem repeats were annotated using TRF (v 4.04)56.
We used the shortest length standards for each transposable element order from Repbase (v 16.03)52 to filter the integrated results. To estimate the expansion time of LTRs in the P. xylostella and B. mori genomes, we investigated the LTRs using LTR_STRUC57. Both 5′ and 3′ LTR regions of the LTR retrotransposons were extracted and aligned to each other using MUSCLE58. Distmat from EMBOSS59 was used to calculate the times since the divergence of the 5′ and 3′ LTRs.
Annotation of non-coding RNA.
We used tRNAscan-s.e.m. (v 1.23)60 to search for tRNA-coding sequences. Invertebrate rRNA from the database61 was used to predict DBM rRNA sequences. Rfam62 (v 9.1) was used in conjunction with INFERNAL63 to predict small nuclear RNAs (snRNAs) and microRNAs (miRNAs).
Gene family construction.
The predicted proteomes in the DBM genome and those from the genomes of 11 insect species13,14,64,65,66,67,68,69,70,71 and 1 Arachnida outgroup species72 were used in BLAST (1 × 10−7). The fragmental alignments of HSPs were joined using Solar36. Clustering was performed to generate gene families using hcluster_sg73. The species-specific genes are those for which we could not find orthologs in the predicted gene repertoires of the compared genomes.
Genome evolution.
We used phase 1 nucleotides of single-copy genes from different genomes and MCMCTREE from PAML74 to estimate the time divergence time of DBM. Sampling was replicated 100,000 times with a frequency of 2 (the first 10,000 trials were disregarded).
Linkage mapping of scaffolds.
RADseq data generated from a cross between DBM strains Pearl-Sel and Geneva88 (ref. 17) were used. Read mapping for each individual was performed using Stampy (v. 1.0.13)75. Polymorphisms were called using the UnifiedGenotyper (v. 1.3-21)76. A custom PERL script identified segregating polymorphic patterns. A genotype file formatted for JoinMap (v. 3.0)77 was produced. Scaffolds were assigned onto corresponding linkage groups on the basis of the alignment result with the RAD alleles (Supplementary Table 9).
Comparison of genomic synteny.
We used a set of lax parameters36 to perform LASTZ (v. 1.01.50) and MCSCAN78 (v. 0.8) to search for syntenic blocks in P. xylostella and B. mori or D. melanogaster.
Genomic variation.
We fragmented the fosmid sequences in silico into 100-bp single-end reads or paired-end reads (insert size of 500 bp). We used SOAPaligner/soap235 to map the reads onto reference sequences and SOAPsnp79 and SOAPIndel35 to annotate SNPs and indels, respectively (with acceptable depths ranging from 3 to 30). On the basis of the sequencing of a single Fuzhou-S individual (Supplementary Table 1, SI), SOAPsv80 was employed for annotating structural variations. We performed whole-genome alignment comparison using LASTZ. The regions that were ≥1 kb with identity of ≥90% were regarded as segmental duplications.
Annotation of genes concerned.
On the basis of available protein sets (Supplementary Table 26) and the predicted proteomes of P. xylostella, B. mori and D. melanogaster, BLASTP was used to search for the homologs in each of the three genomes. We applied cutoffs at 1 × 10−20, bit-score of 100 and coverage of 100 continuous amino acids for gapped alignment. We filtered out the results with total coverage of alignment of <70% for the same species and <40% for different species. We also used InterProScan81 to search for candidate genes on the basis of conserved motifs from InterPro48. The candidates were manually checked against the Conserved Domain Database82 in NCBI to validate the gene searching results and confirm that the method used in our DBM genome was as effective and reliable as the methods used in other insect genomes.
PCR validation.
We randomly selected 20 each of annotated SNPs, structural variations (≥50 bp and ≤200 bp) and transposable elements (≥300 bp and ≤600 bp) within or around the metabolic detoxification genes. PCR primer sets were designed for each of them to amplify an 800-bp region (Supplementary Table 31). Direct Sanger sequencing was performed for PCR products from both ends. Alignments between sequencing results and the reference genome were performed using BLAST or BLAT83.
Quantitative RT-PCR validation.
We used 20 genes for validation of host plant responsiveness, and another 20 genes to examine differential expressions over the life cycle (Supplementary Table 24). We also used a B. thuringiensis strain containing CryIIAd (GenBank DQ358053) to infect the DBM strain and determine the gene expression for sulfate metabolism. Third-instar larvae were treated with CryIIAd (7.589 μg·/ml) by the leaf-soaking method84, with double-distilled water as control or no food supply for starvation. RT-PCR was performed for quantitative gene expression based on the 2−ΔΔCT method85, with the ribosomal protein L32 gene (GenBank AB180441) serving as an internal reference. Each experiment was repeated three times.
Accession codes.
The genome described herein is the first reference genome of P. xylostella, AHIO01000000. Genome assemblies and annotations described here have been deposited at the DNA Data Bank of Japan (DDBJ), the European Molecular Biology Laboratory (EMBL) and GenBank under accession AHIO00000000. Raw sequencing data from the transcriptome have been deposited at the NCBI Short Read Archive (SRA) under accession SRA034927.
References
Whiteman, N.K. & Jander, G. Genome-enabled research on the ecology of plant-insect interactions. Plant Physiol. 154, 475–478 (2010).
Ali, J.G. & Agrawal, A.A. Specialist versus generalist insect herbivores and plant defense. Trends Plant Sci. 17, 293–302 (2012).
Ratzka, A., Vogel, H., Kliebenstein, D.J., Mitchell-Olds, T. & Kroymann, J. Disarming the mustard oil bomb. Proc. Natl. Acad. Sci. USA 99, 11223–11228 (2002).
Talekar, N.S. & Shelton, A.M. Biology, ecology, and management of the diamondback moth. Annu. Rev. Entomol. 38, 275–301 (1993).
Furlong, M.J., Wright, D.J. & Dosdall, L.M. Diamondback moth ecology and management: problems, progress, and prospects. Annu. Rev. Entomol. published online; doi:10.1146/annurev-ento-120811-153605 (27 September 2012).
Zalucki, M.P. et al. Estimating the economic cost of one of the world's major insect pests, Plutella xylostella (Lepidoptera: Plutellidae): just how long is a piece of string? J. Econ. Entomol. 105, 1115–1129 (2012).
Ankersmit, G.W. DDT-resistance in Plutella maculipennis (Curt.) (Lep.) in Java. Bull. Entomol. Res. 44, 421–425 (1953).
Heckel, D.G., Gahan, L.J., Liu, Y.B. & Tabashnik, B.E. Genetic mapping of resistance to Bacillus thuringiensis toxins in diamondback moth using biphasic linkage analysis. Proc. Natl. Acad. Sci. USA 96, 8373–8377 (1999).
Tabashnik, B.E. et al. Efficacy of genetically modified Bt toxins against insects with different genetic mechanisms of resistance. Nat. Biotechnol. 29, 1128–1131 (2011).
Baxter, S.W. et al. Parallel evolution of Bacillus thuringiensis toxin resistance in Lepidoptera. Genetics 189, 675–679 (2011).
He, W. et al. Developmental and insecticide-resistant insights from the de novo assembled transcriptome of the diamondback moth, Plutella xylostella. Genomics 99, 169–177 (2012).
Baxter, S.W. et al. Mis-spliced transcripts of nicotinic acetylcholine receptor α6 are associated with field evolved spinosad resistance in Plutella xylostella (L.). PLoS Genet. 6, e1000802 (2010).
Xia, Q. et al. A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science 306, 1937–1940 (2004).
Zhan, S., Merlin, C., Boore, J.L. & Reppert, S.M. The monarch butterfly genome yields insights into long-distance migration. Cell 147, 1171–1185 (2011).
Zimmer, A. et al. Dating the early evolution of plants: detection and molecular clock analyses of orthologs. Mol. Genet. Genomics 278, 393–402 (2007).
Wang, X. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039 (2011).
Baxter, S.W. et al. Linkage mapping and comparative genomics using next-generation RAD sequencing of a non-model organism. PLoS ONE 6, e19315 (2011).
Heliconius Genome Consortium. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487, 94–98 (2012).
Mutanen, M., Wahlberg, N. & Kaila, L. Comprehensive gene and taxon coverage elucidates radiation patterns in moths and butterflies. Proc. R. Soc. 277, 2839–2848 (2010).
Regier, J.C. et al. Toward reconstructing the evolution of advanced moths and butterflies (Lepidoptera: Ditrysia): an initial molecular study. BMC Evol. Biol. 9, 280 (2009).
Buono, M. & Cosma, M.P. Sulfatase activities towards the regulation of cell metabolism and signaling in mammals. Cell. Mol. Life Sci. 67, 769–780 (2010).
Cosma, M.P. et al. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell 113, 445–456 (2003).
Rausch, T. & Wachter, A. Sulfur metabolism: a versatile platform for launching defence operations. Trends Plant Sci. 10, 503–509 (2005).
Pauchet, Y., Muck, A., Svatos, A., Heckel, D.G. & Preiss, S. Mapping the larval midgut lumen proteome of Helicoverpa armigera, a generalist herbivorous insect. J. Proteome Res. 7, 1629–1639 (2008).
Henniges-Janssen, K., Reineke, A., Heckel, D.G. & Groot, A.T. Complex inheritance of larval adaptation in Plutella xylostella to a novel host plant. Heredity 107, 421–432 (2011).
Eichenseer, H., Mathews, M.C., Powell, J.S. & Felton, G.W. Survey of a salivary effector in caterpillars: glucose oxidase variation and correlation with host range. J. Chem. Ecol. 36, 885–897 (2010).
Wanner, K.W. & Robertson, H.M. The gustatory receptor family in the silkworm moth Bombyx mori is characterized by a large expansion of a single lineage of putative bitter receptors. Insect Mol. Biol. 17, 621–629 (2008).
Tao, X.-Y., Xue, X.-Y., Huang, Y.-P., Chen, X.-Y. & Mao, Y.-B. Gossypol-enhancedP450 gene pool contributes to cotton bollworm tolerance to a pyrethroid insecticide. Mol. Ecol. 21, 4371–4385 (2012).
Li, X., Schuler, M.A. & Berenbaum, M.R. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu. Rev. Entomol. 52, 231–253 (2007).
Labbé, R., Caveney, S. & Donly, C. Genetic analysis of the xenobiotic resistance–associated ABC gene subfamilies of the Lepidoptera. Insect Mol. Biol. 20, 243–256 (2011).
Cui, F. et al. Two single mutations commonly cause qualitative change of nonspecific carboxylesterases in insects. Insect Biochem. Mol. Biol. 41, 1–8 (2011).
Chen, S. & Li, X. Transposable elements are enriched within or in close proximity to xenobiotic-metabolizing cytochrome P450 genes. BMC Evol. Biol. 7, 46 (2007).
Hu, T.T. et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 43, 476–481 (2011).
Kvist, J. et al. Temperature treatments during larval development reveal extensive heritable and plastic variation in gene expression and life history traits. Mol. Ecol. published online; doi:10.1111/j.1365-294X.2012.05521.x (19 March 2012).
Li, R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967 (2009).
Li, R. et al. The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317 (2010).
Xu, X. et al. Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195 (2011).
Boetzer, M., Henkel, C.V., Jansen, H.J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Kiełbasa, S.M., Wan, R., Sato, K., Horton, P. & Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493 (2011).
Mortazavi, A., Williams, B.A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467 (2005).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Elsik, C.G. et al. Creating a honey bee consensus gene set. Genome Biol. 8, R13 (2007).
Trapnell, C., Pachter, L. & Salzberg, S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2009).
Pillai, S. et al. SOAP-based services provided by the European Bioinformatics Institute. Nucleic Acids Res. 33, W25–W28 (2005).
Ogata, H. et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34 (1999).
Apweiler, R. et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 32, D115–D119 (2004).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Price, A.L., Jones, N.C. & Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Edgar, R.C. & Myers, E.W. PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
McCarthy, E.M. & McDonald, J.F. LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics 19, 362–367 (2003).
Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000).
Lowe, T.M. & Eddy, S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Wuyts, J., Perrière, G. & Van de Peer, Y. The European ribosomal RNA database. Nucleic Acids Res. 32, D101–D103 (2004).
Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A. & Eddy, S.R. Rfam: an RNA family database. Nucleic Acids Res. 31, 439–441 (2003).
Nawrocki, E.P., Kolbe, D.L. & Eddy, S.R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
Adams, M.D. et al. The genome sequence of Drosophila melanogaster. Science 287, 2185–2195 (2000).
Holt, R.A. et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science 298, 129–149 (2002).
Richards, S. et al. The genome of the model beetle and pest Tribolium castaneum. Nature 452, 949–955 (2008).
Werren, J.H. et al. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 327, 343–348 (2010).
HoneyBee Genome Sequencing Consortium. . Insights into social insects from the genome of the honeybee Apis mellifera. Nature 443, 931–949 (2006).
Bonasio, R. et al. Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator. Science 329, 1068–1071 (2010).
Kirkness, E.F. et al. Genome sequences of the human body louse and its primary endosymbiont provide insights into the permanent parasitic lifestyle. Proc. Natl. Acad. Sci. USA 107, 12168–12173 (2010).
International Aphid Genomics Consortium. Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol. 8, e1000313 (2010).
Grbić, M. et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature 479, 487–492 (2011).
Li, H. et al. TreeFam: a curated database of phylogenetic trees of animal gene families. Nucleic Acids Res. 34, D572–D580 (2006).
Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Lunter, G. & Goodson, M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 21, 936–939 (2011).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Ooijen, V.A.N.J. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet. Res. 93, 343–349 (2011).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488 (2008).
Li, R. et al. SNP detection for massively parallel whole-genome resequencing. Genome Res. 19, 1124–1132 (2009).
Li, R., Li, Y., Kristiansen, K. & Wang, J. SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–714 (2008).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120 (2005).
Marchler-Bauer, A. et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229 (2011).
Kent, W.J. BLAT—The BLAST-Like Alignment Tool. Genome Res. 12, 656–664 (2002).
Li, A., Yang, Y., Wu, S., Li, C. & Wu, Y. Investigation of resistance mechanisms to fipronil in diamondback moth (Lepidoptera: Plutellidae). J. Econ. Entomol. 99, 914–919 (2006).
Livak, K.J. & Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Acknowledgements
This work was supported through a special project of Research on Diamondback Moth Genomics (grant JB09315) to M.Y. and a Minjiang Scholar Program to L.V., G.M.G., C.J.D. and S.M.S. by the Educational Department of Fujian Province and through a key project (grant 31230061) to M.Y. from the National Natural Science Foundation of China. Insect rearing and sampling, as well as some of the DNA extractions, were conducted at the Fujian Provincial Key Laboratory of Biodiversity and Eco-safety and the Key Laboratory of Integrated Pest Management for Fujian-Taiwan Crops, the Ministry of Agriculture, China. We are grateful to A.D. Briscoe (University of California–Irvine) for her help in organizing and for providing ORs, OBPs and CSPs from Danaus plexippus and Heliconius melpomene and to G.L. Lövei for his comments and suggestions on the manuscript. We appreciate J. Liao and M. Zou for providing the Bt-treated P. xylostella larvae used for quantitative gene expression analysis. We thank H. Wang, J. Luo, Y. Hong, S. Pan, L. Yang, Y. Weng, Y. Hong and Y. Liu for their technical assistance in rearing insects and preparing samples.
Author information
Authors and Affiliations
Contributions
M.Y., G.Y. and Jun Wang managed the project. W.H., M.X., J.B., C.L., B.W., X.L., X.X., F.K., D.Z., Y.H., F.S., Y.Y., X.M., Y. Liang, Y.C., L.Y., Y. Liu, L.P., Y. Zheng and Y. Zhang prepared insects and DNA samples and created the figures. M.Y., G.Y., W.H., M.X., X.Y., D.Z., S.W.B., L.V., P.W., X.L., K.C., S.H. and X.X. designed experiments and analysis. Z.Y., D.Z. and Q.C. performed genome assembly. W.H., X.Y., Q.Z., Z.W., C.L., Q.C., M.H., Y. Luo and C.G. performed genome annotation, comparative genomics and genomic variation analysis. Q.C., Z.W., S.W.B. and J.W.D. performed genetic mapping. Q.C., W.T. and L.Y. performed data submission and database construction. M.Y., G.Y., W.H., G.L., L.F., J.L., X.Z., Junyi Wang, Jian Wang, H.Y. and Jun Wang provided coordination. W.H., M.Y., D.Z., G.Y., S.W.B., L.V., G.M.G., C.J.D. and P.W. wrote the manuscript. M.Y., W.H., D.Z., G.Y., S.W.B., L.V., G.M.G., C.J.D., P.W., X.L., J.W.D., S.M.S., M.C., S.H. and X.X. revised the manuscript. X.M., Y.Y., D.Z., X.Y., J.L., B.W., F.K., F.S., Y.C., W.H. and M.Y. performed experimental validation and analysis.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Note, Supplementary Figures 1–25 and Supplementary Tables 1–32 (PDF 11747 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
You, M., Yue, Z., He, W. et al. A heterozygous moth genome provides insights into herbivory and detoxification. Nat Genet 45, 220–225 (2013). https://doi.org/10.1038/ng.2524
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.2524
