
Abstract
We sequenced eight melanoma exomes to identify new somatic mutations in metastatic melanoma. Focusing on the mitogen-activated protein (MAP) kinase kinase kinase (MAP3K) family, we found that 24% of melanoma cell lines have mutations in the protein-coding regions of either MAP3K5 or MAP3K9. Structural modeling predicted that mutations in the kinase domain may affect the activity and regulation of these protein kinases. The position of the mutations and the loss of heterozygosity of MAP3K5 and MAP3K9 in 85% and 67% of melanoma samples, respectively, together suggest that the mutations are likely to be inactivating. In in vitro kinase assays, MAP3K5 I780F and MAP3K9 W333* variants had reduced kinase activity. Overexpression of MAP3K5 or MAP3K9 mutants in HEK293T cells reduced the phosphorylation of downstream MAP kinases. Attenuation of MAP3K9 function in melanoma cells using siRNA led to increased cell viability after temozolomide treatment, suggesting that decreased MAP3K pathway activity can lead to chemoresistance in melanoma.
Similar content being viewed by others


Main
There are no long-lasting effective treatments for disseminated melanoma; however, recent developments in molecularly targeted therapies have shown success in short-term, progression-free survival and the reduction of tumor burden1. The recent advent of next-generation sequencing (NGS) has enabled the identification of cancer-associated mutations in an unbiased manner. The mutation catalogs generated with this method provide enormous potential for understanding the molecular basis of disease and identifying novel therapeutic targets. Further characterizing the pathways involved in the etiology of melanoma will help guide the development of new treatments for this disease.
Using whole-exome capture, we sequenced eight melanoma cell lines (Supplementary Table 1) and matched normal lymphoblastoid cell lines (LCLs) using two NGS platforms (Illumina Genome Analyzer II and Life Technologies SOLiD) and mapped reads with platform-appropriate alignment programs (Online Methods). The schematic of how the analysis was performed is shown in Supplementary Figure 1. To maximize the chance of finding bona fide mutations, we applied strict filtering criteria (described in Supplementary Fig. 1 and in the Online Methods) to minimize the false positive rate without overly inflating the false negative rate. Comparing Illumina SNP array data2 with variant calls from the exome data yielded >99.5% concordance for each sample, thus achieving a mean false negative rate of ∼0.45%. Overall, 3,215 somatic alterations were identified, with a range of 243–523 per sample (Supplementary Table 2). Of these, 1,076 were synonymous (silent) mutations, and 2,139 were predicted to alter protein structure (range: 175–326 per sample), comprising 1,925 missense, 122 nonsense and 32 splice-site mutations and 64 small insertions and/or deletions (indels) (Supplementary Table 2). The ratio of nonsynonymous to synonymous changes (N:S ratio) was 1.9:1, which is not higher than the N:S ratio of 2.5:1 predicted for nonselected, passenger mutations2, indicating that most of the mutations are likely to be passengers rather than drivers in melanoma. Recent exome analysis of 14 metastatic melanomas found a similarly low N:S ratio (2.0:1)3. Analysis of the mutation spectrum showed that the proportion of C>T/G>A transitions was greater than that of other nucleotide substitutions (4.1:1) (Supplementary Fig. 2). We observed 17 tandem mutations, including 10 CC>TT/GG>AA alterations, which, taken together, is consistent with the mutation signature associated with exposure to UV light4.
Of the 1,740 genes found to have protein-altering changes (Supplementary Table 3), 446 were reported to be mutated in a recent exome analysis of melanoma3, and 166 have mutations documented in the Catalogue of Somatic Mutations in Cancer (COSMIC) database5. The overlap between our data set and these two other sources revealed 58 genes commonly mutated in melanoma, suggesting that many of these are potentially drivers of melanoma pathogenesis (Supplementary Table 3). We verified mutations in key melanoma-associated genes, including GRIN2A5 (mutated in 2/8 samples), TRRAP5 (2/8 samples), ADAM29 (ref. 6) (2/8 samples), ADAMTS18 (ref. 7) (2/8 samples) and ERBB4 (ref. 8) (2/8 samples) (Supplementary Tables 3 and 4). We also observed the common occurrence of mutations in many G protein–coupled receptor family members9 (Supplementary Table 3). Genes newly identified as being involved in melanoma included SLC2A12 (mutated in 3/8 samples) and RGSL1 (3/8 samples), both of which were mutated with high frequency (38%) in the discovery screen. Mutations were found in 22 genes lying in previously described regions of homozygous deletion (Supplementary Table 5), including in PTPRD10 (5 mutations in 4 samples), a putative tumor suppressor gene for melanoma and glioblastoma11.
The MAPK pathway has an important role in the development of melanoma12,13, and BRAF, encoding a MAP3K, is the most commonly mutated gene in melanoma14. Considering the importance of BRAF, along with the impact that mutation of this kinase has on the efficacy of some new molecularly targeted therapies for melanoma, we focused our attention on other mutated MAP3K family members.
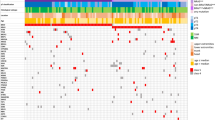
MAP3K5, MAP3K8 and MAP3K9 each showed somatic mutations in one out of eight melanoma samples. We validated the mutations in MAP3K5 and MAP3K9, but the MAP3K8 mutation was found to be a false positive. A prevalence screen revealed that 8 of 85 melanoma cell lines had nonsynonymous somatic mutations in MAP3K5 (total 8 mutations), and 13 of 85 cell lines had mutations in MAP3K9 (18 mutations) (Fig. 1). Overall, MAP3K5 and MAP3K9 were mutated in 9% and 15% of melanoma cell lines, respectively, and mutation of at least one of these genes occurred in 24% of samples. DNA from tumors matched to the cell lines was sequenced, and MAP3K5 and MAP3K9 mutations were validated in all but one sample (Supplementary Table 6), indicating that the mutations were not the result of growth in cell culture. For the one discordant pair, it is possible that the encoded p.Trp333* (c.999A>G; NM_033141) substitution in MAP3K9 arose in the cell line in vitro or that the cell line was derived from a subpopulation of tumor cells that carried the mutation and that was present at levels too low for detection by sequencing of the tumor. With the exception of one cell line, mutations in MAP3K5 and MAP3K9 were mutually exclusive (Supplementary Table 6), suggesting they may target the same pathway. Four MAP3K9 mutations (chromosome 14) and five MAP3K5 mutations (chromosome 6) were homozygous, indicating somatic loss of heterozygosity (LOH) at these loci. Analysis of SNP array data for these and other melanoma cell lines12 (K.D.-R. and N.K.H, unpublished data) confirmed this supposition and showed that 98 of 115 (85%) and 77 of 115 (67%) samples had LOH for MAP3K5 and MAP3K9, respectively (Supplementary Fig. 3). LOH included loss of chromosome 6 (15 of 98 samples; 15%), loss of the whole chromosome 6 q-arm encompassing MAP3K5 (18 of 98; 18%) or loss on chromosome 14 of MAP3K9 (46 of 77; 60%). Areas of regional LOH that included up to 5 Mb on either side of MAP3K5 (39 of 98 samples; 40%) and MAP3K9 (9 of 77; 12%) also occurred. Additionally, MAP3K5 (26/98; 27%) and MAP3K9 (22/77; 29%) were often found in focal areas of LOH encompassing only a small number of genes. The high rate of LOH, the distribution of mutations along the entire length of each gene and the identification of a nonsense mutation in MAP3K9 suggest that the mutations are inactivating. Of note, along with homozygous mutations, a number of heterozygous mutations were identified. We propose that, as with TP53, PTEN15 and CDKN1B16, these heterozygous mutations result in reduced gene function via haploinsufficiency and reduction of the amount of wild-type protein. Mutations in MAP3K5 and MAP3K9 were not correlated with mutations and/or deletions in BRAF, NRAS, CDKN2A, PTEN or TP53 (Supplementary Table 6). The expression levels of the MAP3K5 and MAP3K9 transcripts did not correlate with mutation status (Supplementary Fig. 4 and Supplementary Table 7), suggesting that the mutations do not grossly affect mRNA expression or stability.

Schematics of MAP3K5 and MAP3K9 depicting the domain structure, phosphorylation sites (below each protein) and position of validated somatic nonsynonymous alterations identified by whole-exome sequencing (above each gene).
We analyzed the available three-dimensional protein structures of the MAP3K proteins to gain insight into whether the alterations in MAP3K5 and MAP3K9 may have an effect on structure or kinase activity (Fig. 2). This modeling showed that the MAP3K5 p.Ile780Phe (c.2338A>T; NM_005923) alteration is likely to affect the packing of helices in the kinase domain (Fig. 2a). The MAP3K9 p.Trp333* alteration results in the production of a truncated kinase domain that is unlikely to be functional. The Arg160 residue in MAP3K9 forms hydrogen bonds with Glu167, which would be disrupted by the change of this residue to cysteine (p.Arg160Cys, c.478C>T; NM_033141), and the MAP3K9 p.Pro263Leu (c.788C>T; NM_033141) alteration is predicted to increase conformational flexibility in the corresponding loop (Fig. 2b). The MAP3K9 Glu319 residue forms hydrogen bonds with Arg393, and its change to lysine would disrupt this interaction, potentially altering the kinase domain surface and affecting interactions with other molecules.

(a) Top, MAP3K5 kinase domain (PDB ID 2CLQ). The kinase domain was aligned with PDB ID 2PHK (not shown) to determine approximate positions for amino acid residues binding to ATP (yellow) and substrate (light green). Bottom left, the wild-type Ile780 residue (green) and its surroundings; bottom right, the mutant Phe780 residue and its surroundings (green). (b) Top left, the MAP3K9 (PDB ID 3DTC) kinase domain. Top right, the same domain rotated by 180° to show the location of all residues of interest. The kinase domain was aligned with PDB ID 2PHK (not shown) to determine the approximate positions for residues that bind ATP (yellow) and substrate (green). Middle, wild-type residues (Arg160, Pro263 and Glu319; green) and their surroundings; bottom, mutant residues (Cys160, Leu263 and Lys319; green). The disruption of hydrogen bonds can be seen for the p.Arg160Cys and p.Glu319Lys alterations. The p.Asp176Asn alteration in MAP3K9 is in a disordered loop, and the Glu663 residue in MAP3K5 is not resolved in the crystal structure; therefore, they are not shown in the diagrams. Only side chains within 5 Å of the residue of interest are depicted.
To confirm these predicted changes, we performed in vitro kinase assays using cDNA constructs containing mutations that were suggested to confer decreased kinase activity in the modeling analysis. We tested mutations for both MAP3K5 (coding for p.Glu663Lys, c.G1987A, NM_005923 and p.Ile780Phe substitutions) and MAP3K9 (coding for p.Trp333*, as well as the previously described kinase-dead MAP3K9 Lys171Ala variant17). We overexpressed these mutants in mammalian cells and, after immunoprecipitation, assayed kinase activity. Wild-type MAP3K9 and MAP3K5 were seen to have robust kinase activity for the phosphorylation of myelin basic protein (MBP) as well as substantial autophosphorylation (Fig. 3a,b). The MAP3K9 Lys171Ala and Trp333* mutants had a 92% and 95% reduction in kinase activity for MBP relative to the wild-type enzyme, respectively (P < 0.001). Additionally, the p.Ile780Phe substitution in MAP3K5 almost completely abolishes kinase activity (93% reduction relative to wild type; P < 0.01), whereas the p.Glu663Lys alteration had a weaker, yet statistically significant, effect on MBP phosphorylation (30% reduction relative to wild type; P < 0.05).

(a,b) HEK293T cells were transfected with wild-type or mutant MAP3K9 (K171A and W333* variants) or MAP3K5 (E663K and I780F variants) to assess the effect of mutation on kinase activity in vitro. Autophosphorylation of MAP3K9 (a) or MAP3K5 (b) and phosphorylation of the kinase substrate MBP were measured by the incorporation of [32P]γATP and quantified with a phosphor imager. Error bars, s.d. from four independent quantitations; *, P < 0.05; **, P < 0.01; ***, P < 0.001 by two-tailed Student's t test.
Cellular stresses, including UV radiation and oxidative stress, activate MAP3K5, leading to downstream activation of the c-Jun NH2-terminal kinase (JNK) and p38 MAPK effector pathways and context-dependent induction of apoptosis, differentiation, survival or senescence18,19 (Supplementary Fig. 5). Although MAP3K9 is less intensively studied, its transcript is widely expressed20 and activation results in downstream signaling through MAP2K4 and JNK, with the effect on other MAPK effector modules currently unknown17. To further investigate the functional consequences of MAP3K5 and MAP3K9 mutations, we assessed key components of the downstream signaling pathways after introduction of either the wild-type or mutant kinases into cells. Given the well-established difficulty in transfecting melanoma cell lines in addition to their often perturbed MAPK signaling, we chose HEK293T cells as a model system because of their high inherent transfection efficiency and low constitutive MAPK signaling. Exogenous expression of MAP3K5 led to activation of this protein by phosphorylation, with downstream activation of the MAP2K4-MAP2K7 and JNK and p38 pathways compared to the effect of expressing control enhanced green fluorescent protein (EGFP) from the same vector (Fig. 4a). Similar levels of MAP3K9 and MAP3K5 expression were achieved, as detected using an antibody that recognizes the Myc epitopes expressed on both proteins in a protein blot (data not shown). However, we observed differential activation of downstream MAPKs by these MAP3Ks, as expression of MAP3K9 led to activation of MAP2K4-MAP2K7 and JNK and not the p38 module (Fig. 4b). Expression of MAP3K9 and, to a lesser degree, MAP3K5, also activated MEK1/2 and ERK (Fig. 4a,b). We engineered MAP3K5 constructs that contained the two mutations that were most likely to affect protein function, one in the kinase domain (coding for p.Ile780Phe; Fig. 2a) and one adjacent to the kinase domain that introduces a charge change (p.Glu663Lys; Fig. 1). The expression of the I780F variant resulted in reduced phosphorylation of MAP3K5 when expression levels were normalized to the amount of total MAP3K5 expressed, and the expression of both constructs caused a reduction in JNK signaling and phosphorylation of MEK1/2 (P-MEK1/2) (Fig. 4a). Expression of either mutant MAP3K9 protein (W333* or control K171A17) resulted in decreased signaling through MAP2K4-MAP2K7 and JNK and MEK1-MEK2 and ERK (Fig. 4b), consistent with the reduction in kinase activity of these mutants in vitro (Fig. 3).

(a,b) HEK293T cell lines were transfected with expression plasmids for EGFP or with wild-type or mutant MAP3K5 (a) or MAP3K9 (b) for 48 h before analysis of protein expression and MAPK signaling by protein blot using the indicated antibodies. Results are representative of three independent repeats of this experiment.
Until recently, melanoma was notoriously refractory to chemotherapeutic intervention, and understanding the mechanisms of this chemoresistance is vital to improving the outlook for individuals with metastatic disease. Temozolomide (TMZ) is an alkylating agent that has been reported to modestly increase progression-free survival as a single agent in randomized phase 3 trials18,21. In a high-throughput siRNA screen for sensitizers to TMZ treatment (data not shown), we noted that knockdown of MAP3K9 resulted in increased resistance, rather than sensitivity, to TMZ treatment. As the mutations that we have examined seem to decrease MAP3K5 and MAP3K9 activity and downstream signaling, we examined the effect of attenuating wild-type MAP3K5 and MAP3K9 levels in melanoma cells, specifically determining whether decreased expression of wild-type kinases can contribute to chemoresistance to TMZ. Transfection of the melanoma cell lines UACC903 and UACC647 with three independent siRNAs targeting either MAP3K5 (data not shown) or MAP3K9 (Fig. 5a) decreased target mRNA expression by at least 60% of the levels observed in cells transfected with scrambled control siRNA. Exposure of control siRNA–transfected melanoma cells to TMZ resulted in substantially decreased cell viability (Fig. 5b), consistent with apoptosis being the predominant form of cell death in melanoma cell lines after TMZ exposure22. For the two cell lines tested, siRNA knockdown of MAP3K9 alone reduced cell viability in UACC903 cells, whereas siRNA-mediated knockdown by itself had little if any effect in UACC647 cells. However, with the combination of both siRNA knockdown and TMZ exposure, both cell lines showed a resistance phenotype, as shown by increased cell viability in the presence of both agents compared with exposure to TMZ alone (Fig. 5b). Similar experiments conducted for MAP3K5 did not show a statistically significant increase in cell viability following combined siRNA and TMZ exposure (data not shown). These findings suggest that attenuated MAP3K9 activity can contribute to chemotherapeutic resistance in melanoma.

(a) Melanoma cell lines (UACC903 and UACC647) were transfected with siRNA targeting GFP (control) or with three independent siRNAs targeting MAP3K9 (siRNAs 1–3) for 5 d. Expression of MAP3K9 is shown as determined by quantitative RT-PCR (qRT-PCR), normalized to GAPDH levels and expressed relative to control siRNA. Experiments were performed in triplicate. Error bars, s.d. All three independent siRNAs resulted in decreased expression of MAP3K9. (b) UACC903 (left) and UACC647 (right) cells were transfected as in a for 24 h before treatment with the indicated dose of TMZ. Cell viability was determined after 5 d, and measurements indicated that reduced MAP3K9 expression in the presence of TMZ resulted in a resistant phenotype. Data are representative of three independent experiments performed in triplicate. Error bars, s.d.; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
In conclusion, we have shown almost mutually exclusive mutation of MAP3K5 and MAP3K9 in approximately 24% of melanomas, which occurs independently of activating mutations in BRAF or NRAS. Together with the high rates of LOH at these two loci, aberrations in these genes represent a very common event in melanoma. Given that homozygous deletion of MAP3K5 results in increased tumor initiation in a carcinogen-induced model of skin cancer19, that MAP3K9 regulates apoptosis through MAP2K4-MAP2K7 and JNK23 and that diminished activation of JNK1 can result in enhanced survival of tumor cells24, our data indicate that abrogation of this signaling axis is important for melanoma development (Supplementary Fig. 4). Reactivation of the signaling pathways downstream of mutant MAP3K5 or MAP3K9 might thus be therapeutically advantageous in treating this disease.
URLs.
COSMIC, http://www.sanger.ac.uk/genetics/CGP/cosmic/; Geneious v5.3, http://www.geneious.com/; Illumina, http://www.illumina.com/; Roche-NimbleGen, http://www.nimblegen.com/products/seqcap/ez/index.html; Applied Biosystems, http://www.lifetechnologies.com/; Novoalign, http://www.novocraft.com/; Picard, http://picard.sourceforge.net/index.shtml/; Depth of Coverage v3.0, http://www.broadinstitute.org/gsa/wiki/index.php/Depth_of_Coverage_v3.0; 1000 Genomes Project, http://www.1000genomes.org/.
Methods
Samples and cell culture.
All human tissue was taken with informed consent under a protocol approved by the Queensland Institute of Medical Research Human Research Ethics Committee (HREC, approval number H0311-084; P726). All melanoma cell lines were established from fresh tumor biopsies and cultured as described25. A small proportion of each fresh-frozen tumor from which the cell lines were derived was retained in order to confirm mutations were tumor derived. Human embryonic kidney (HEK293T) cells were routinely maintained in RPMI medium (Sigma) supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin and glutamine. The endogenous MAP3K5 and MAP3K9 coding sequences were verified to be wild type in HEK293T cells.
DNA extraction and quantitation.
DNA was extracted from cultured cells using a QIAamp DNA Mini kit (Qiagen) according to the manufacturer's instructions, quantified using a NanoDrop 1000 (ThermoScientific) and quality assessed with a Bioanalyzer DNA 12000 chip (Agilent).
Illumina DNA library preparation.
Genomic DNA (2 μg) was randomly fragmented using NEBNext dsDNA Fragmentase (NEB) for 1 h. After purification, fragments were end repaired according to standard instructions in the Preparing Samples for Sequencing Genomic DNA protocol (see URLs). End-repaired fragments were A tailed before ligation with Illumina single-end adaptor oligonucleotide mix. All reagents used, with the exception of the adaptor oligonucleotide mix, were part of the NEBNext DNA Sample Prep Reagent Set 1 (NEB). Adaptor-ligated DNA fragments were purified using a QIAquick PCR purification kit (Qiagen) before agarose gel size selection of a 200- to 300-bp product. This product was further purified using the QIAquick Gel Extraction kit (Qiagen) before being used as template in the SeqCap EZ Exome Library SR protocol (see URLs). To test the integrity of the adaptor-ligated DNA fragments, 1 μl of purified product was PCR amplified using adaptor-specific oligonucleotides (Illumina) and then verified with a Bioanalyzer DNA 1000 kit (Agilent).
SOLiD DNA library preparation.
Genomic DNA (5 μg) was sheared to a mean size of 100–110 bp using a Covaris S2 and the recommended shearing protocol in the Applied Biosystems SOLiD 3 Plus system manual, with the modification of 6 cycles and cycles/burst set to 200 (see URLs). After purification with the QIAquick PCR Purification kit, ends were repaired and 3′ adenylated using the NEBNext DNA Sample Prep Reagent Set 1. Adaptors were ligated using the NEB Quick Ligation kit, and products were purified by Agencourt AMPure SPRI bead purification. Products were assessed using a Bioanalyzer DNA 7500 chip (Agilent) before nick translation and subsequent amplification using Platinum PCR Supermix HiFi (Invitrogen) according to the SOLiD 3 System Library Preparation protocol. Samples were purified using a QIAquick PCR Purification kit, assessed by NanoDrop 1000 and Quant-iT Picogreen (Invitrogen) quantification and analyzed with a Bioanalyzer DNA 7500 chip.
NimbleGen SeqCap EZ Exome library short-read preparation.
The exome library was generated according to the manufacturer's instructions (see URLs). Amplified product (1 μg (Illumina) or 2 μg (SOLiD)) was added to the exome library along with hybridization buffers, COT DNA (Roche) and an excess (1,000 pM) of SE-HE1 and SE-HE2 oligonucleotides, for Illumina, or hybridization-enhancing oligonucleotides SOLiD-A and SOLiD-B, for SOLiD chemistry, to block nonspecific hybridization. The hybridization mix was incubated at 47 °C for 64–72 h. Exome-captured DNA was washed and recovered with magnetic Streptavidin Dynabeads (Invitrogen). This process recovers only exome-hybridized DNA from the biotinylated exon library. As the exonic content of the original starting material is only a fraction of the genome, it was necessary to PCR enrich the captured fragments using adaptor-specific oligonucleotides to generate enough material to sequence. The captured DNA was amplified with platform-specific oligonucleotides using Phusion High-Fidelity PCR Mastermix (Finnzymes). The amplified product was purified with a QIAquick PCR Purification kit before being verified on a DNA High-Sensitivity chip using an Agilent Bioanalyzer. The captured enriched DNA was assessed by quantitative PCR (qPCR) to estimate the relative fold enrichment by measuring the relative abundance of control targets in the amplified captured DNA.
Illumina cluster generation and SBS sequencing.
Each exome-captured DNA library was denatured with 2 N NaOH to a final concentration of 1 nM. This was achieved by adding 2 μl of a 10 nM stock library to 1 μl of 2 N NaOH and 17 μl of elution buffer (EB; Qiagen). The denatured libraries were diluted in hybridization buffer to the desired picomolar concentration. Using an Illumina Cluster Station, the diluted exome-captured library was hybridized to an eight-lane flow cell, followed by cluster generation by isothermal amplification with the Illumina Single-Read Cluster Generation kit v4. To ensure sufficient coverage for SNP detection, four lanes of the flow cell were used for each sample library. The flow cell was then subjected to a 72-bp sequencing-by-synthesis (SBS v4) protocol using a Genome Analyzer II, according to the manufacturer's instructions (see URLs).
SOLiD-templated bead preparation and sequencing by ligation.
Emulsion PCR (ePCR) was carried out according to the SOLiD 3 Plus System Bead Preparation instructions (see URLs). Briefly, an oil phase, SOLiD P1 DNA beads and an aqueous phase were prepared using a dilution of the exome-captured library at a final concentration of 500 pM in 1× Low TE Buffer. The ePCR mix was prepared in conjunction with the ULTRA-TURRAX Tube Drive (IKA) before being dispensed in a 96-well plate and cycled on a GeneAmp PCR System 9700 (Applied Biosystems) using standard ePCR cycling parameters. An emulsion break reaction to release templated beads was followed by a wash and bead enrichment and isolation. The 3′ ends of the templates from enriched beads were modified using terminal transferase, and beads were quantified using the SOLiD Bead Concentration Chart and NanoDrop 1000. Sequencing by ligation was performed for each sample using three quadrants of a 50-bp slide flow cell on the SOLiD 3 Plus sequencing platform.
Image extraction and base calling.
For Illumina sequencing, images were analyzed using RTA v1.6 software (Illumina) during the SBS sequencing run, and the bases were called after completion of the run. FastQ files were generated using the Illumina CASAVA v1.6 software package. For SOLiD sequencing, the SOLiD Software suite (Applied Biosystems) including SOLiD Analysis Tools (SAT) was used to analyze and generate the csfasta files for alignment.
Mismatch and gapped alignment.
For samples (A02, A06, D05 and D14), which were sequenced on the Illumina platform, we aligned all reads to the hg19 reference genome26. For single-base substitutions, we used Bowtie v0.11.3 (ref. 27), and for indels we used Novoalign (see URLs). For samples (D35, D36, D41 and D49), which were sequenced on the SOLiD platform, we aligned all reads with BFAST28. Sequencing error rates were determined in two samples chosen at random. The pass-filter error rate ranged from 0.3–0.8%.
Variation detection using SAMtools.
The alignment files were exported in SAM format to facilitate the calling of variations using SAMtools29.
Variation annotation and SNP filtering.
The pileup file of all variations detected in each sample was first compared to all variations annotated in dbSNP132 along with data from the 1000 Genomes Project (see URLs). After this analysis, all newly identified variations were fully annotated. All filtering and annotation was performed using ANNOVAR30. To predict the effect the nonsynonymous mutations might have on the encoded proteins, we used dbNSFP31, which collates the outputs from the prediction programs SIFT32, Polyphen2 (ref. 33), LRT34, MutationTaster35 and PhyloP36.
Coverage statistics.
The percent on-target statistics and mean coverage were generated using Picard (see URLs) and Depth of Coverage v3.0 from the GATK package (see URLs).
Determination of false negative rate using stringent filtering criteria.
All samples sequenced had either been previously assayed with the Illumina Hap300 SNP array11 (A02, A06, D05, D14, D35, D36, D41 and D49) or recently assayed using the Illumina OmniQuad array (A02-LCL, A06-LCL, D05-LCL, D14-LCL, D35-LCL, D36-LCL, D41-LCL and D49-LCL). These data were used as the baseline for the detection of SNPs present in each sample. Inclusion of all reads with Phred SNP quality of 20 resulted in a false negative rate (FNR) of ∼20%. As this was an unacceptable level of error, we devised a set of criteria which would bring the FNR rate to a minimum and thereby reduce the overall number of mutations to a more manageable level. First, the minimum depth had to be ≥10 reads. On average, this covered 88% of all bases in each sample. Next, the minimum mutation call had to be ≥20% of all reads covering that genomic position. Lastly, the minimum Phred SNP quality score for the mutant base had to be ≥100. With these stringent criteria, we reduced the overall FNR to 0.45%.
Detection of known mutations.
In order to test the appropriateness of the filtering parameters, we assessed a list of mutations that we had previously identified in some of the exome-sequenced samples. We found six of seven BRAFV600E mutations, with one sample (D41) being filtered out because it had an SNP quality score of 84. Two of three TP53 mutations were confirmed; one sample with a TP53 mutation was filtered out because it had an SNP quality score of 91. We also confirmed the PPP3R2 and PRKAR1A mutations, both seen in one sample each. CDKN2A had a high degree of LOH, as previously shown in melanoma11, and, as such, was a problematic region for successful exome-probe hybridization. Previously identified mutations were not all present in the exome-sequencing data for a number of reasons: one alteration (encoding p.Pro114Leu, c.341C>T; NM_000077) was present at an SNP quality of 94 and, hence, did not make the cutoff; one deletion (8-bp del, causing a frameshift in codon 11, c.32_39del) was present but was represented by a single read and therefore did not make the cutoff; and an additional pair of mutations were not detected at any depth due to poor coverage at the corresponding locus.
Verification of mutations using Sanger sequencing.
We found 70 genes that each harbored three or more mutations, the majority of which have previously been reported to be mutated in melanoma (Supplementary Table 4). Fifteen genes had apparent recurrent or 'hotspot' mutations, with the most common mutation being BRAFV600E, which occurred in 6 of 8 samples (Supplementary Table 8). We verified the BRAFV600E mutations by Sanger sequencing, but none of the other 14 putative recurrent mutations were verified. This result was most likely caused by misalignment of sequence tags to the human reference genome, especially in genes with repetitive regions or paralogs, and highlights a problem in NGS data, that false positives are present even with the use of strict filtering criteria. In light of this finding, we took steps to verify ∼7% (142 mutations) of the mutations presented and confirmed 69% of these (98 mutations). The false positive rate for Illumina was 2% and was 29% for SOLiD, a discrepancy that is most likely a result of differences in the specific alignment parameters used for the different sequencing platforms.
PCR primer design.
All primers were designed using the automated primer design software PrimerZ37. PCR and sequencing were performed as previously described25. The sequences for the primers used for PCR amplification are listed in Supplementary Tables 9–11. All chromatograms were analyzed and variants annotated using Geneious Pro software (see URLs). All but one of the MAP3K5 and MAP3K9 mutations identified in the cell line discovery screen were confirmed to be tumor derived using DNA from the tumors from which the cell lines were established.
Analysis of variant amino acids in kinase domain structures.
We performed a BLAST38 search against the PDB39 to identify available structures for MAP3K5 and MAP3K9. PDB ID 2CLQ40 is an exact match for the kinase domain of MAP3K5; however, there were no significant hits for the other parts of the protein. PDB ID 3DTC41 is an exact match for the kinase domain of MAP3K9. MAP3K9 also has an SH3 domain, but there was no available structure for this domain. Amino acid alterations were mapped onto the corresponding structures, and each protein was structurally aligned with the structure of another kinase domain that is bound to ATP and a substrate (PDB ID 2PHK42), using MUSTANG43 to provide an approximate location for the ATP- and substrate-binding pockets. Structures incorporating single–amino acid changes were created, and the geometry of these models was minimized to idealize bond lengths, bond angles, planarities, chiralities, dihedrals and non-bonded interactions using PHENIX44.
Protein blot analysis.
Subconfluent HEK293T cells in six-well plates were transfected with 1 μg of expression plasmid for control EGFP or wild-type or mutant MAP3K5 or MAP3K9 using Lipofectamine 2000 (Invitrogen) at a 3:1 Lipofectamine:DNA ratio and harvested 48 h after transfection. Cells were lysed in ice-cold lysis buffer containing 20 mM HEPES, pH 7.8, 0.42 M NaCl, 0.5% NP40, 25% glycerol, 0.2 mM EDTA, 1.5 mM MgCl2, 0.1 mM PMSF and protease and phosphatase inhibitors. Samples (20 μg total protein) were resolved on 4–15% Mini-PROTEAN TGX gels (Bio-Rad) and transferred to PVDF membranes. The following antibodies from Cell Signaling Technology were used for detection of total and phosphorylated protein at a 1:1,000 dilution, except the antibody to MAP3K9, which was used at a 1:10,000 dilution: MAP3K5 (3762) and P-MAP3K5 (3761); MAP3K9 (5029), MAP2K4 (9152) and P-MAP2K4 (9156); MEK1 (9124), MEK2 (9125) and P-MEK1/2 (9121); ERK (4695) and P-ERK (4376); p38 (9212) and P-p38 (9215); and JNK (9258) and P-JNK (4671). Antibodies were also used to detect Myc (Upstate, 05-724; 1:1,000 dilution) and GAPDH (Trevigen, 2275-PC-1; 1:5,000 dilution). Enhanced chemiluminescence (ECL) detection of antibody binding was quantified using the GelDoc EZ System (Bio-Rad).
Kinase assays.
HEK293T cells were transfected with 5 μg of vector encoding either wild-type or mutant MAP3K9 or MAP3K5 for 48 h. Cells were lysed in ice-cold lysis buffer (10 mM KPO4, 1 mM EDTA, 10 mM MgCl2, 5 mM EGTA, 50 mM β-glycerophosphate, 0.5% NP40, 0.1% Brij35, 0.1% sodium deoxycholate, 1 mM NaVO4, 5 mM NaF, 2 mM DTT and Complete protease inhibitors (Roche)) and MAP3K5 proteins immunoprecipitated using 2 μg of antibody to FLAG (Sigma) for 2 h at 4 °C. Immunoprecipitated proteins were captured by adding protein G beads (Invitrogen) for 1 h at 4 °C, followed by three washes in lysis buffer. Immunocaptured protein-bead complexes were resuspended in 50 μl of kinase reaction buffer (25 mM Tris-HCl, 10 mM MgCl2, 5 mM β-glycerophosphate, 100 μM NaVO4, 1 mM dithiothreitol (DTT), 150 μM ATP (unlabeled), 10 μCi [32P]γATP and 1 μg of MBP). Kinase reactions were performed at room temperature for 30 min before termination by the addition of 5× sample buffer. Half of each reaction was loaded onto and resolved on an SDS-PAGE gel, and the gel was dried and exposed to film. For quantitation, dried gels were exposed to a Fuji phosphor imaging plate and quantified using a Fuji FLA-5000 phosphor imager with accompanying software. Quantification of the kinase activity of each of the mutant enzymes was normalized to that of the corresponding wild-type kinase.
siRNA and TMZ sensitization assays.
The melanoma cell lines (UACC903 and UACC647) were reverse transfected with three siRNA sequences targeting MAP3K9, in addition to a scrambled siRNA control (AllStars Non-Silencing, Qiagen). siRNA (1 μl of 0.667 μM) was printed into each well of barcoded 384-well plates with a solid-white bottom (Corning 8749) using a Biomek FX Laboratory Automation Workstation (Beckman Coulter). Transfection reagent, RNAiMAX (Invitrogen), was used to introduce the siRNA sequences into the cells. A mixture of RNAiMAX and serum-free RPMI medium was added to the plates (20 μl per well) using a BioTek μFill Dispenser (BioTek). Plates were then incubated for 30 min at room temperature to allow the formation of transfection reagent–nucleic acid complexes. Melanoma cells were trypsinized, quantified and resuspended in 10% FBS–RPMI assay medium and dispensed into the plates (20 μl per well) containing the siRNA using a μFill Dispenser (500 cells per well for UACC903; 750 cells per well for UACC647). Cells were incubated at 37 °C for 24 h before treatment with varying concentrations of TMZ (ranging from 1.2 μM to 2.5 mM) or vehicle alone in medium with 5% FBS by dispensing a 10-μl volume per well. The plates were further incubated at 37 °C for 5 d before cellular viability was measured using CellTiter-Glo luminescent reagent (Promega) and an Analyst GT Multimode Microplate Reader (Molecular Devices). The final assay volume (per well) contained a 13 nM concentration of each siRNA, 40 nl of RNAiMAX transfection reagent and 5% FBS (20 μl serum-free medium + 20 μl of medium with 10% FBS + 10 μl of medium with 5% FBS).
Quantification of MAP3K9 expression after siRNA transfection by qRT-PCR.
Cells were reverse transfected with three siRNA sequences targeting MAP3K9 in a 96-well plate and incubated for 5 d. RNA was extracted using an RNeasy MiniPrep kit according to the manufacturer's instructions (Qiagen), and RNA concentration was measured using a NanoDrop. QuantiTect primers for MAP3K9 and GAPDH were purchased (Qiagen), and cDNA was generated using a Power SYBR Green RNA-to-CT 1-Step kit (Applied Biosystems). All samples were run in triplicate on a 384-well PCR plate using an Applied Biosystems 7900HT Fast Real-Time PCR machine. GAPDH expression was measured as a control for sample normalization.
Quantification of MAP3K5 and MAP3K9 expression in melanoma cell lines by qRT-PCR.
Total RNA was prepared from melanoma cell lines using an RNeasy MiniPrep kit according to the manufacturer's instructions. For mRNA analyses, 0.5 μg of total RNA was reverse transcribed using Superscript III according to the manufacturer's instructions (Invitrogen), and MAP3K5, MAP3K9 and GAPDH expression was analyzed using SYBR Green PCR Master Mix (Applied Biosystems). Primer sequences were MAP3K5 (forward) 5′-cacgtgatgacttaaaatgcttg-3′ and MAP3K5 (reverse) 5′-agtcaatgatagccttccacagt-3′; MAP3K9 (forward) 5′-ggctgaagctcaaggatgg-3′ and MAP3K9 (reverse) 5′-gtgaacttgtgctggaaatcag-3′; and GAPDH (forward) 5′-ggctctccagaacatcatccctgc-3′ and GAPDH (reverse) 5′-gggtgtcgctgttgaagtcagagg-3′. qRT-PCR was performed using a 7900HT Fast Real Time PCR System.
Accession codes.
Nucleotide sequences are available from Entrez PubMed, including for MAP3K5 (NM_005923), MAP3K8 (NM_005204) and MAP3K9 (NM_033141). For three-dimensional modeling, structures were obtained from the Protein Data Bank (PDB) for the MAP3K5 kinase domain (ID 2CLQ) and the MAP3K9 kinase domain (3DTC) and kinase domain with ATP and a substrate bound (2PHK).
References
Flaherty, K.T. et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 363, 809–819 (2010).
Sjöblom, T. et al. The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274 (2006).
Wei, X. et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat. Genet. 43, 442–446 (2011).
Greenman, C. et al. Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 (2007).
Bamford, S. et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 91, 355–358 (2004).
Wei, X. et al. Analysis of the disintegrin-metalloproteinases family reveals ADAM29 and ADAM7 are often mutated in melanoma. Hum. Mutat. 32, E2148–E2175 (2011).
Wei, X. et al. Mutational and functional analysis reveals ADAMTS18 metalloproteinase as a novel driver in melanoma. Mol. Cancer Res. 8, 1513–1525 (2010).
Prickett, T.D. et al. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat. Genet. 41, 1127–1132 (2009).
Prickett, T.D. et al. Exon capture analysis of G protein–coupled receptors identifies activating mutations in GRM3 in melanoma. Nat. Genet. 43, 1119–1126 (2011).
Stark, M. & Hayward, N. Genome-wide loss of heterozygosity and copy number analysis in melanoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 67, 2632–2642 (2007).
Solomon, D.A. et al. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 68, 10300–10306 (2008).
Dhillon, A.S., Hagan, S., Rath, O. & Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 26, 3279–3290 (2007).
Johannessen, C.M. et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468, 968–972 (2010).
Davies, H. et al. Mutations of the BRAF gene in human cancer. Nature 417, 949–954 (2002).
Berger, A.H. & Pandolfi, P.P. Haplo-insufficiency: a driving force in cancer. J. Pathol. 223, 137–146 (2011).
Paige, A.J. Redefining tumour suppressor genes: exceptions to the two-hit hypothesis. Cell. Mol. Life Sci. 60, 2147–2163 (2003).
Durkin, J.T. et al. Phosphoregulation of mixed-lineage kinase 1 activity by multiple phosphorylation in the activation loop. Biochemistry 43, 16348–16355 (2004).
Takeda, K., Noguchi, T., Naguro, I. & Ichijo, H. Apoptosis signal–regulating kinase 1 in stress and immune response. Annu. Rev. Pharmacol. Toxicol. 48, 199–225 (2008).
Iriyama, T. et al. ASK1 and ASK2 differentially regulate the counteracting roles of apoptosis and inflammation in tumorigenesis. EMBO J. 28, 843–853 (2009).
Bisson, N. et al. Mice lacking both mixed-lineage kinase genes Mlk1 and Mlk2 retain a wild type phenotype. Cell Cycle 7, 909–916 (2008).
Middleton, M.R. et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J. Clin. Oncol. 18, 158–166 (2000).
Naumann, S.C. et al. Temozolomide- and fotemustine-induced apoptosis in human malignant melanoma cells: response related to MGMT, MMR, DSBs, and p53. Br. J. Cancer 100, 322–333 (2009).
Xu, Z., Maroney, A.C., Dobrzanski, P., Kukekov, N.V. & Greene, L.A. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol. Cell. Biol. 21, 4713–4724 (2001).
She, Q.B., Chen, N., Bode, A.M., Flavell, R.A. & Dong, Z. Deficiency of c-Jun-NH2-terminal kinase-1 in mice enhances skin tumor development by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 62, 1343–1348 (2002).
Pavey, S. et al. Microarray expression profiling in melanoma reveals a BRAF mutation signature. Oncogene 23, 4060–4067 (2004).
Fujita, P.A. et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 39, D876–D882 (2011).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Homer, N., Merriman, B. & Nelson, S.F. BFAST: an alignment tool for large scale genome resequencing. PLoS ONE 4, e7767 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Liu, X., Jian, X. & Boerwinkle, E. dbNSFP: a lightweight database of human non-synonymous SNPs and their functional predictions. Hum. Mutat. 32, 894–899 (2011).
Kumar, P., Henikoff, S. & Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Adzhubei, I.A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Chun, S. & Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561 (2009).
Schwarz, J.M., Rodelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576 (2010).
Siepel, A., Pollard, K.S. & Haussler, D. New methods for detecting lineage-specific selection. in Proceedings of the 10th International Conference on Research in Computational Molecular Biology (RECOMB 2006) 3909, 190–205 (Springer, 2006).
Tsai, M.F. et al. PrimerZ: streamlined primer design for promoters, exons and human SNPs. Nucleic Acids Res. 35, W63–W65 (2007).
Altschul, S.F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Berman, H.M. et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000).
Bunkoczi, G. et al. Structural and functional characterization of the human protein kinase ASK1. Structure 15, 1215–1226 (2007).
Hudkins, R.L. et al. Mixed-lineage kinase 1 and mixed-lineage kinase 3 subtype-selective dihydronaphthyl[3,4-a]pyrrolo[3,4-c]carbazole-5-ones: optimization, mixed-lineage kinase 1 crystallography, and oral in vivo activity in 1-methyl-4-phenyltetrahydropyridine models. J. Med. Chem. 51, 5680–5689 (2008).
Lowe, E.D. et al. The crystal structure of a phosphorylase kinase peptide substrate complex: kinase substrate recognition. EMBO J. 16, 6646–6658 (1997).
Konagurthu, A.S., Whisstock, J.C., Stuckey, P.J. & Lesk, A.M. MUSTANG: a multiple structural alignment algorithm. Proteins 64, 559–574 (2006).
Adams, P.D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Acknowledgements
The authors would like to acknowledge M. Aziz for data processing, M. Kassner for technical support and J.-C. Deza for graphics support. This work was funded through grants from the National Health and Medical Research Council of Australia, the Australian Centre for Vaccine Development, the US National Cancer Institute (5U01CA129447 to J.T.), the US National Cancer Institute, Division of Cancer Epidemiology and Genetics (to K.M.B.), and a charitable donation by Francis Najafi (to K.M.B. and J.T.).
Author information
Authors and Affiliations
Contributions
N.K.H., K.M.B., R.G. and J.T. devised the study. M.S.S., M.G.G., K.D.-R., S.T., J.R., D.M., I.N. and Y.W. performed the exome sequencing and analyzed the sequencing data. S.L.W., M.G.G., V.F.B., B.R.B., D.C., N.M.N., J.P.M., T.P. and H.Y. produced and analyzed the functional data. M.S.S., M.G.G., L.G.A., K.D.-R., V.Z. and D.Y. carried out confirmatory sequencing. C.W.S. and C.L. established the melanoma cell line panel and provided the fresh melanoma tumors. J.M.P. extracted and collated clinical records for the melanoma patients. M.S.S., S.L.W., M.G.G., V.F.B., J.P.M., H.Y. and N.K.H. wrote the manuscript. J.J.E. and B.K. performed protein modeling. D.C., C.S., N.T. and H.Y. performed the TMZ and siRNA sensitization studies and RT-PCR. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–5 and Supplementary Tables 1,2 and 4–11. (PDF 2797 kb)
Supplementary Table 3
Somatic mutations identified via exome sequencing (XLSX 464 kb)
Rights and permissions
About this article
Cite this article
Stark, M., Woods, S., Gartside, M. et al. Frequent somatic mutations in MAP3K5 and MAP3K9 in metastatic melanoma identified by exome sequencing. Nat Genet 44, 165–169 (2012). https://doi.org/10.1038/ng.1041
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.1041
This article is cited by
-
Genomic profiling of idiopathic peri-hilar cholangiocarcinoma reveals new targets and mutational pathways
Scientific Reports (2023)
-
DKK3 ameliorates neuropathic pain via inhibiting ASK-1/JNK/p-38-mediated microglia polarization and neuroinflammation
Journal of Neuroinflammation (2022)
-
Promoter methylation-regulated miR-148a-3p inhibits lung adenocarcinoma (LUAD) progression by targeting MAP3K9
Acta Pharmacologica Sinica (2022)
-
Inhibition of apoptosis signal-regulating kinase by paeoniflorin attenuates neuroinflammation and ameliorates neuropathic pain
Journal of Neuroinflammation (2019)
-
Mycobacterium bovis BCG in metastatic melanoma therapy
Applied Microbiology and Biotechnology (2019)
