
Abstract
The Psychiatric Genomics Consortium-Posttraumatic Stress Disorder group (PGC-PTSD) combined genome-wide case–control molecular genetic data across 11 multiethnic studies to quantify PTSD heritability, to examine potential shared genetic risk with schizophrenia, bipolar disorder, and major depressive disorder and to identify risk loci for PTSD. Examining 20 730 individuals, we report a molecular genetics-based heritability estimate (h2SNP) for European-American females of 29% that is similar to h2SNP for schizophrenia and is substantially higher than h2SNP in European-American males (estimate not distinguishable from zero). We found strong evidence of overlapping genetic risk between PTSD and schizophrenia along with more modest evidence of overlap with bipolar and major depressive disorder. No single-nucleotide polymorphisms (SNPs) exceeded genome-wide significance in the transethnic (overall) meta-analysis and we do not replicate previously reported associations. Still, SNP-level summary statistics made available here afford the best-available molecular genetic index of PTSD—for both European- and African-American individuals—and can be used in polygenic risk prediction and genetic correlation studies of diverse phenotypes. Publication of summary statistics for ∼10 000 African Americans contributes to the broader goal of increased ancestral diversity in genomic data resources. In sum, the results demonstrate genetic influences on the development of PTSD, identify shared genetic risk between PTSD and other psychiatric disorders and highlight the importance of multiethnic/racial samples. As has been the case with schizophrenia and other complex genetic disorders, larger sample sizes are needed to identify specific risk loci.
Similar content being viewed by others

Introduction
Posttraumatic stress disorder (PTSD) is a common and debilitating mental disorder that occurs in some individuals following a traumatic event. It includes symptoms such as reexperiencing the event, avoidance of event-related stimuli and chronic hyperarousal. In the United States, 1 in 9 women and 1 in 20 men will meet criteria for the diagnosis at some point in their lives.1 The societal impact of PTSD is large, with increased rates of suicide, hospitalization and substance use.2 Regarding etiology, the role of nature versus nurture in response to traumatic events has been debated for over a century.3 Psychiatrists who treated ‘shell shock’ in soldiers returning from World War I argued over whether soldiers who succumbed to the stressors of war were ‘moral invalids’ or whether such breakdowns could occur in any man who was ‘buried in a trench or saw his friend’s brains scattered before him’.4 This historical controversy has been reignited as the age of genomic medicine has reached PTSD. Genomic research is given the highest priority in the US National Research Action Plan on PTSD that was established in response to an executive order by President Obama aimed at improving mental health care and services for veterans, military service members and their families.5 However, fundamental questions remain as to the role of genetic factors in PTSD etiology.
Twin study estimates of PTSD heritability range from 24 to 72% following trauma,6, 7, 8, 9 with female heritability two to three times higher than that in males.8 Twin studies have also documented that genetic influences on PTSD are shared with other mental disorders, particularly major depression.10, 11, 12 Such studies have made important contributions beyond heritability estimation; for example the discordant twin study by Gilbertson et al.13 showed that smaller hippocampal volume is likely a risk factor for PTSD rather than a consequence of the disorder. Despite this excellent work, the importance of genetic influences for PTSD etiology is not universally accepted among mental health clinicians or researchers. This may be in part because the latent modeling of genetic variation in twin studies is not easily interpretable to those not immersed in the methodology of behavioral genetics.
To more definitively establish the contribution of genetic variants to PTSD risk, to examine the genetic overlap between PTSD and other mental disorders and to identify risk loci, the Psychiatric Genomics Consortium-Posttraumatic Stress Disorder group (PGC-PTSD) group employed the consortium science approach of aggregating genomic (genome-wide association study (GWAS)) data from multiple contributing groups. In recent years, the consortium approach has delivered groundbreaking results for many complex genetic phenotypes—including height,14 diabetes15 and schizophrenia16—and here we report initial findings on the genetic architecture of PTSD.
Materials and methods
Overview
Eleven groups contributed data for analysis; nine contributed individual-level data and two groups could only share single-nucleotide polymorphism (SNP)-level summary statistics. These two groups implemented the quality control (QC) and GWAS analyses outlined below before sharing summary statistics. Individual-level genotypes are necessary for genome-wide complex trait analysis (GCTA) and polygenic risk scoring (PRS), and hence these two data sets are not included in the polygenic analyses. For all data sets, the first analytical step was ancestry assignment. As described below, this yielded 19 ancestry-assigned data sets for analysis across the 11 contributing studies. Next, QC procedures and imputation were performed and then single SNP (GWA and meta-analyses) and polygenic analyses (GCTA, PRS, and linkage disequilibrium (LD) score regression (LDSC)) were completed.
Participants
All participants were adults. Contributing studies provided individual-level genotype data or summary statistics consistent with their institutional review board-approved protocols. Descriptions of each of the 11 contributing studies are provided in the Supplementary Information. In many cohorts included in the consortium, all the controls were trauma exposed (for example, Nurses Health Study II PTSD substudy). Thus, in the combined analysis, the vast majority of controls were trauma exposed (N controls=15 548, N trauma-exposed controls=13 638, 87.7% of controls were trauma exposed). Case and control numbers along with other data set information is provided in Supplementary Table S1, and Figure 1 illustrates the study and analytical design.

Study design for phase 1 PGC-PTSD: (a) single variant GWAS and meta-analyses (b) polygenic analyses. (a) Each of the 19 gray boxes represents one ancestry-assigned data set/GWAS. Within-ancestry meta-analyses were followed by the transethnic meta-analysis. (b) Blue boxes denote polygenic analyses. LDSC could not be applied to the African-American (AA) subsample. Latino/Hispanic (LA) and South African samples were deemed too small for polygenic analyses. GWAS, genome-wide association study; LDSC, linkage disequilibrium score regression; PGC-PTSD, Psychiatric Genomics Consortium-Posttraumatic Stress Disorder group.
Ancestry assignment
SNPweights software17 was used to assign ancestry, applied to uncleaned data because the use of Hardy–Weinberg equilibrium filters can remove ancestry informative SNPs in samples with mixed ancestry background, and is therefore not compatible with this protocol. Ancestry assignment is separate from controlling for ancestry analytically, described below. Briefly, SNPweights uses external genomic reference panels to derive ancestry informative weights for individual SNPs. We used the four-group, continental ancestry panel to quantify individual participants’ proportions of: African (YRI), European (CEU), Asian (ASI) and Native American (NAT)17 ancestry. The specific cutoffs implemented in this report for assigning ancestry—which were selected after inspection of principal components (PC) plots — and adjusting cutoffs—were as follows: European American (EA) was defined as having ⩾90% European ancestry. African American (AA) was defined as ⩾90% for the combination of African and European ancestry, and also <3% Asian and Native American ancestry. Individuals who self-identify as Latino or Hispanic (LA) in the United States may have predominately European ancestry and may have relatively recent admixture with Native American ancestry or both Native American and African ancestry. In this report we focused on LA individuals with two-way admixture; cutoffs used were ⩾85% for the combination of European and Native American ancestry, <10% African ancestry and <3% Asian Ancestry. With exception of the South African data (see below), individuals not falling into the EA, AA and LA categories were excluded from analysis (on the basis of low case and/or control numbers).
Data contributed from South Africa were handled separately because the African ancestry panel used in SNPweights (YRI) is a West African sample. For the South African sample, the typical GWAS method of conducting principal components analysis (PCA), followed by visual inspection of data, was used. In doing so, we identified two populations, which are the two South African data sets in this report. For comparison of PGC-PTSD ancestry to previously published meta-analyses of psychiatric phenotypes see Figure 2a. Recent admixture is evident for AA and LA participants in this study (Figure 2b).

Ancestral composition for phase 1 of PGC-PTSD and principal components (PC) plot of individuals’ data. (a) PGC-PTSD (left) compared with the largest psychiatric meta-analyses (center) and estimated world ancestry (right). (b) Plot of first two principal components and assigned ancestry according to the protocol described in the text. Each dot is one individual. GWAS, genome-wide association study; PGC-PTSD, Psychiatric Genomics Consortium-Posttraumatic Stress Disorder group.
QC methods, relatedness testing and imputation
QC procedures were performed sequentially on each of the 19 data subsets as follows: monomorphic SNPs and SNPs with missingness >0.05 were removed, and individuals with missingness >0.02 were removed. Individuals with heterozygosity >|0.2| and individuals failing sex checks were removed. SNPs with missingness >0.02 were removed (a more stringent SNP missingness filter was applied after individual level filters). SNPs with differential missingness between cases and controls >0.02 were removed. SNPs failing Hardy–Weinberg equilibrium: controls (P<1 × 10−6) and cases (P<1 × 10−10) were removed. All analyses were performed using second-generation PLINK.18
PCA was performed within each data set and then across all data sets using FastPCA.19 PCA was conducted on high-quality SNPs with low LD passing filters: SNP directly genotyped in all data sets; minor allele frequency (MAF) >0.05; Hardy–Weinberg equilibrium P>1 × 10−4; not strand ambiguous (i.e. no AT or GC SNPs); not in high LD region (MHC chr6:25–35 Mb, chr8 inversion chr8:7–13 Mb); and r2 between SNPs <0.2 (i.e., the PLINK option: ‘—indep-pairwise 200 100 0.2’, applied twice). Within each data set, scatterplots of PCs were visually examined and outliers removed. This process was repeated until cases and controls appeared evenly interspersed across all PC pairs.
Imputation to the 1000 Genomes20 phase 1 reference was performed within the PGC pipeline16 using SHAPEIT for phasing21 and IMPUTE2 for imputation.22 Imputation was performed with a chunk size of 3 Mb with default parameters on the full set of 2186 phased haplotypes (August 2012, 30 069 288 variants, release ‘v3.macGT1’). Samples were then combined (within ancestry groups) for relatedness testing and calculation of PC covariates. The same filters as above were employed and we removed one individual from each pair of related or duplicate individuals (pi-hat value >0.2), preferentially retaining cases.
Single variant analyses, gene and pathway analyses
Single variant analysis (GWAS within each of the 19 data subsets) was performed using an additive model in PLINK, with the first 10 PCs as covariates, on dosage data. Fixed-effects meta-analysis was accomplished using METAL23, 24 with inverse variance weighting. Plotting was performed in R.25 Analyses were completed with both study-specific PCs and with PCs computed within each ancestry group (‘generic’ PCs for AA, EA, LA and separately the two South African data sets), with similar results. Final results in Manhattan, QQ plot, top hits tables and Supplementary Information online use study-specific PCs.
Gene and pathway analyses were completed using MAGMA (Multi-marker Analysis of GenoMic Annotation)26 and default parameters as in the manual (version 1.06). Gene and pathway analyses were conducted on the 16 data sets with individual-level genotype data (versus SNP-level P-values) so that we could control for ancestry using PCs. This was particularly important for the AA, LA and South African samples given poorer external sequence data resources that are needed for the option that uses summary statistics. Thus, we performed gene-based analyses on each of the 16 data sets with genotype data and PCs, followed by pathway analyses on the gene-level results (as per MAGMA procedures). For completeness we also then used the summary statistic method on the three remaining data sets for which raw data were not available, and meta-analyzed (at the gene level, per MAGMA procedures) with the other 16 data sets. Like de Leeuw et al.,26 we used the MSigDB Canonical Pathways because this list contains a wide variety of gene sets, drawn from different gene-set databases, thus providing results that are not overly dependent on the choice of a narrow set of gene sets.
SNP-chip heritability estimation with GCTA
GCTA27, 28, 29 was used to estimate SNP-chip heritability (h2SNP) in the EA and AA subsamples (separately) as follows: following QC, data sets were combined using PLINK18, 30 (that is, the 7 EA bed/bim/fam file sets were combined into one EA7 bed/bim/fam file set). Genetic relationship matrices (GRMs) were made (one chromosome at a time for computational efficiency) using ‘—chr n’ for chromosomes 1–22, ‘—maf 0.01’ to restrict to SNPs with MAF >1%, ‘—make-grm-bin’ to make the GRMs and then combined with ‘—mgrm’ command. Heritability estimation with ‘—reml-no-constrain’ command, specifying a GRM with ‘—grm-bin’, a phenotype file with ‘—pheno’, prevalence with ‘—prevalence’ quantitative covariates (here, PCs 1–10) with ‘—qcovar' and binary covariates (here, sex and study indicator covariates) with ‘—covar’. Prevalence was specified as 11% (females), 5% (males) and 8% (combined).
Polygenic risk scoring
PRS was conducted using PLINK18, 30 for the three major adult psychiatric disorders (schizophrenia (SCZ),16 bipolar disorder (BIP)31 and major depressive disorder (MDD)32) for which GWAS results from large studies are publicly available. SNP-level summary statistics from each of the three ‘discovery’ disorders were used to create the ‘score’ files. Each individual in this study was scored for genetic risk by weighting risk alleles according to the natural log of the odds ratio (OR) from each of the discovery disorder meta-analyses. The following commands were used: to specify the bed/bim/fam fileset from this study to be scored (—bfile), specify the file with logORs (—score), stop the default behavior of mean imputation (no-mean-imputation), specify the file with P-values from the discovery disorder (—q-score-file) and provide specified ranges of P-values to be scored (—q-score-range). See Supplementary Table S2 for 38 P-value bins (12 bins per disorder plus one additional bin for genome-wide significant loci for both SCZ and BIP). After scoring, the significance and magnitude of polygenic PTSD prediction was calculated for each of the 38 P-value bins. Two logistic regressions were run. The first regressed PTSD on polygenic risk score, 10 PCs and study indicator covariates. The second regression was the same as the first, but with the polygenic risk score term removed. Nagelkerke’s r2 was calculated for both models, and the difference was the r2 for the polygenic risk score term.
SNP-chip heritability and genetic correlation estimation with LDSC
LDSC33, 34 was used on the SNP-level summary statistics from the 7 EA data sets for which raw genotype data were available. With raw data we could rule out population stratification and the presence of related individuals, and consequently the constrained version of LDSC could be used, affording greater power for heritability estimation. By constraining the LDSC regression intercept to be 1 (that is, the expected χ2 for a single SNP with LD score equal to zero and with no influence from population stratification), there is one less parameter to estimate in the LDSC regression and standard error of the heritability estimate is reduced. For genetic correlation, we report both constrained and unconstrained results. As with GCTA, we use population prevalence estimates of 11% (female), 5% (male) and 8% (combined). Separate male and female heritability estimates were calculated using sex-specific subsamples of the data. General instructions for LDSC are provided here: https://github.com/bulik/ldsc
Results
Single variant (GWAS and meta-analyses), gene and pathway analyses
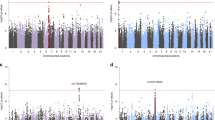
No variants achieved genome-wide significance in either the transethnic or EA meta-analyses. For Manhattan plots, QQ plots and top hits tables for each of the three meta-analyses, see Supplementary Figures S1 and S2 and Supplementary Tables S3–S5. Though not currently informative about individual risk loci, summary statistics are useful for polygenic predictions and cross-disorder analyses, and are available for download: www.med.unc.edu/pgc.
In the AA meta-analysis, one variant on chromosome 13 exceeded genome-wide significance (rs139558732, OR=2.19, P=3.33 × 10−8). This SNP and nearby variants were not present for analysis in the EA and LA studies because of low frequency (MAF ⩽1%) in those populations. The variant was present in the two South African data sets, and was no longer genome-wide significant in the transethnic meta-analysis (OR=2.05, P=1.31 × 10−07, only AA and South African data sets contributing data). To further investigate the possibility that this chromosome 13 locus in the KLHL1 gene (see Supplementary Figure S3 for regional plot in AA meta-analysis) was associated with PTSD in AA individuals, we requested data from the Army STARRS consortium, and meta-analyzed results from their data and ours.35 Doing so also resulted in loss of genome-wide significance for rs139558732 (OR=1.90, P=1.0 × 10−06). Thus, despite nominally achieving genome-wide significance in the AA meta-analyses, we do not report rs139558732 as a PTSD risk variant.
We also conducted meta-analyses of our data with the two SNPs reported as genome-wide significant by the Army STARRS consortium.35 STARRS reported one locus for AA (rs159572) and one for EA (rs11085374).35 Results after meta-analysis for the reported SNPs were not genome-wide significant (rs159572, P=0.2744; rs11085374, P=1.74 × 10−05), perhaps unsurprisingly given lack of consistent association within the Army STARRS report itself.35
Gene-based and pathway analyses using MAGMA26 yielded no significant results after correction for multiple testing, consistent with observation from other GWAS analyses in which gene and pathway methods did not yield significant findings until the primary GWAS was well powered enough to identify specific risk loci. Nevertheless, we present the top gene and pathway results in Supplementary Tables S7 and S8. Top results for genes did not include PTSD candidate genes, though GRINA, a glutamate receptor, was among the top 20 results. The top pathway was the neurotrophic factor-mediated Trk receptor signaling pathway,36 which includes BDNF (brain-derived neurotrophic factor), NGF (nerve growth factor) and other neurotrophin-related genes, that collectively regulate synaptic strength and plasticity in the nervous system of mammals.37 Given the primacy of learning in the PTSD phenotype, this is an intriguing pathway result that awaits follow-up in a better-powered analysis.
Heritability estimation from molecular genetic (SNP) data
LDSC and GCTA were used to estimate SNP-chip heritability (h2SNP). Using both methods, female heritability estimates (29% average of LDSC=0.36 s.e.=0.12, P=0.003 and GCTA=0.21, s.e.=0.09, P=0.019) were comparable to those for other psychiatric disorders (see Figure 3). In contrast, in males, the point estimate was not significantly different from zero and was lower than previously reported estimates for major psychiatric disorders38 (7% average of LDSC=0.05, s.e.=0.13, P=0.69 and GCTA=0.08, s.e.=0.10, P=0.43). Analyzing males and females together—as has been done in published reports—the point estimate was 15% (average of LDSC=0.18, s.e.=0.06, P=0.003 and GCTA=0.12, s.e.=0.05, P=0.016). Partitioned heritability estimation39 will be conducted on future releases of PGC-PTSD data when the z-score for overall heritability is higher. The current z-score is 3.0, and a z-score of 7 was deemed adequate for partitioned heritability analysis in the primary publication.39

PTSD SNP-chip heritability (h2SNP) overall and for males and females separately and comparison with other psychiatric disorders. Gray bars denote PTSD heritability estimates. Slashed bars reflect SCZ, BIP and MDD heritability estimates calculated using LDSC as applied to published data.16, 31, 32 Red lines denote twin study heritability estimates, see Discussion. European-American (EA) samples only per description in text; error bars reflect s.e. BIP, bipolar disorder; LDSC, linkage disequilibrium score regression; MDD, major depressive disorder; PTSD, posttraumatic stress disorder; SCZ, schizophrenia; SNP, single-nucleotide polymorphism.
In our sample, the point estimate for heritability among AAs was much lower and not statistically different from zero (GCTA=−0.005, s.e.=0.04, P=0.45). Unlike our EA samples, we could not compare this estimate with one from LDSC because LDSC is currently not suitable for use in populations with recent admixture (for example, AA and LA).
Cross-disorder genetic effects between PTSD and SCZ, BIP and MDD
Because of the relatively low power for heritability estimation for PTSD with our current samples, we decided to limit the number of genetic correlations tested to focus on three major adult psychiatric disorders (SCZ,16 BIP31 and MDD32) for which GWAS results from large studies are publicly available. As shown in the top (EA) portion of Table 1, PRS suggested overlap with both SCZ and BIP even after overly stringent (because of correlated tests) Bonferroni correction. Moreover, 58% (22/38) of PRS tests among EA individuals were nominally significant. For variance explained by discovery disorder P-value bins and associated statistics, see Supplementary Table S6. In contrast, PRS revealed no evidence of overlap with MDD, but this could be because of low power in both the MDD40 and the present PTSD analysis.41 This possibility is consistent with evidence of PTSD-MDD genetic overlap found with the more powerful (constrained) version of LDSC. Constrained LDSC results further supported the PRS finding of shared PTSD-SCZ genetic effects.
Discussion
We believe the present report provides the first molecular genetic evidence of PTSD heritability and extends previous findings about shared genetic effects between PTSD and other disorders.8, 10, 11, 12 These results portend future success in identifying specific PTSD risk loci when viewed in the context of the trajectories of genetic discoveries for other complex genetic disorders16, 42 that yielded significant polygenic results before identification of robust single variant associations. Specifically, examining 20 730 individuals, we found a molecular genetics-based heritability estimate for EA females of 29% that is similar to h2SNP for schizophrenia and is substantially higher than h2SNP in EA males (estimate not distinguishable from zero). We found strong evidence of overlapping genetic risk between PTSD and schizophrenia along with more modest evidence of overlap with bipolar and major depressive disorder. The combination of informative polygenic results and lack of robust single variant analyses (in our data and in the evaluation of previously reported PTSD loci35, 43, 44, 45, 46) strongly suggests the need for better powered analyses.
Regarding polygenic results, a distinction must be drawn between the EA and AA components of this study. For reasons discussed below, the present report was far more informative for PTSD genetics in EA individuals than AA individuals, despite comparable sample sizes (both ∼10 000 with 25% cases). Among EA individuals, these molecular genetic results parallel twin study results6, 7, 8, 9 that showed moderate heritability for PTSD overall and higher heritability in females than males.
There are several possible reasons why female heritability may be higher than male heritability, including differences in trauma exposure47, 48 that itself is heritable,49, 50 sex-based biology and sex-based symptom differences. Given that heritability estimates convey the relative importance of genetic and environmental influences, sex-based differences in either would affect heritability. We can assume comparable genetic variation in female and male subjects because we only used genetic variants that conformed to Hardy–Weinberg equilibrium expectations (thus implying no selective removal of particular alleles). This leaves sex-based differences in relevant environmental influences as one explanation and, indeed, rates of exposure to various types of trauma are known to vary by sex.51 As well, particular trauma types vary in the degree to which they are associated with PTSD.52 Greater variability in the profile of PTSD-inducing environmental influences in males, as compared with females, would lead to greater relative importance of environmental factors in the development of PTSD in males, and this could explain observed lower heritability estimates in males. This possibility could be tested via detailed study of trauma and environmental histories of males and females, combined with mathematical quantification of PTSD risk based on the frequency and magnitude of PTSD-inducing effects of relevant variables.
Second, female and male sex may be viewed as environmental variables, in that genetic variation is expressed in the context of a particular individual, and there are differences in female and male biology (for example, sex hormone levels). To the extent that the female biological ‘environment’ is more conducive to the expression of genetic variation, and the male biological ‘environment’ tends to dampen genetically influenced variability in PTSD liability, sex itself may account for different heritability estimates in females vs males. Biological differences have been reported between sexes regarding responses to trauma51 and responses to environmental variables thought to mediate the later development of PTSD.53 Thus, there may be sex-based biological differences in trauma liability. In order for such effects to explain the observed greater heritability in females compared with males, the specified biological processes would need to lead to greater expression of genetically influenced liability to PTSD in females compared with males.
The most pedestrian explanation for sex-based heritability differences for PTSD is that reliability and/or validity of PTSD diagnosis differs by sex. Lower male heritability could be a result of lower reliability and validity of PTSD diagnosis in males, because heritability estimates are always capped by reliability and validity of measurement. Regarding this possibility, it would be worthwhile to examine reasons why particular populations might systematically over- or under-report PTSD (thereby decreasing reliability and validity). Perhaps cultural factors that are more permissive for accurate reporting of PTSD symptoms in females contribute to more precise measurement of PTSD in females, and this would permit higher heritability estimates. The same issue of reliability and validity applies if diagnostic nosology is more appropriate for females than males. If the current PTSD diagnosis ‘carves nature at it’s joints’ better for females than males, there will be a lower bound on male heritability estimates compared with females.
In sum, greater measurement precision of phenotypic and environmental variables relevant to PTSD, as well as PTSD diagnosis, will aid accurate estimation of heritability. It will also increase power in primary GWAS analyses. PGC-PTSD is pursuing these strategies for continued GWAS efforts, in addition to increasing sample size.
A common misconception about SNP-chip heritability estimates calculated with GCTA and LDSC is that they should be similar to twin study estimates, when in reality twin studies have the advantage of capturing all genetic effects—common, rare and those not genotyped by available methods. Thus, the assumption should be that h2SNP<h2TWIN when using GCTA and LDSC, and this is what we observe for PTSD, as has been observed for many other phenotypes.54 Though somewhat limited by power in the present study, strong evidence of shared genetic effects between PTSD and SCZ, and more modest evidence of shared effects with both MDD and BIP, is consistent with recent reports of partially shared genetic effects across nearly all psychiatric disorders34, 38 and with twin study evidence of shared genetic influences on MDD and PTSD.10, 11, 12 No evidence of overlap was found with attention deficit hyperactivity disorder and autism. However, this could be because of low power and should be reexamined when PGC-PTSD is substantially expanded.
In contrast, there were no significant findings for heritability or shared genetic effects in AA individuals. This should not be taken as evidence that genetic effects in AA individuals differ from those in EA individuals. Rather, this report highlights an interpretational disparity between EA and AA individuals. There are no twin studies conducted in primarily non-EA populations, and hence there is no prior information indicating whether heritability might differ across ancestry groups. Second, polygenic methods like PRS and LDSC rely on external data resources that are far less common generally, and nonexistent for many, non-EA populations. Third, newer methods, such as LDSC, have not been adapted for recently admixed genomes, and thus they cannot be applied to AA and many LA individuals. Other factors responsible for lower power in the AA PTSD analysis are Eurocentric bias on genotyping arrays and the inherently greater genetic variation in the African portions of AA individuals’ chromosomes, necessitating more markers to achieve the same proportion of genomic coverage. Given these factors, publication of this relatively large sample of AA individuals is particularly important.
Our findings suggest that a sample of ∼10 000 individuals (with 25% cases) is not sufficient—in EAs or in AAs—to identify robust risk loci. A larger sample size and greater genotyping coverage (particularly in AA individuals) will afford greater power in future single variant association analyses. As well, it is possible that highly standardized phenotyping and/or ascertainment of more specific populations may increase power by decreasing phenotypic heterogeneity, as is arguably the explanation for success of the CONVERGE consortium.55 Future work needs to more carefully consider trauma exposure, a necessary though not sufficient condition for the development of PTSD. Although the majority of controls (87.7%) in the current analysis were trauma exposed, the inclusion of nontrauma-exposed controls may have reduced power to detect PTSD loci. Moreover, sex differences in both type and quantity of trauma exposure are well documented56 and will be important to consider in future research examining sex differences in heritability. In the interim, the successful polygenic analyses discussed above (that is, significant heritability estimates and genetic correlations) mean that the summary statistics made available with this report will be informative for PTSD studies when used with appropriate polygenic methods (for example, PRS and LDSC).
In summary, we find that PTSD—a disorder that by definition requires an environmental exposure, trauma—is also partly genetic in origin. This result comes as a foregone conclusion for researchers steeped in the behavioral genetics literature, but for those unfamiliar with twin studies or skeptical of their results, heritability based on molecular genetic data is compelling. Reassuringly, these molecular genetic heritability results are consistent with twin studies. All available evidence suggests that PTSD heritability among females is higher than males. Overall, PTSD heritability is comparable to that of MDD, but female heritability is close to that of SCZ and BIP, two of the most genetically influenced psychiatric disorders. Male PTSD SNP-chip heritability, in contrast, was lower than any recorded for a major psychiatric disorder and not distinguishable from zero with current sample sizes. Although large in comparison with previous reports, the current sample size of over 20 000 was still a limiting factor in single variant analyses. The strong evidence of shared genetic effects between PTSD and SCZ is worth further investigation and consistent with data from birth cohort studies that have found early childhood factors such as low IQ57, 58 and psychotic symptoms59 to be risk factors for both syndromes, but not for MDD or BIP. More variable evidence suggests that larger samples are needed to precisely resolve genetic overlap for BIP and MDD. Finally, the distribution of individuals of different ancestries in this analysis illuminated analytical and interpretational disparities for individuals of all ancestries as compared with EA individuals, and highlights the need for large genetic studies in non-European populations. Data from the largest genetic examination of PTSD to date demonstrate significant heritability for the development of PTSD, suggest at least partial genetic mediation of comorbidities, and outline important areas for future progress in understanding the genomic architecture of PTSD.
References
Kessler RC, Demler O, Frank RG, Olfson M, Pincus HA, Walters EE et al. Prevalence and treatment of mental disorders, 1990 to 2003. N Engl J Med 2005; 352: 2515–2523.
Davidson JR . Trauma: the impact of post-traumatic stress disorder. J Psychopharmacol Oxf Engl 2000; 14 (2 Suppl 1): S5–S12.
Herman J . Trauma and Recovery: The Aftermath of Violence—From Domestic Abuse to Political Terror. Basic Books: New York, 1997.
Myers C . A final contribution to the study of shell shock - being a consideration of unsettled points needing investigation. Lancet 1915; 185: 316–330.
Department of Veterans Affairs. National Research Action Plan: Responding to the Executive Order Improving Access to Mental Health Services for Veterans, Service Members and Military Families. CreateSpace Independent Publishing Platform, 2014.
True WR, Rice J, Eisen SA, Heath AC, Goldberg J, Lyons MJ et al. A twin study of genetic and environmental contributions to liability for posttraumatic stress symptoms. Arch Gen Psychiatry 1993; 50: 257–264.
Stein MB, Jang KL, Taylor S, Vernon PA, Livesley WJ . Genetic and environmental influences on trauma exposure and posttraumatic stress disorder symptoms: a twin study. Am J Psychiatry 2002; 159: 1675–1681.
Sartor CE, McCutcheon VV, Pommer NE, Nelson EC, Grant JD, Duncan AE et al. Common genetic and environmental contributions to post-traumatic stress disorder and alcohol dependence in young women. Psychol Med 2011; 41: 1497–1505.
Wolf EJ, Mitchell KS, Koenen KC, Miller MW . Combat exposure severity as a moderator of genetic and environmental liability to post-traumatic stress disorder. Psychol Med 2014; 44: 1499–1509.
Wolf EJ, Miller MW, Krueger RF, Lyons MJ, Tsuang MT, Koenen KC . Posttraumatic stress disorder and the genetic structure of comorbidity. J Abnorm Psychol 2010; 119: 320–330.
Koenen KC, Fu QJ, Ertel K, Lyons MJ, Eisen SA, True WR et al. Common genetic liability to major depression and posttraumatic stress disorder in men. J Affect Disord 2008; 105: 109–115.
Sartor CE, Grant JD, Lynskey MT, McCutcheon VV, Waldron M, Statham DJ et al. Common heritable contributions to low-risk trauma, high-risk trauma, posttraumatic stress disorder, and major depression. Arch Gen Psychiatry 2012; 69: 293–299.
Gilbertson MW, Shenton ME, Ciszewski A, Kasai K, Lasko NB, Orr SP et al. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nat Neurosci 2002; 5: 1242–1247.
Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 2010; 467: 832–838.
Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 2010; 42: 579–589.
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014; 511: 421–427.
Chen C-Y, Pollack S, Hunter DJ, Hirschhorn JN, Kraft P, Price AL . Improved ancestry inference using weights from external reference panels. Bioinforma Oxf Engl 2013; 29: 1399–1406.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ . Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015; 4: 7.
Galinsky KJ, Bhatia G, Loh P-R, Georgiev S, Mukherjee S, Patterson NJ et al. Fast Principal-Component Analysis Reveals Convergent Evolution of ADH1B in Europe and East Asia. Am J Hum Genet 2016; 98: 456–472.
Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA et al. A map of human genome variation from population-scale sequencing. Nature 2010; 467: 1061–1073.
Delaneau O, Marchini J, Zagury J-F . A linear complexity phasing method for thousands of genomes. Nat Methods 2012; 9: 179–181.
Howie BN, Donnelly P, Marchini J . A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009; 5: e1000529.
Sanna S, Jackson AU, Nagaraja R, Willer CJ, Chen W-M, Bonnycastle LL et al. Common variants in the GDF5-UQCC region are associated with variation in human height. Nat Genet 2008; 40: 198–203.
Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet 2008; 40: 161–169.
Ihaka R, Gentleman R . R: a language for data analysis and graphics. J Comput Graph Stat 1996; 5: 299–314.
de Leeuw CA, Mooij JM, Heskes T, Posthuma D . MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 2015; 11: e1004219.
Lee SH, Wray NR, Goddard ME, Visscher PM . Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet 2011; 88: 294–305.
Yang J, Lee SH, Goddard ME, Visscher PM . GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011; 88: 76–82.
Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet 2010; 42: 565–569.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Psychiatric GWAS Consortium - Bipolar Disorder. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 2011; 43: 977–983.
Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium, Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 2013; 18: 497–511.
Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, et al. Schizophrenia Working Group of the Psychiatric Genomics Consortium. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 2015; 47: 291–295.
Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh P-R et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 2015; 47: 1236–1241.
Stein MB, Chen C, Ursano RJ, Cai T, Gelernter J, Heeringa SG et al. Genome-wide association studies of posttraumatic stress disorder in 2 cohorts of us army soldiers. JAMA Psychiatry 2016; 73: 695–704.
Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P . The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015; 1: 417–425.
Nestler E, Hyman S, Malenka R . Molecular Neuropharmacology: A Foundation for Clinical Neuroscience, Second Edition. McGraw-Hill Professional: New York, 2008.
Cross-Disorder Group of the Psychiatric Genomics Consortium, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 2013; 45: 984–994.
Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 2015; 47: 1228–1235.
Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium, Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 2013; 18: 497–511.
Dudbridge F . Power and predictive accuracy of polygenic risk scores. PLoS Genet 2013; 9: e1003348.
Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460: 748–752.
Nievergelt CM, Maihofer AX, Mustapic M, Yurgil KA, Schork NJ, Miller MW et al. Genomic predictors of combat stress vulnerability and resilience in U.S. Marines: a genome-wide association study across multiple ancestries implicates PRTFDC1 as a potential PTSD gene. Psychoneuroendocrinology 2015; 51: 459–471.
Guffanti G, Galea S, Yan L, Roberts AL, Solovieff N, Aiello AE et al. Genome-wide association study implicates a novel RNA gene, the lincRNA AC068718.1, as a risk factor for post-traumatic stress disorder in women. Psychoneuroendocrinology 2013; 38: 3029–3038.
Xie P, Kranzler HR, Yang C, Zhao H, Farrer LA, Gelernter J . Genome-wide association study identifies new susceptibility loci for posttraumatic stress disorder. Biol Psychiatry 2013; 74: 656–663.
Logue MW, Baldwin C, Guffanti G, Melista E, Wolf EJ, Reardon AF et al. A genome-wide association study of post-traumatic stress disorder identifies the retinoid-related orphan receptor alpha (RORA) gene as a significant risk locus. Mol Psychiatry 2012; 18: 937–942.
Benjet C, Bromet E, Karam EG, Kessler RC, McLaughlin KA, Ruscio AM et al. The epidemiology of traumatic event exposure worldwide: results from the World Mental Health Survey Consortium. Psychol Med 2016; 46: 327–343.
Tolin DF, Foa EB . Sex differences in trauma and posttraumatic stress disorder: a quantitative review of 25 years of research. Psychol Bull 2006; 132: 959–992.
Kremen WS, Koenen KC, Afari N, Lyons MJ . Twin studies of posttraumatic stress disorder: differentiating vulnerability factors from sequelae. Neuropharmacology 2012; 62: 647–653.
Afifi TO, Asmundson GJG, Taylor S, Jang KL . The role of genes and environment on trauma exposure and posttraumatic stress disorder symptoms: a review of twin studies. Clin Psychol Rev 2010; 30: 101–112.
Olff M, Langeland W, Draijer N, Gersons BPR . Gender differences in posttraumatic stress disorder. Psychol Bull 2007; 133: 183–204.
Breslau N, Chilcoat HD, Kessler RC, Peterson EL, Lucia VC . Vulnerability to assaultive violence: further specification of the sex difference in post-traumatic stress disorder. Psychol Med 1999; 29: 813–821.
Francis DD, Young LJ, Meaney MJ, Insel TR . Naturally occurring differences in maternal care are associated with the expression of oxytocin and vasopressin (V1a) receptors: gender differences. J Neuroendocrinol 2002; 14: 349–353.
Visscher PM, Brown MA, McCarthy MI, Yang J . Five years of GWAS discovery. Am J Hum Genet 2012; 90: 7–24.
CONVERGE Consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 2015; 523: 588–591.
Benjet C, Bromet E, Karam EG, Kessler RC, McLaughlin KA, Ruscio AM et al. The epidemiology of traumatic event exposure worldwide: results from the World Mental Health Survey Consortium. Psychol Med 2016; 46: 327–343.
Koenen KC, Moffitt TE, Roberts AL, Martin LT, Kubzansky L, Harrington H et al. Childhood IQ and adult mental disorders: a test of the cognitive reserve hypothesis. Am J Psychiatry 2009; 166: 50–57.
Koenen KC, Moffitt TE, Poulton R, Martin J, Caspi A . Early childhood factors associated with the development of post-traumatic stress disorder: results from a longitudinal birth cohort. Psychol Med 2007; 37: 181–192.
Fisher HL, Caspi A, Poulton R, Meier MH, Houts R, Harrington H et al. Specificity of childhood psychotic symptoms for predicting schizophrenia by 38 years of age: a birth cohort study. Psychol Med 2013; 43: 2077–2086.
Acknowledgements
We thank study participants and research groups contributing to PGC-PTSD for sharing their data and their time to make this work possible. We thank One Mind (LED), Cohen Veterans Bioscience (LED and PGC-PTSD group) and the Stanley Center for Psychiatric Research for their financial support. Analysis and other work on this project was supported by 3U01MH094432-03S1 (MJD) and 5U01MH094432-04 (MJD). Finally, we acknowledge Steve Hyman of the Stanley Center for Psychiatric Research and Harvard University for comments on various versions of this manuscript. For study-specific acknowledgments, see Supplementary Information.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
Dr Kranzler has been an advisory board member, consultant or CME speaker for Indivior, Lundbeck and Otsuka. He is also a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative that is supported by AbbVie, Alkermes, Ethypharm, Indivior, Lilly, Lundbeck, Otsuka, Pfizer and XenoPort. Dr Ressler is a founding member of Extinction Pharmaceuticals to develop D-Cycloserine to augment the effectiveness of psychotherapy. He has received no equity or income from this relationship within the past 3 years. Dr Ressler is also on the Scientific Advisory Boards for Resilience Therapeutics, Sheppard Pratt-Lieber Research Institute, Laureate Institute for Brain Research, The Army STARRS Project and the Anxiety and Depression Association of America. He holds patents for use of D-cycloserine and psychotherapy, targeting PAC1 receptor for extinction, targeting tachykinin 2 for prevention of fear and targeting angiotensin to improve extinction of fear. He receives or has received research funding from NIMH, HHMI, NARSAD, and the Burroughs Wellcome Foundation. In the past 3 years, Dr Dan J Stein has received research grants and/or consultancy honoraria from AMBRF, Biocodex, Cipla, Lundbeck, National Responsible Gambling Foundation, Novartis, Servier and Sun. Dr Murray Stein reports receiving in the past 3 years consultant fees from Actelion, Janssen, Dart Neuroscience, Healthcare Management Technologies and Pfizer. He also has an equity interest in Resilience Therapeutics and Oxeia Biopharmaceuticals. He has also received editorial honoraria from Biological Psychiatry and Up-To-Date. Dr Bierut is listed as an inventor on Issued US Patent 8,080,371, ‘Markers for Addiction’ covering the use of certain SNPs in determining the diagnosis, prognosis and treatment of addiction. In the past 3 years, Dr Kessler received support for his epidemiological studies from Sanofi Aventis; was a consultant for Johnson & Johnson Wellness and Prevention, Shire and Takeda; and has served on an advisory board for the Johnson & Johnson Services Lake Nona Life Project. Kessler is a co-owner of DataStat, a market research firm that carries out health-care research. In the past 3 years, Dr Liberzon has been a consultant for ARMGO Pharmaceutical, Sunovion Pharmaceutical and Trimaran Pharma. The remaining authors declare no conflicts of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Duncan, L., Ratanatharathorn, A., Aiello, A. et al. Largest GWAS of PTSD (N=20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol Psychiatry 23, 666–673 (2018). https://doi.org/10.1038/mp.2017.77
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2017.77
This article is cited by
-
Relationship between SLC6A2 gene polymorphisms and brain volume in Han Chinese adults who lost their sole child
BMC Psychiatry (2024)
-
No replication of Alzheimer’s disease genetics as a moderator of the association between combat exposure and PTSD risk in 138,592 combat veterans
Nature Mental Health (2024)
-
Genetic overlap between multivariate measures of human functional brain connectivity and psychiatric disorders
Nature Mental Health (2024)
-
Characterizing the phenotypic and genetic structure of psychopathology in UK Biobank
Nature Mental Health (2024)
-
The impact of assortative mating, participation bias and socioeconomic status on the polygenic risk of behavioural and psychiatric traits
Nature Human Behaviour (2024)
