
Abstract
Progress in elucidating the molecular and cellular pathophysiology of neuropsychiatric disorders has been hindered by the limited availability of living human brain tissue. The emergence of induced pluripotent stem cells (iPSCs) has offered a unique alternative strategy using patient-derived functional neuronal networks. However, methods for reliably generating iPSC-derived neurons with mature electrophysiological characteristics have been difficult to develop. Here, we report a simplified differentiation protocol that yields electrophysiologically mature iPSC-derived cortical lineage neuronal networks without the need for astrocyte co-culture or specialized media. This protocol generates a consistent 60:40 ratio of neurons and astrocytes that arise from a common forebrain neural progenitor. Whole-cell patch-clamp recordings of 114 neurons derived from three independent iPSC lines confirmed their electrophysiological maturity, including resting membrane potential (−58.2±1.0 mV), capacitance (49.1±2.9 pF), action potential (AP) threshold (−50.9±0.5 mV) and AP amplitude (66.5±1.3 mV). Nearly 100% of neurons were capable of firing APs, of which 79% had sustained trains of mature APs with minimal accommodation (peak AP frequency: 11.9±0.5 Hz) and 74% exhibited spontaneous synaptic activity (amplitude, 16.03±0.82 pA; frequency, 1.09±0.17 Hz). We expect this protocol to be of broad applicability for implementing iPSC-based neuronal network models of neuropsychiatric disorders.
Similar content being viewed by others


Introduction
A detailed knowledge of the pathophysiology underlying the majority of human neuropsychiatric disorders remains largely enigmatic. However, functional genomic studies have begun to offer novel insights into many forms of neurological and psychiatric illness.1, 2, 3, 4, 5 There is widespread consensus that validated and robust human cellular models for brain disorders would be of considerable benefit.6, 7
The discovery of induced pluripotent stem cells (iPSCs) has provided the opportunity to investigate the physiology of living human neurons derived from individual patients.8 Several protocols have been reported for generating iPSC-derived neurons based on a variety of different methods. One of the most commonly employed approaches is neural induction through embryoid body (EB) formation.9 Another widely implemented method for neural induction is inhibition of the transforming growth factor-β/SMAD signaling pathway by Noggin and SB431542.10, 11 More recently, Zhang et al.12 reported a novel method utilizing forced expression of neurogenin-2 (NGN2) with puromycin selection to generate highly pure networks of glutamatergic neurons from human embryonic stem cells and iPSCs. In addition, protocols have been developed for generating three-dimensional neural cultures using cerebral organoids cultured in a spinning bioreactor,13 cortical spheroids in free-floating conditions14 or three-dimensional Matrigel culture.15
In establishing optimized and standardized methods for neural differentiation of iPSCs, one of the most important questions is the functional maturity of the resulting neuronal networks. The design of optimized neural differentiation protocols is critical for the reliable generation of functional neurons that can form active networks and demonstrate mature electrophysiological properties. Bardy et al.16 recently reported a significant advance in achieving functionally mature iPSC-derived neuronal networks. However, the major limitation with this approach is the requirement for nonstandard culture medium and extracellular recording solution during the differentiation process and electrophysiological recording.
Neuron–astrocyte interactions are critical both during early neurodevelopment and in the adult brain.17 Astrocytes are involved in the guidance of neuronal precursors and for increasing the length of neuronal fiber projections during development.18 Moreover, astrocytes dynamically modulate synaptic transmission.19, 20 Consequently, the functional maturation of human pluripotent stem cell-derived neurons is substantially improved by the presence of astrocytes.14, 21 For the derivation of iPSC-derived neuronal networks, astrocytes can either be introduced through co-culture22, 23, 24 or differentiated from a common neural progenitor that gives rise to both neurons and astrocytes as occurs in vivo.9 The co-culture approach allows more flexibility in having experimental control over the neuron-to-astrocyte ratio and the source of the co-cultured astrocytes. The major drawback, however, is the potential for introducing a source of variability, especially concerning species differences when using co-cultures of rodent astrocytes with human iPSC-derived neurons. In contrast, differentiation protocols based on a common progenitor giving rise to both neurons and astrocytes proceed more similarly to in vivo neurodevelopment.9
Using the latter approach, we now report a simplified differentiation protocol for deriving functionally mature neuronal networks from iPSCs without the need for astrocyte co-culture or specialized media.
Materials and methods
Human iPSC lines
Reprogramming of human primary skin fibroblasts from two adult donors (line 1: male, age 57 years; line 2: female, age 54 years) was performed as described previously using a single, multicistronic lentiviral vector encoding OCT4, SOX2, KLF4 and MYC.25 Donors provided written informed consent in accordance with the Medical Ethical Committee of the Erasmus University Medical Center. Quality control of iPSC clones was performed by karyotyping, real-time quantitative PCR and EB differentiation.26 Line 3 (male, newborn) was reprogrammed from cord blood CD34+ cells using episomal reprogramming (Axol Biosciences, Cambridge, UK).
Differentiation of human iPSCs to neuronal networks
Generation of NPCs
Human iPSC lines 1 and 2 were dissociated from mouse embryonic fibroblasts with collagenase (100 U ml−1, Thermo Fisher Scientific, Waltham, MA, USA) for 7 min at 37 °C/5% CO2. EBs were generated by transferring dissociated iPSCs to non-adherent plates in human embryonic stem cell medium (Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Thermo Fisher Scientific), 20% knockout serum (Thermo Fisher Scientific), 1% minimum essential medium/non-essential amino acid (Sigma-Aldrich, St Louis, MO, USA), 7 nl ml−1 β-mercaptoethanol (Sigma-Aldrich), 1% L-glutamine (Thermo Fisher Scientific) and 1% penicillin/streptomycin (Thermo Fisher Scientific)) on a shaker in an incubator at 37 °C/5% CO2. EBs were grown for 2 days in human embryonic stem cell medium, changed into neural induction medium (DMEM/F12, 1% N2 supplement (Thermo Fisher Scientific), 2 μg ml−1 heparin (Sigma-Aldrich) and 1% penicillin/streptomycin) on day 2 (d2) and cultured for another 4 days in suspension (d3–d6). For generation of neural precursor cells (NPCs), EBs were slightly dissociated at d7 by trituration and plated onto laminin-coated 10 cm dishes (20 μg ml−1 laminin (Sigma-Aldrich) in DMEM for 30 min at 37 °C), initially using neural induction medium (d7–14), and then from d15 in NPC medium (DMEM/F12, 1% N2 supplement, 2% B27-RA supplement (Thermo Fisher Scientific), 1 μg ml−1 laminin, 20 ng ml−1 basic fibroblast growth factor (Merck-Millipore, Darmstadt, Germany) and 1% penicillin/streptomycin). On d15, cells were considered pre-NPCs (passage 1) and able to be passaged (1:4) and cryopreserved when confluent. From passage 5, cells were considered NPCs and used for neural differentiation.
Line 3 NPCs were derived using the protocol reported by Shi et al.9 with modifications (Axol Biosciences, line ax0015) to examine the generalizability of our neural differentiation protocol.
Neural differentiation
NPCs (passages 5–11) were plated on sterile coverslips in 6- or 12-well plates and coated with poly-L-ornithine (Sigma-Aldrich) for 1 h at room temperature. Coated coverslips were washed 3 times with sterile water and dried for 30 min. Subsequently, a 100 μl drop of laminin solution (50 μg ml−1 in water) was placed in the middle of each coverslip, incubated for 15–30 min at 37 °C/5% CO2 and then replaced with a 100 μl drop of DMEM until plating of NPCs. Immediately before plating, NPCs were washed with Dulbecco’s phosphate-buffered saline and dissociated with collagenase (100 U ml−1). One fully confluent 10 cm dish of NPCs was divided over a 12-well plate. A 100 μl drop of NPC cell suspension was placed on the laminin-coated spot for 1 h to allow for attachment of NPCs on coverslips in neural differentiation medium (Neurobasal medium, 1% N2 supplement, 2% B27-RA supplement, 1% minimum essential medium/non-essential amino acid, 20 ng ml−1 brain-derived neurotrophic factor (ProSpec Bio, Rehovot, Israel), 20 ng ml−1 glial cell-derived neurotrophic factor (ProSpec Bio), 1 μM dibutyryl cyclic adenosine monophosphate (Sigma-Aldrich), 200 μM ascorbic acid (Sigma-Aldrich), 2 μg ml−1 laminin and 1% penicillin/streptomycin). After 1 h, 900 μl of neural differentiation medium was added to each well. Cells were refreshed with medium 3 times per week. During weeks 1–4, medium was fully refreshed. After 4 weeks of neural differentiation, only half of the volume of medium per well was refreshed. Electrophysiology and confocal imaging were performed between 8 and 10 weeks after plating of NPCs.
Immunocytochemistry and quantification
Cell cultures were fixed using 4% formaldehyde in phosphate-buffered saline. Primary antibodies were incubated overnight at 4 °C in labeling buffer containing 0.05 M Tris, 0.9% NaCl, 0.25% gelatin and 0.5% Triton-X-100 (pH 7.4). The following primary antibodies were used: SOX2, Nestin, MAP2, TBR1, GAD67, NeuN and glial fibrillary acidic protein (GFAP) (Merck-Millipore); FOXG1 (ProSci, Poway, CA, USA); Vimentin (Santa Cruz Biotechnology, Dallas, TX, USA); AFP (R&D Systems, Minneapolis, MN, USA); TRA-1-81 and Nanog (Beckton Dickinson, Franklin Lakes, NJ, USA); OCT4, BRN2, SATB2, CUX1, CUX2 and CTIP2 (Abcam, Cambridge, UK); Synapsin, MAP2 (Synaptic Systems, Göttingen, Germany); and PSD95 (Thermo Fisher Scientific). The following secondary antibodies were used: Alexa-488, Alexa-546, Alexa-555 and Cy3 antibodies (Jackson ImmunoResearch, West Grove, PA, USA). Samples were imbedded in Mowiol 4-88 (Sigma-Aldrich), after which confocal imaging was performed with a Zeiss LSM700 confocal microscope using ZEN software (Zeiss, Oberkochen, Germany).
Electrophysiology
Whole-cell patch-clamp recordings
Culture slides were collected from 12-well culture plates. Whole-cell patch-clamp recordings were performed at 8–10 weeks following the initiation of NPC differentiation. Recording micropipettes (tip resistance 3–6 MΩ) were filled with internal solution composed of (in mM): 130 K-gluconate, 0.1 EGTA, 1 MgCl2, 2 MgATP, 0.3 NaGTP, 10 HEPES, 5 NaCl, 11 KCl and 5 Na2-phosphocreatine (pH 7.4). Recordings were made at room temperature using a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA). Signals were sampled and filtered at 10 and 3 kHz, respectively. The whole-cell capacitance was compensated and series resistance was monitored throughout the experiment in order to confirm the integrity of the patch seal and the stability of the recording. Voltage was corrected for liquid junction potential (−14 mV). The bath was continuously perfused with oxygenated artificial cerebrospinal fluid (ACSF) composed of (in mM): 110 NaCl, 2.5 KCl, 2 CaCl2, 10 glucose and 1 NaH2PO4, 25 NaHCO3, 0.2 ascorbic acid and 2 MgCl2 (pH 7.4). For voltage-clamp recordings, cells were clamped at −80 mV. Spontaneous postsynaptic currents were recorded for 3 min. Fast sodium and potassium currents were evoked by voltage steps ranging from −80 to +50 mV in 10 mV increments. Capacitance was derived from the Clampex 10.2 (Molecular Devices) membrane-test function. For current-clamp recordings, voltage responses were evoked from a holding potential of −75 mV using 500 ms steps ranging from −20 to +150 pA in 10 pA intervals delivered at 0.5 Hz. Single action potential (AP) properties were calculated from the first evoked AP in response to a depolarizing step. Repetitively firing neurons were defined as those capable of firing ⩾3 mature APs without significant accommodation in response to a depolarizing current step.
Spontaneous AP activity was measured for 3 min using the minimum hyperpolarizing holding current in which spiking was evident (0–10 pA), from an initial holding potential of −80 mV. AP threshold was calculated as the point at which the second derivative of the AP waveform exceeded baseline. AP rise and decay times were calculated between 10% and 90% of the AP amplitude. Data analysis was performed by Clampfit 10.2 (Molecular Devices). Spontaneous postsynaptic currents were analyzed by MiniAnalysis software (Synaptosoft, Fort Lee, NJ, USA).
Equilibration procedure from cell culture medium to ACSF
Before initiating whole-cell recordings, cell culture medium was gradually replaced with oxygenated ACSF in order to minimize the impact of the relative difference in osmolarity (culture medium, 220 mOsm l−1; ACSF, 305 mOsm l−1). Into the 1 ml volume of culture medium per well, 300 μl of oxygenated ACSF was added for 5 min, after which 300 μl was removed. This replacement procedure was repeated 5 times at room temperature. Slides were placed immediately thereafter into the recording chamber with continuous perfusion of oxygenated ACSF.
Biocytin labeling
Juxtasomal labeling of neurons was performed using biocytin (5% w/v internal solution) at 8 weeks following the initiation of NPC differentiation. With a GΩ seal on the cell soma, neurons were subjected to 15–20 min of 100–150 pA square-wave current pulses delivered at 2 Hz. Cultures were fixed using 4% formaldehyde in phosphate-buffered saline. Secondary staining with Alexa-488-streptavidin (Jackson ImmunoResearch) was performed in labeling buffer overnight at 4 °C, after which slides were mounted in Mowiol 4-88 and imaged with a Zeiss LSM700 confocal microscope using ZEN software (Zeiss). Sholl analysis and dendrite length quantification were performed using Fiji (ImageJ, National Institutes of Health, Bethesda, MD, USA) software.27
Electron microscopy
Fixation was performed for 1 h in 2% glutaraldehyde and 0.1 M sodium cacodylate (NaCac). After rinsing in 0.1 M NaCac, cells were pelleted in 2% agar and postfixed in 2% glutaraldehyde for 15 min. Subsequently, cells were osmicated for 1 h with 1% OsO4, dehydrated with EtOH and propylene oxide, followed by embedding in Durcupan Plastic (Fluka, Buchs, Switzerland) for 72 h. Ultrathin sections (60 nm) were cut using an ultramicrotome (Leica, Wetzlar, Germany), mounted on nickel grids and counterstained with uranyl acetate and lead citrate. Imaging was performed with a CM100 Transmission Electron Microscope (Philips, Eindhoven, The Netherlands).
Statistical analysis
Statistical comparisons of continuous variables were performed using analysis of variance with post hoc Tukey’s test, using SPSS (Version 21, IBM, Armonk, NY, USA). For categorical parameters, Fisher’s exact test was used. The threshold for statistical significance was set at P<0.01 in order to correct for the 17 different electrophysiological parameters measured.
Results
Generation of forebrain-patterned NPCs from iPSCs
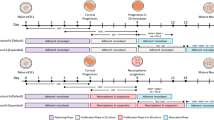
NPCs are capable of generating a diversity of neural lineages, including both neurons and astrocytes. To generate iPSC-derived NPCs (lines 1 and 2), iPSCs were detached from feeder cells using collagenase and suspended colonies were transferred to non-adherent plates (Supplementary Figure 1). Suspended colonies were cultured on a shaker that promoted the formation of spherical EBs (Figure 1a). EBs were cultured for 6 days (d1–d6), of which the first 2 days (d1–d2) were in human embryonic stem cell medium (knockout serum based) and then 4 days (d3–d6) in neural induction medium (advanced DMEM with heparin and N2 supplement). On day 7 of differentiation (d7), EBs were gently dissociated and plated onto laminin-coated dishes in neural induction medium for 8 days (d7–d14), resulting in a population of pre-NPCs (passage 1). At d15, pre-NPCs were dissociated by collagenase and replated onto laminin-coated dishes in NPC medium (advanced DMEM with N2, B27 supplement and laminin) containing basic fibroblast growth factor to promote selection and proliferation of precursor cells. The medium was changed every other day. Once confluent, cells were passaged 1:4 and could be cryopreserved in liquid nitrogen. From passage five, the cells exhibited a homogeneous morphology and marker profile of mature NPCs, expressing SOX2, Nestin, Vimentin and the forebrain-specific NPC marker FOXG1 (Figure 1b).

Generation and characterization of NPCs and neuronal networks from iPSCs. (a) Scheme illustrating the major developmental stages of the protocol for generating NPCs and neuronal networks. (b) Immunostaining for NPC markers Nestin, SOX2, Vimentin and FOXG1 (scale bars=30 μm). (c) Proportion of NeuN+ and GFAP+ cells (days 56–70). (d) Immunostaining for glial marker GFAP, and mature neuronal markers MAP2 and NeuN (top, scale bar=20 μm; bottom, scale bar=10 μm). (e) Co-labeling of pre- and postsynaptic marker proteins, Synapsin and PSD95 (scale bar=2 μm). (f) Quantification of Synapsin+, PSD95+ and double-labeled puncta density (n=20 neurons). EB, embryoid body; GFAP, glial fibrillary acidic protein; iPSC, induced pluripotent stem cells; NPC, neural precursor cells.
Differentiation of to neuronal network cultures
NPCs were utilized between passages 5 and 11 for neural differentiation. NPCs were plated onto poly-L-ornithine/laminin-coated coverslips in neural differentiation medium (neurobasal medium with N2, B27-RA) supplemented with growth factors brain-derived neurotrophic factor, glial cell-derived neurotrophic factor, dibutyryl cyclic adenosine monophosphate and ascorbic acid. Throughout the entire period of neural differentiation, medium was replaced 3 times per week. During weeks 1–4, the medium was fully exchanged. From week 5 onwards, only half of the medium was replaced per exchange. Electrophysiological recordings and confocal imaging were performed at 8–10 weeks following the initiation of NPC differentiation. Neurons were positive for the neuron-specific cytoskeletal marker β-III-tubulin, nuclear marker NeuN, dendritic marker MAP2, presynaptic marker Synapsin and postsynaptic marker PSD95 (Figures 1d and e). Quantification of Synapsin and PSD95 puncta confirmed their frequent colocalization, consistent with synaptic network connectivity, of which ~70% were glutamatergic PSD95-labeled synapses (Figures 1e and f). Moreover, electron microscopy confirmed a classical synaptic morphology, including presynaptic vesicle pools and postsynaptic density (Supplementary Figures 2a and b). Furthermore, the majority of neurons were CTIP2+, consistent with a glutamatergic lineage identity, and mutually exclusive of neurons exhibiting GAD67 labeling (Supplementary Figure 2c). Both glutamatergic and GABAergic synapses were immunohistochemically confirmed by labeling for VGLUT1 and GAD67, respectively (Supplementary Figure 2d). The proportion of immature neurons, mature neurons and astroglia was quantified by staining for doublecortin (DCX), NeuN and GFAP, respectively. Overall, NeuN+ cells constituted 15.9% of all DAPI+ nuclei, and 10.8% expressed the astrocyte marker GFAP. The ratio of NeuN+ (mature neurons) to GFAP+ (astrocytes) was 59.5 to 40.5% (Figure 1c). The remaining cells were SOX2-expressing NPCs (59.7%) and DCX-expressing immature neurons (13.6%) (Supplementary Figure 3).
We next studied the expression of cortical layer-specific markers in the differentiated neurons (Figure 2).28, 29 Subsets of neurons were positive for the transcription factor BRN2 that is expressed in late cortical progenitors and upper layer neurons (II-IV) (Figure 2a), the cortical-layer marker TBR1 that is expressed in deep layer neurons (V and VI) and the subplate (Figure 2b), FOXP2 that is expressed in layers V and VI (Figure 2c), CUX1 and CUX2 expressed in upper layer neurons (II–IV), SATB2 expressed in layers II-V, FOXG1 expressed in forebrain neural progenitors and widely in neurons of the developing telencephalon, and CTIP2 expressed in glutamatergic projection neurons from layers V and VI (Figures 2d–f). Juxtasomal neuronal biocytin labeling demonstrated an elaborate axonal and dendritic morphology. Sholl analysis was performed to quantify dendritic branching and total dendritic length (Supplementary Figure 4).

Cortical layer markers in neuronal networks. Cultures were stained at day 56 following the initiation of NPC differentiation for (a) BRN2 marker of late cortical progenitors and upper layer (II-IV) neurons, and mature dendritic marker MAP2, (b) TBR1 that is expressed by deep layer neurons (V and VI) and in the subplate, (c) FOXP2 expressed in deep layer (V and VI) neurons, (d) CUX1 marker of upper layer (II–IV) neurons and telencephalic marker FOXG1 and (e) CUX2 marker of upper layer (II–IV) neurons and SATB2 expressed in corticocortical projection neurons from layer V and upper layers. (f) CTIP2 expression in deep layer glutamatergic projection neurons. NPC, neural precursor cells.
Electrophysiology results
Whole-cell patch-clamp recordings were performed to characterize the functional maturity of the iPSC-derived neuronal networks. Electrophysiological recordings were compared across three independent lines (Figures 3,4,5).

Active and passive electrophysiological properties. (a) Representative traces from a neuron firing repetitive mature APs during depolarizing constant-current injections. Current steps are shown in the bottom panel (Vm=−75 mV). The lowest depolarizing step indicates the minimal current needed to evoke an action potential, and the highest step corresponds to the current at which the response frequency became saturated. (b) Percentage of repetitive versus nonrepetitively firing neurons. (c) Frequency–current (F-I) plot among repetitively firing neurons. (d–k) Active and passive membrane properties. AP parameters were calculated from the first evoked spike. (d) Input resistance (F=3.65, P=0.03), (e) resting membrane potential (F=0.82, P=0.44), (f) capacitance (F=0.18, P=0.84), (g) AP threshold (F=1.25, P=0.29), (h) AP amplitude (F=1.01, P=0.37), (i) AP half-width (F=4.70, P=0.012), (j) AP rise time (F=1.23, P=0.30) and (k) decay time (F=4.62, P=0.013). AP, action potential.

Spontaneous action potentials. (a) Representative current-clamp recording from a spontaneously active neuron (Vm=−68 mV). (b) Percentage of neurons with spontaneous AP firing. (c) Voltage responses of the same neuron in (a) to hyperpolarizing or depolarizing current injections (bottom panel), before (top panel) and after (middle panel) TTX application (Vm=−75 mV). (d) Sodium currents were abolished by TTX (before, top panel; after, bottom panel) (Vm=−80 mV). (e) Voltage dependence of the peak amplitude of the sodium current.

Neuronal network synaptic activity. (a) Representative voltage-clamp recording from a neuron with spontaneous synaptic input (Vm=−80 mV). (b) Zoom-in of the region in (a) marked by the red asterisk, containing two postsynaptic events. (c) Percentage of neurons exhibiting spontaneous synaptic input. (d–g) Spontaneous postsynaptic currents: (d) frequency (F=2.55, P=0.09), (e) amplitude (F=7.25, P=0.001; post hoc Tukey: P=0.01 for line 1 vs 2, P=0.004 for line 2 vs 3 and P=0.52 for line 1 vs 3), (f) rise time (F=1.24, P=0.30) and (g) decay time (P=0.023, F=4.01).
Most protocols that have been reported for neuronal differentiation of human pluripotent stem cells employ a semidefined culture medium, whereas electrophysiological recordings are performed either in the same culture medium or after transferring from the culture medium directly into a defined ACSF. Importantly, the use of culture medium for electrophysiological recordings of neurons has previously been found to impair spontaneous and evoked firing of action potentials, network-level spontaneous calcium activity and synaptic activity.16 Notably however, those experiments involved an immediate switch from culture medium to ACSF, for which the substantial acute increase in extracellular osmolarity (from 220 mOsm kg−1 in culture medium to 305 mOsm kg−1 in ACSF) has previously been reported as highly stressful for neurons.30 Therefore, we implemented a gradual transition from the culture medium to the ACSF recording medium over 25 min using 5 serial partial exchanges (see Materials and methods section for details).
Mature APs were defined as being those that reached a membrane potential above 0 mV, with a fast depolarization (⩽5 ms rise time) and rapid repolarization (⩽10 ms decay time). Nearly all recorded cells were capable of firing mature APs in response to depolarizing current injections (111/114 cells, 97.4%). Among these cells, 79.3% (88/111) exhibited repetitive firing of mature APs (Figures 3a and b), with a peak frequency of ~12 Hz (Figure 3c). The remaining 20.7% (23/111 neurons) fired an initial mature AP followed by a sequence of APs that exhibited rapid accommodation and no longer met the criteria for AP maturity.
Detailed electrophysiological measurements of intrinsic properties were performed among the group of neurons that were defined as mature based on their ability to fire mature APs repetitively in response to current injection. Passive and active membrane properties were quantified and compared in order to evaluate both the electrophysiological maturity of the neurons and the variability between lines. The mean input resistance was 1.28±0.05 GΩ (Figure 3d). Resting membrane potential was −58.2±1.0 mV (Figure 3e). The average capacitance was 49.1±2.9 pF (Figure 3f). AP threshold was −50.9±0.5 mV (Figure 3g). AP amplitude, measured from voltage threshold to peak, was 66.5±1.3 mV (Figure 3h). AP half-width was 3.18±0.11 ms (Figure 3i). AP rise and decay times were 1.9±1.0 ms (Figure 3j) and 3.36±0.16 ms (Figure 3k), respectively.
Another important aspect of neuronal network maturity is spontaneous AP firing.31, 32, 33 The majority of neurons exhibited spontaneous APs (59.1%, 52/88 neurons) (Figures 4a and b). Importantly, sustained high-quality whole-cell recordings could be maintained for >30 min (longest recording time examined) with a stable membrane potential and AP waveform, confirming that the presence of spontaneous APs was not the result of declining cell health (Supplementary Figure 5a). Moreover, spontaneous firing of APs was also evident in non-permeating cell-attached recordings, thereby establishing that the presence of spontaneous APs was not an artifact of the whole-cell configuration (Supplementary Figure 5b).
In order to confirm that the observed APs were driven by active sodium channel conductance, we blocked voltage-gated sodium channels by applying tetrodotoxin to the bath solution in a subset of recordings. As expected, action potentials were completely abolished (Figure 4c). Voltage-clamp recordings demonstrated the presence of fast sodium currents, as evident from the rapid inward current observed in response to depolarized membrane potentials (Figure 4d, upper panel, and Figure 4e). Inward voltage-gated sodium currents were also completely blocked by TTX (Figure 4d, lower panel).
Another important aspect of neuronal maturity is synaptic connectivity. Spontaneous synaptic activity was evident in 73.8% of neurons (Figures 5a–c). The frequency and amplitude of synaptic events was 1.09±0.17 Hz (Figure 5d) and 16.03±0.82 pA (Figure 5e), respectively. Line 2 exhibited significant pairwise differences in the amplitude of synaptic events compared with lines 1 and 3 (F=7.25, P=0.001; post hoc Tukey: P=0.01 for line 1 vs 2, P=0.004 for line 2 vs 3 and P=0.52 for line 1 vs 3). The kinetics of these events resembled those typically observed from neuronal recordings in acute ex vivo neocortical tissue slices, with an average rise time of 1.66±0.65 ms (Figure 5f) and decay time of 5.59±0.48 ms (Figure 5g). Blockade of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-Methyl-D-aspartic acid (NMDA) receptors using 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 50 μM) and (2R)-amino-5-phosphonovaleric acid (APV, 50 μM) confirmed the dominant contribution of glutamatergic transmission to the synaptic network activity (Supplementary Figure 6).
Discussion
We describe the results of a robust simplified protocol for neuronal network differentiation from human iPSCs with a particular focus on electrophysiological maturity. The observed electrophysiological maturity was achieved using a common iPSC-derived neural progenitor to obtain both neurons and astrocytes, thereby obviating the need for exogenous glial cell co-culture. We observed a consistent 60:40 ratio of neurons to glia, which included neurons representative of both upper and deep cortical layers, in addition to a substantial population of NPCs and DCX+ immature neurons. The robustness of the resulting neuronal networks was further evident by reducing the volume of medium changes over the course of differentiation, following the rationale that the emerging neuronal networks become increasingly self-sufficient.
This protocol requires no specialized media to obtain high-quality whole-cell patch-clamp recordings from iPSC-derived neurons with mature electrophysiological properties. We implemented a gradual equilibration procedure to transition cultures from standard neural differentiation medium to ACSF. The significance of the osmotic environment to the electrophysiological properties of iPSC-derived neurons was recently demonstrated by Bardy et al.,16 who introduced a specialized medium for neural cell culture and electrophysiological recordings. We now demonstrate the feasibility of using standard neural differentiation media while minimizing the physiological response to acute osmotic changes through a gradual equilibration from culture medium to ACSF.
Electrophysiological properties are a defining property of neuronal maturation. Many neuronal electrophysiological parameters exhibit significant alterations over the course of neurodevelopment.34, 35, 36 Resting membrane potential (Vm) tends to become progressively more hyperpolarized during neurodevelopment and stabilizes at approximately −70 mV in human neocortical ex vivo tissue slices,37 for which our protocol generated neurons with a comparable average Vm of −58 mV. Input resistance also decreases throughout neurodevelopment, as a result of both a higher ion channel density and a more complex cell morphology.35, 36 Neurons from adult human neocortex have an input resistance on the order of 50–150 MΩ,37 whereas that of second-trimester human neocortical neurons is ∼2 GΩ.34 Our protocol generated neurons with an average input resistance of 1.3 GΩ, consistent with a late gestational or early postnatal neurodevelopmental period. As neurons mature, their AP firing threshold becomes increasingly hyperpolarized, and the AP waveform exhibits more rapid kinetics with larger amplitudes.36, 37 Consistent with our measurements of input resistance, we observed that both AP threshold and AP half-width were also comparable to neurons recorded from ex vivo mid-to-late gestational human neocortical tissue.34
The emergence of synaptic transmission is another defining aspect of neuronal network maturation that is continuously and dynamically regulated by short- and long-term forms of plasticity, and considered among the latest developing aspects of neuronal physiology.38 Consistent with the estimated neurodevelopmental stage of the passive membrane properties and active AP characteristics in neurons derived using the current protocol, the synaptic parameters we measured are also comparable to those observed in mid-to-late gestational human neocortex.34 However, in contrast to the low variability that we observed across different lines regarding passive membrane and AP characteristics, synaptic properties exhibited a generally higher variance. Synapse formation and synaptic function develop over an extended period in neurodevelopment and are governed by a sizeable proportion of the genome, with ~9% of all protein-coding genes expressed at mammalian excitatory synapses.39, 40 Accordingly, for iPSC-based functional genomic studies of neurophysiology, isogenic controls may be particularly important for investigating synaptic function. In contrast, AP parameters and passive membrane properties appear to be more robust across differing genetic backgrounds.
In summary, we have developed a simplified differentiation protocol for generating electrophysiologically mature iPSC-derived neuronal networks without the need for astrocyte co-culture or specialized media. Moreover, our findings provide a quantitative basis for considering the variability of distinct electrophysiological parameters for iPSC-based disease modeling. We envision this protocol to be of considerable utility for implementing cellular modeling approaches to the study of human neuropsychiatric disease pathophysiology.
References
Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016; 530: 177–183.
Sztainberg Y, Chen H, Swann JW, Hao S, Tang B, Wu Z et al. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature 2015; 528: 123–126.
Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, Kootar S et al. η-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 2015; 526: 443–447.
Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015; 347: 1436–1441.
Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F . Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 2014; 518: 409–412.
Kelava I, Lancaster MA . Stem cell models of human brain development. Cell Stem Cell 2016; 18: 736–748.
Wen Z, Christian KM, Song H, Ming GL . Modeling psychiatric disorders with patient-derived iPSCs. Curr Opin Neurobiol 2016; 36: 118–127.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S . Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131: 861–872.
Shi Y, Kirwan P, Livesey FJ . Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc 2012; 7: 1836–1846.
Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011; 8: 267–280.
Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L . Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 2009; 27: 275–280.
Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013; 78: 785–798.
Lancaster Ma, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME et al. Cerebral organoids model human brain development and microcephaly. Nature 2013; 501: 373–379.
Paşca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods 2015; 12: 671–678.
Kim YH, Choi SH, D’Avanzo C, Hebisch M, Sliwinski C, Bylykbashi E et al. A 3D human neural cell culture system for modeling Alzheimer’s disease. Nat Protoc 2015; 10: 985–1006.
Bardy C, van den Hurk M, Eames T, Marchand C, Hernandez RV, Kellogg M et al. Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proc Natl Acad Sci USA 2015; 112: E2725–E2734.
Zuchero JB, Barres BA . Glia in mammalian development and disease. Development 2015; 142: 3805–3809.
Sultan S, Li L, Moss J, Petrelli F, Cassé F, Gebara E et al. Synaptic integration of adult-born hippocampal neurons is locally controlled by astrocytes. Neuron 2015; 88: 957–972.
Chung W-S, Allen NJ, Eroglu C . Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol 2015; 7: a020370.
Clarke LE, Barres BA . Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci 2013; 14: 311–321.
Johnson MA, Weick JP, Pearce RA, Zhang S-C . Functional neural development from human embryonic stem cells: accelerated synaptic activity via astrocyte coculture. J Neurosci 2007; 27: 3069–3077.
Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011; 473: 221–225.
Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012; 482: 216–220.
Wen Z, Nguyen HN, Guo Z, Lalli MA, Wang X, Su Y et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 2014; 515: 414–418.
Warlich E, Kuehle J, Cantz T, Brugman MH, Maetzig T, Galla M et al. Lentiviral vector design and imaging approaches to visualize the early stages of cellular reprogramming. Mol Ther 2011; 19: 782–789.
De Esch CEF, Ghazvini M, Loos F, Schelling-Kazaryan N, Widagdo W, Munshi ST et al. Epigenetic characterization of the FMR1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem Cell Rep 2014; 3: 548–555.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9: 676–682.
Gaspard N, Bouschet T, Herpoel A, Naeije G, van den Ameele J, Vanderhaeghen P . Generation of cortical neurons from mouse embryonic stem cells. Nat Protoc 2009; 4: 1454–1463.
Espuny-Camacho I, Michelsen KA, Gall D, Linaro D, Hasche A, Bonnefont J et al. Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron 2013; 77: 440–456.
Pasantes-Morales H . Volume regulation in brain cells: cellular and molecular mechanisms. Metab Brain Dis 1996; 11: 187–204.
Spitzer NC . Electrical activity in early neuronal development. Nature 2006; 444: 707–712.
Khazipov R, Luhmann HJ . Early patterns of electrical activity in the developing cerebral cortex of humans and rodents. Trends Neurosci 2006; 29: 414–418.
Luhmann HJ, Sinning A, Yang J-W, Reyes-Puerta V, Stüttgen MC, Kirischuk S et al. Spontaneous neuronal activity in developing neocortical networks: from single cells to large-scale interactions. Front Neural Circuits 2016; 10: 40.
Moore AR, Filipovic R, Mo Z, Rasband MN, Zecevic N, Antic SD . Electrical excitability of early neurons in the human cerebral cortex during the second trimester of gestation. Cereb Cortex 2009; 19: 1795–1805.
Frick A, Feldmeyer D, Sakmann B . Postnatal development of synaptic transmission in local networks of L5A pyramidal neurons in rat somatosensory cortex. J Physiol 2007; 585: 103–116.
Oswald A-MM, Reyes AD . Maturation of intrinsic and synaptic properties of layer 2/3 pyramidal neurons in mouse auditory cortex. J Neurophysiol 2008; 99: 2998–3008.
Testa-Silva G, Verhoog MB, Linaro D, de Kock CPJ, Baayen JC, Meredith RM et al. High bandwidth synaptic communication and frequency tracking in human neocortex. PLoS Biol 2014; 12: e1002007.
Moore AR, Zhou W-L, Jakovcevski I, Zecevic N, Antic SD . Spontaneous electrical activity in the human fetal cortex in vitro. J Neurosci 2011; 31: 2391–2398.
Morciano M, Beckhaus T, Karas M, Zimmermann H, Volknandt W . The proteome of the presynaptic active zone: from docked synaptic vesicles to adhesion molecules and maxi-channels. J Neurochem 2009; 108: 662–675.
Bayés A, van de Lagemaat LN, Collins MO, Croning MDR, Whittle IR, Choudhary JS et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci 2011; 14: 19–21.
Acknowledgements
Funding was provided by ZonMw Vidi (017.106.384), Middelgroot (40-00506-98-10026) and ALW (834.12.002) from the Netherlands Organization for Scientific Research, Dutch Technology Foundation STW, Applied Science Division of NWO and the Technology Programme of the Ministry of Economic Affairs (Project 12197), NeuroBasic PharmaPhenomics consortium to SAK and Hersenstichting Fellowship (F2012(1)-39) to FMSdV. We thank Gerard Borst for helpful discussions and Elize Haasdijk for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Gunhanlar, N., Shpak, G., van der Kroeg, M. et al. A simplified protocol for differentiation of electrophysiologically mature neuronal networks from human induced pluripotent stem cells. Mol Psychiatry 23, 1336–1344 (2018). https://doi.org/10.1038/mp.2017.56
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2017.56
This article is cited by
-
Pathological Deficit of Cystatin B Impairs Synaptic Plasticity in EPM1 Human Cerebral Organoids
Molecular Neurobiology (2024)
-
ANDA: an open-source tool for automated image analysis of in vitro neuronal cells
BMC Neuroscience (2023)
-
Suspension culture improves iPSC expansion and pluripotency phenotype
Stem Cell Research & Therapy (2023)
-
In vitro and in vivo differences in neurovirulence between D614G, Delta And Omicron BA.1 SARS-CoV-2 variants
Acta Neuropathologica Communications (2022)
-
Utilizing machine learning algorithms to predict subject genetic mutation class from in silico models of neuronal networks
BMC Medical Informatics and Decision Making (2022)

{kind=link}