
Abstract
Neuroinflammation is proposed as one of the mechanisms by which Alzheimer’s disease pathology, including amyloid-β plaques, leads to neuronal death and dysfunction. Increases in the expression of markers of microglia, the main neuroinmmune cell, are widely reported in brains from patients with Alzheimer’s disease, but the literature has not yet been systematically reviewed to determine whether this is a consistent pathological feature. A systematic search was conducted in Medline, Embase and PsychINFO for articles published up to 23 February 2017. Papers were included if they quantitatively compared microglia markers in post-mortem brain samples from patients with Alzheimer’s disease and aged controls without neurological disease. A total of 113 relevant articles were identified. Consistent increases in markers related to activation, such as major histocompatibility complex II (36/43 studies) and cluster of differentiation 68 (17/21 studies), were identified relative to nonneurological aged controls, whereas other common markers that stain both resting and activated microglia, such as ionized calcium-binding adaptor molecule 1 (10/20 studies) and cluster of differentiation 11b (2/5 studies), were not consistently elevated. Studies of ionized calcium-binding adaptor molecule 1 that used cell counts almost uniformly identified no difference relative to control, indicating that increases in activation occurred without an expansion of the total number of microglia. White matter and cerebellum appeared to be more resistant to these increases than other brain regions. Nine studies were identified that included high pathology controls, patients who remained free of dementia despite Alzheimer’s disease pathology. The majority (5/9) of these studies reported higher levels of microglial markers in Alzheimer’s disease relative to controls, suggesting that these increases are not solely a consequence of Alzheimer’s disease pathology. These results show that increased markers of microglia are a consistent feature of Alzheimer’s disease, though this seems to be driven primarily by increases in activation-associated markers, as opposed to markers of all microglia.
Similar content being viewed by others

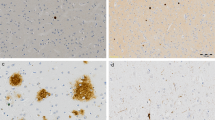
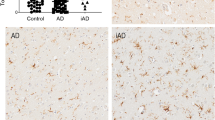
Introduction
Elevations in neuroinflammatory markers are widely reported in Alzheimer’s disease (AD), both in animal models1, 2, 3 and human subjects.4, 5, 6, 7 This has contributed to the development of the neuroinflammatory hypothesis of AD that suggests that aberrant activation of immune cells may drive neuronal death and dysfunction in AD.8 This is supported by genome-wide association studies that have identified polymorphisms in inflammation-associated genes as risk factors for the development of AD.9, 10, 11
Microglia are the resident immune cells of the brain, and are thought to be the main cells responsible for initiating the immune response to AD pathology. Several of the inflammation-associated genetic risk factors for AD, including human leukocyte antigen (HLA)-DRB1/B5,9 cluster of differentiation (CD) 33,12 triggering receptor expressed on myeloid cell-2 (TREM2)10 and phospholipase C γ2,10 are highly expressed in microglia where they are involved in cell function and activation. This suggests that aberrant microglial activation is a causal factor in the development of AD, as opposed to a consequence of AD pathology. Although it is commonly accepted that there are increased microglia markers in the brains of patients with AD relative to controls, no one has yet systematically synthesized the literature to see whether this is supported by the totality of the evidence. Here, we describe the results of a systematic review examining microglia in post-mortem human brain samples from patients with AD and healthy controls. We find that some markers associated with cell activation, such as major histocompatibility complex- II (MHCII) and CD68, are consistently increased in the AD brain, but that studies using other common microglial markers that stain both resting and activated cells, such as ionized calcium-binding adaptor molecule-1 (Iba1) and CD11b, are heterogeneous and do not demonstrate a consistent elevation. We further identify brain regions, such as the white matter and the cerebellum, that appear to be more resistant to inflammation in AD.
Methods
The systematic search was conducted in Medline, Embase and PsychINFO covering articles published up to 23 February 2017. The search protocol was developed based on Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) and World Health Organization (WHO) Review Protocol Template Guidelines where applicable for a systematic review of descriptive (non-interventional) data, and is provided in Appendix 1 of the Supplementary Materials section. The search queried the following terms with numerous synonyms and related words as both MeSH/Emtree terms (where applicable) and as keywords (for title, abstract and keyword searches): Alzheimer’s Disease AND brain AND inflammation. The additional term ‘AND post-mortem’ with its synonyms and related words was included for the Embase and PsychINFO searches. A full list of terms used for each search can be found in Appendix 2.
Studies were included if they used human brain samples from patients with AD, included a measure of inflammation, were conducted post-mortem and included a comparison to a control group without AD or a confounding neurological disease. This review initially set out to include all inflammatory markers, including for astrocytes, complement, cytokines, lipid mediators and other immune cells, but the title and abstract screening returned 757 eligible papers, making it unfeasible to summarize all the evidence in a single paper. It was decided to proceed with a review of microglia, as these are the main immune effector cells in the brain, and are the source of many of the other inflammatory mediators measured in other studies. Microglia terms in the initial search include the MeSH term: neuroglia and neurogenic inflammation, the Emtree terms: neurogenic inflammation, glia and leukocyte antigen, and keywords: microglia, HLA, MHC, CD11b, CD68, Iba1, OX-42 and CD45 along with their synonyms and alternate spellings (see Appendix 2 of the Supplementary Materials for the full list of terms). In addition, any other papers returned in the full search that used other markers identified by the study authors as being specific to microglia or their activation were included. Studies were excluded if they were not in English or not published in full in a peer-reviewed journal. Papers that measured markers associated with an M1 or M2 phenotype, such as interleukin (IL)-1β, tumour necrosis factor-α, IL-4 or IL-10 but that did not localize these markers specifically to microglia, were not included.
Data from included studies were extracted by at least two independent reviewers (KEH and DM, VG or MT), with a third reviewer employed in the case of a conflict. Extracted data included origin of brain samples, number of subjects, sex, age, apolipoprotein E (APOE) genotype, histological confirmation of AD status, Braak stage, control history of neurological or psychiatric disease, post-mortem interval (length of time between death and retrieval of brain), brain regions examined, medications at the time of death, anti-inflammatory drug use at death, technique for measuring microglia, marker of microglia used and the results of the comparison between the AD subjects and healthy controls. Where available, information on relationship between labelled microglia and amyloid-β plaques or neurofibrillary tangles was also extracted. The terms ‘higher’ or ↑ and ‘lower’ or ↓ are used in the text and tables referring to significantly higher or lower levels of the microglial marker used in the study relative to the nonneurological aged control group. As the type of outcome reporting was extremely heterogeneous, results were reported as higher, lower or unchanged for AD relative to control as identified by the study authors. Meta-analysis or other summary statistics were not used because of the large variability in assessment techniques and brain regions examined between studies. Data were listed as ‘Not Reported’ if the relevant information could not be found in the article, or in a previous article specifically referred to by the authors in the Methods section. In a few instances where information in the article was unclear, the corresponding authors were contacted to provide clarification or additional detail.
Results
A total of 22 229 articles were screened, of which 757 met inclusion for full text review for inflammatory markers, including 224 that examined microglia (Figure 1). A total of 113 papers that quantified microglia and compared the results statistically between control and AD groups were fully extracted and are presented in the tables below (see Figure 2 for a visual summary of the results). Fifty-three papers that made a nonquantitative comparison between AD and control, often as a qualitative assessment of the intensity of immunohistochemistry staining, were not fully extracted but are summarized under the ‘Non-quantitative comparisons’ heading.

Flow diagram of systematic search.

Summary results of systematic search. Size of circle is proportional to the number of identified studies, whereas the colour and position on the graph illustrates the percentage of studies that identified an increase in AD in at least one brain region. AD, Alzheimer’s disease; CD, cluster of differentiation; Iba1, ionized calcium-binding adapter molecule 1; IL, interleukin; MHC, major histocompatibility complex; RCA-1, Ricinus communis agglutin-1; TSPO, translocator protein; TREM2, triggering receptor expressed on myeloid cells 2.
The microglial markers analysed in this review all serve distinct functions within the cell, and as such, the interpretation of an up- or down-regulation of expression will vary depending on the marker used. The functions of all major markers included in this review, their association with polarization and their expression on other cell types besides microglia is shown in Supplementary Table 1. M1 and M2 polarization is increasingly regarded as an oversimplification of many diverse functions of activated microglia; however, it is used here to provide a reference for whether a marker is associated with the pro-inflammatory or phagocytic microglial phenotype. As shown in the table, it is important to note that all the major markers in this review are also expressed on other cell types, particularly on macrophages, and hence it is possible that other infiltrating or perivascular immune cells may have contributed to the results.
Major histocompatibility complex II
MHCII is expressed on the surface of antigen-presenting cells and is responsible for antigen recognition and the activation of the adaptive immune system. Within the brain, MHCII is primarily expressed on microglia, where it is generally considered a marker of activated cells, though it may have weaker expression in resting cells.14 Forty-three papers were identified that quantitatively compared markers of MHCII between AD and control in post-mortem human brains (Table 1). The majority (41/43) used immunohistochemistry for HLA-DR or for multiple isoforms of HLA (HLA-DR-DQ-DP) quantified by cell counts, scoring or staining area, whereas other techniques included gene expression by quantitative PCR (qPCR) or protein quantification by western blot.
Thirty-six papers reported higher MHCII in AD relative to control brains in at least one of the measured brain regions,7, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49 whereas 7 identified no difference between AD and control,50, 51, 52, 53, 54, 55, 56 and 5 identified no difference in at least one brain region.21, 32, 34, 41, 44 Increased MHCII staining, counts or expression was noted in: the entorhinal cortex,7, 18, 20, 35, 43, 47 frontal and temporal gyri,7, 19, 24, 32, 33, 39, 45, 56 hippocampus20, 25, 27, 30, 32, 33, 34, 35, 36, 37, 38, 40, 42, 43, 44, 47 and frontal,18, 21, 25, 40, 41, 44, 46, 48, 49, 52 temporal16, 17, 18, 28, 30, 35, 43, 44, 48, 52 and occipital cortices.25, 35, 41, 43, 44 HLA-DR was found to increase with AD plaque stage and clinical dementia rating in the entorhinal cortex, hippocampus and occipital and temporal cortices.43, 47 HLA-DR-stained microglia were reported to take on an activated morphology in AD,19, 23, 45, 48, 49 indicating that AD pathology may stimulate the activation of microglia and upregulation of MHCII. In contrast, seven papers identified no difference in MHCII between AD and control in several regions including the hippocampus,21, 55 frontal, temporal and parietal grey matter,52 the temporal polar association cortex54 and the subventricular zone of the lateral ventricle.53
Of the five studies examining HLA-DR immunoreactivity in the cerebellum, four found no differences between AD and control,25, 34, 44, 51 suggesting that regional differences may explain some of the null findings. Overmyer et al.52 noted that women had higher HLA-DR reactivity than men in AD, whereas the reverse was true in the controls, suggesting that sex differences in the AD and control groups could also contribute to between-study variability. HLA-DR reactivity increased in both AD and control patients >75 of age in one study,52 but decreased in AD patients >80 of age in another,28 making the effect of age on HLA-DR expression unclear. APOE genotype can also impact the results, with the ɛ4 risk allele increasing HLA-DR-positive cells or area in the frontal and temporal cortices48 and middle frontal gyrus.56
Ionized calcium-binding adaptor molecule 1
Iba1 is a cytoplasmic protein expressed in monocyte lineage cells and in the brain and is primarily restricted to microglia.57 Although its expression is thought to increase with microglial activation,58 where it may be involved in membrane ruffling and phagocytosis,59 it is considered a marker of all microglia, rather than an activated subset.60 Twenty papers quantitatively compared Iba1 between AD and control post-mortem human brains (Table 2). Ten studies identified increases in Iba1 cell counts, staining intensity or expression in AD compared with control samples in at least one brain region,20, 60, 61, 62, 63, 64, 65, 66, 67, 68 whereas 10 studies identified no difference from controls32, 54, 69, 70, 71, 72, 73, 74, 75, 76 and 5 reported lower Iba1 in AD than controls32, 54, 56, 75, 76 in at least one of the regions measured.
The results of studies measuring Iba1 are relatively heterogeneous. Higher Iba1 was noted in the AD frontal cortex in four studies,20, 63, 64, 68 but was the same as control in three others32, 70, 74 and lower in the white matter of the frontal lobe in one study.32 Iba1-positive cell density72 and gene expression74 was the same in AD and control in the temporal cortex in two studies, but greater by western blot or immunocytochemistry in three other studies.20, 61, 66 Similarly, although Iba1 was higher in AD than control in the hippocampus in three studies,20, 62, 67 it was unchanged in five studies32, 69, 71, 73, 75 and reduced in one study.76 Iba1 was higher in the entorhinal cortex20, 62 and inferior parietal cortex65 in AD compared with control, but this is based on a limited number of studies. All but 1 of the 10 studies that identified increases in Iba1 in AD used expression-based assays. The remaining positive study quantified plaque-associated microglia.20 In contrast, 6 of the 10 null studies used cell counting, 2 of which used stereology, the gold standard for quantifying cells without bias. Expression-based assays like qPCR, western blot, or intensity of immunohistochemistry staining indicate the amount of Iba1 in a sample. An increase could reflect a change in cell numbers, cell size or function, as Iba1 expression is thought to increase with microglial activation. In contrast, the studies using cell counting for Iba1, which is expressed by all microglia,58, 77 assess the absolute number of microglia in the samples. The discrepancy between the studies using expression-based assays and those that used cell counting suggests that Iba1 expression, and thus microglial activation, increases in AD without affecting the absolute number of microglia.
Cluster of differentiation 68
CD68 is a common marker for macrophage lineage cells, primarily localized to microglia within the brain parenchyma, and perivascular macrophages in the cerebral blood vessels and, occasionally, parenchyma.78 Although there is some CD68 expression on resting microglia,14 it labels the lysosome and is therefore commonly considered a marker of activated phagocytic microglia.60 Twenty-one studies were identified that compared CD68 between AD and control post-mortem brain samples (Table 3). Twenty of these studies used immunohistochemistry to visualize CD68-positive cells and measured cell counts or staining area, whereas one study measured CD68 gene expression by qPCR. Seventeen identified an increase in CD68 expression, staining or positive cell counts in AD relative to control samples in at least one region,17, 18, 20, 28, 41, 56, 69, 76, 78, 79, 80, 81, 82, 83, 84, 85, 86 whereas four found no difference between AD and control,51, 87, 88, 89 and four reported no difference in at least one of the brain regions measured.20, 41, 69, 80
CD68-positive cell counts, staining area or gene expression were measured in the hippocampus in eight studies: six identified higher levels in AD20, 69, 76, 78, 79, 86 and two identified no differences.69, 87 CD68 was higher in AD than control in the frontal cortex in three studies,18, 41, 82 though for white matter only in one of the studies.41 Elevations in CD68 were also reported in the temporal cortex,20, 28 the olfactory bulb,80 the calcarine cortex,69, 86 the superior temporal sulcus,83 the orbitofrontal cortex,86 parahippocampal gyrus76 and temporal association isocortex.84 No difference between AD and control was reported in the caudate nucleus,88 combined cerebellum and cerebral cortex,51 mediodorsal nucleus of the thalamus88 and the middle frontal gyrus.56 CD68 immunoreactivity appears to increase with age in control subjects, but decreases with age in patients with AD.28 It also increases with APOE ɛ4 genotype.56 Characteristics of AD and control groups could therefore contribute to between-study variability. On balance, CD68 appears to be increased in the brains of patients with AD, though there is some variation between studies and brain regions.
Cluster of differentiation 11b
CD11b forms part of complement receptor 3 that aids in the recognition and phagocytosis of antigens, including amyloid-β.90 Like Iba1, CD11b is expressed by both resting and activated microglia, though it too is inducible with activation.14 Five studies were identified that compared CD11b between AD and control post-mortem human brain samples (Supplementary Materials Table 2). Two papers reported an increase in CD11b,15, 38 whereas three reported no differences between AD and control.74, 76, 91
Like Iba1, the results of studies measuring CD11b in AD and control post-mortem human brain samples are heterogeneous. Of two studies in the hippocampus, one identified an increase in CD11b gene expression,38 whereas the other identified no difference in expression between AD and controls.76 Similarly, Akiyama et al. reported an increase in CD11b-positive cells in the temporal lobe of AD cases,15 whereas Lee et al.74 found no difference in gene expression relative to controls in the same region. Two studies that compared CD11b gene expression in the prefrontal cortex identified no difference between AD and control.91 Interestingly, two of the studies that identified increases in CD11b also measured MHCII, and both reported greater increases in MHCII than CD11b in the AD brain.15, 38 Based on these studies, CD11b does not appear to be consistently increased in the AD brain.
Cluster of differentiation 45
CD45 is a cell surface antigen that is expressed on most hematopoietic lineage cells, where it is involved in the regulation of numerous processes, including cell division and differentiation. CD45 is expressed by both resting and activated microglia, but it does not appear to be inducible with activation in humans.92 Six papers were identified that compared CD45 in AD and control human brains (Supplementary Materials Table 3). Two used immunohistochemistry with cell counting, three measured gene expression and one used a combination of western analysis and immunohistochemistry with cell counting. Four studies reported higher CD45 in AD in at least one brain region,15, 76, 93, 94 whereas two reported no difference relative to control71, 95 and one reported no difference in at least one region.94
All three of the studies that used immunohistochemistry with cell counting reported an increase in CD45-positive cells in AD patients in the temporal lobe,15 midfrontal cortex,93 frontal cortex94 and hippocampus molecular and pyramidal layers,94 respectively. In contrast, the polymorphous layer of the hippocampus did not demonstrate increased CD45-positive cells.94 Studies using gene expression were more heterogeneous: Sanchez-Mejias et al.76 detected higher CD45 gene expression in the hippocampus and parahippocampal gyrus in AD, whereas Magistri et al.71 identified no such increase in the hippocampus relative to controls. No difference in expression was also reported in the frontal cortex.95 The evidence suggests that there are more CD45-positive cells in the brains of patients with AD, but that this is not necessarily accompanied by increased CD45 gene expression.
Ferritin
Iron is stored in the brain as heavy (H) or light (L) subunits. Neurons and other cells primarily contain H ferritin, which is the less reactive form, whereas glial cells contain the L subunit, which can be used to generate free radicals as part of the inflammatory response.96, 97 Although not specific to microglial cells, ferritin immunohistochemistry combined with morphological identification can be used to visualize these cells in the central nervous system, and increases in ferritin levels with inflammation are thought to be caused by increases in microglial number and activation. Seven papers measured ferritin in post-mortem human brain samples from patients with AD and controls (Supplementary Materials Table 4). Five of these papers identified greater levels of ferritin, ferritin-positive microglia or microglia-associated plaques in AD in at least one of the brain regions measured,96, 98, 99, 100, 101 whereas two found no difference in cell counts relative to control,33, 51 and one identified no change in ferritin-associated plaques in one region and a decrease in another.100
Three papers that measured ferritin-positive cell counts or protein levels in the hippocampus identified higher levels in AD,96, 99, 101 whereas one that combined counts from the hippocampus with the amygdala, superior frontal gyrus and the superior, middle and inferior temporal gyri found no significant difference relative to control, though a nonsignificant increase was noted.33 Higher ferritin-positive microglia was also reported in AD relative to control in the amygdala, entorhinal cortex, and frontal, occipital, parietal and temporal neocortices.99 No difference between AD and control was reported in one study that counted microglia over 12 slices of combined cerebellum and cerebral cortex.51
Cluster of differentiation 33
CD33 is a myeloid cell surface marker that is involved in the regulation of the innate immune system. In microglia, CD33 seems to regulate phagocytosis of amyloid-β, with a reduction in CD33 associated with increased phagocytosis.70 Two polymorphisms in the CD33 gene have recently been identified that modulate AD risk,102, 103 with the protective allele associated with reduced CD3312, 66, 70 and increased phagocytosis.12 Four papers were identified that used qPCR, western blot or immunohistochemistry to compare levels of CD33 in the brains of patients with AD to controls, three of which reported elevations in AD, whereas one reported no change (Supplementary Materials Table 5).
Higher expression of CD33 genes or of CD33 immunolabelled cells was noted in three studies in the superior and middle temporal gyri, frontal cortex and temporal cortex of patients with AD relative to controls.66, 70, 104 In contrast, Sanchez-Mejias et al.76 reported no difference in CD33 gene expression between patients with AD and controls in the hippocampus cornu ammonis 1 (CA1), CA3 and parahippocampal gyrus.76 CD33 expression was shown to correlate with pan-microglial markers Iba1 or CD11b, supporting its microglial localization.66, 70, 104 As CD33 increases when adjusting for Iba1 in most of studies, it is likely a marker of microglial function or activity, and not of the number of microglia. Though few studies are currently available, the evidence indicates that CD33 is likely increased in the brains of patients with AD.
Triggering receptor expressed on myeloid cells 2
TREM2 is a regulatory protein under the control of γ-secretase that controls toll-like receptor 4 signalling. In microglia, TREM2 seems to be important for microglial activation and phagocytosis of apoptotic neurons.105 Polymorphisms in the R47H allele of the TREM2 gene have recently been identified as a strong genetic risk factor for the development of AD,13 though the actual impact of these polymorphisms on microglial function remain unclear. Four papers were identified that compared levels of TREM2 in the brains of patients with AD and controls (Supplementary Materials Table 6). Two identified an increase of TREM2 in AD,61, 106 whereas one reported no difference relative to control76 and one reported opposing results for qPCR and western analysis.107
Both papers that examined the temporal cortex identified an increase in TREM2, either for the protein,61, 106 staining intensity by immunohistochemistry61 or gene expression.106 Roussos et al.107 examined the impact of R47H genotype on TREM2 mRNA and protein in the superior temporal gyrus. They also reported higher TREM2 gene expression in AD R47H carriers relative to controls and no difference in TREM2 between AD noncarriers and control. Their protein measurements, however, indicated decreased TREM2 in AD R47H carriers relative to controls with no difference between AD noncarriers and control. The authors speculate that the discrepancy between gene expression and protein quantity may be explained by increased immature TREM2 and increased degradation. In contrast, Sanchez-Mejias et al.76 identified no difference in TREM2 gene expression relative to control in the hippocampus CA1, CA3 and the parahippocampal gyrus. As with CD33, TREM2 expression is increased even when adjusting for Iba1,106 and hence it can be considered a marker of microglial function as opposed to number.
Cluster of differentiation 11c
CD11c is a transmembrane protein expressed on the surface of various immune cells, including microglia, macrophages and neutrophils. Four studies were identified that compared CD11c in AD and control post-mortem brains, all of which used immunohistochemistry and reported higher staining scores or cell counts in AD in at least one brain region15, 108, 109, 110 (Supplementary Materials Table 7).
Three studies examined CD11c in the hippocampus. Two reported higher levels of staining quantified by a semiquantitative scoring system in AD in the CA1 and subiculum;108, 110 however, Paulus et al.109 quantified CD11c by unbiased cell counts and identified no difference in CA1 or the granular layer of the dentate gyrus relative to control. Elevated CD11c was also reported in the temporal lobe,15 frontal cortex,109 entorhinal cortex110 and superior temporal gyrus.108 No difference between AD and control was detected in the frontal white matter.109 Based on the limited number of studies, it appears that CD11c is increased in brain of patients with AD relative to controls, but this may be influenced by differences in methodology between the studies.
IL-1α-expressing microglia
IL-1α is a proinflammatory cytokine that plays a central role in the induction of an immune response. Although IL-1α is not a microglia marker, three papers measuring it were included in this review because they used immunohistochemistry and cell counting to examine IL-1α-positive microglia as an indication of activation (Supplementary Materials Table 8). All three papers are from the same research group and reported higher IL-1α-positive microglia in AD in at least one of the measured brain areas, but two reported no difference relative to control in one region.
Elevated IL-1α-positive microglia were reported in the parahippocampal cortex,111 the hippocampus112 and the frontal,112 occipital112 and temporal lobes.112, 113 Within the temporal cortex, layers III–VI were found to be enriched in IL-1α-expressing microglia in AD, whereas layers I–II were no different from controls.113 No difference between AD and control brains was detected in the cerebellum.112 Based on the available evidence, IL-1α-expressing microglia seem to be increased in the brains of patients with AD.
Ricinus communis agglutinin-1
Microglia appear to be the only resident brain cells that express the lectin Ricinus communis agglutinin-1 (RCA-1).114 Two papers were identified that used RCA-1 immunohistochemistry to compare microglia counts between AD and control post-mortem human brain samples (Supplementary Materials Table 9). Both reported significantly more RCA-1-labelled microglia in AD in the inferomedial temporal lobe115 and the association cortex, periallocortex/allocortex and primary cortex, respectively.116
Translocator protein
Translocator protein (TSPO), also known as the peripheral benzodiazepine receptor, is a mitochondrial protein expressed on activated microglia in the brain. It is the ligand for (11)C-PK11195 that is commonly used in positron emission tomography imaging of activated microglia in vivo. Two papers returned in the systematic search compared in situ PK11195 binding or expression of TSPO in control and AD post-mortem human brain samples (Supplementary Materials Table 10). Kravitz et al.117 identified higher [3H]-PK11195 binding in the entorhinal cortex, the subiculum, the striatum and various areas of the hippocampus. In contrast, Marutle et al.118 identified no difference in PK11195 binding in the hippocampus between control and AD brains, though they reported higher levels in the frontal cortex. Kravitz et al.117 also measured TSPO mRNA and identified higher expression in the hippocampus in AD relative to controls, but no difference in the striatum. Because these two studies have somewhat opposing results for the brain region they have in common, the hippocampus, it is unclear whether TSPO is increased in post-mortem brain samples from patients with AD. PK11195 has a lower affinity for TSPO and a lower signal-to-noise ratio than newer ligands which may explain some of the discrepancy. A recent review of positron emission tomography imaging of microglia in AD in vivo found that of the five papers using the PK11195 ligand, three identified increases in AD relative to controls and two identified no differences, whereas for the second-generation radioligands, 5/6 reported increases in AD in various brain regions.119
Cluster of differentiation 163
CD163 is a scavenger receptor expressed on monocyte/macrophage lineage cells. Two papers identified higher CD163 in AD than control post-mortem human brains (Supplementary Materials Table 11). Dal Bianco et al.20 identified a greater number of CD163-positive cells in the cortical areas of the temporal lobe, including the hippocampus and entorhinal and temporal cortices. Like Dal Bianco et al.20 Pey et al.120 also found higher levels of CD163 staining in the hippocampus, as well as the frontal and occipital cortices. Based on this small number of studies, CD163 seems to be upregulated in AD relative to control post-mortem brains.
Microglia identified by morphology
Two studies used nonspecific cell stains to visualize microglia that were identified based on morphology (Supplementary Materials Table 12). Shefer121 identified both a greater absolute number of glia and a greater number of microglia per volume in the subiculum of the archicortex in the hippocampal fissure. This combination of relative and absolute counts provides evidence that the apparent increase in microglia was not just a function of tissue shrinkage. In contrast, Pelvig et al.122 also identified microglia based on morphology and quantified them using stereology, but found no difference in the total number of cells in the neocortex.
Other
Seventeen papers returned in the systematic search used markers other than those discussed in previous sections (Supplementary Materials Table 13). Some of these, such as Ox-42 and GLUT-5, are known microglial markers, whereas others were identified by the study authors as being generalizable either to microglia or their activation. Most (16/17) reported changes in markers consistent with increased numbers or activation of microglia in AD.
High-throughput techniques: microarray and proteomics
Microarrays and proteomic studies that specifically discussed microglia or their markers were included in the interest of comprehensiveness, but they are presented separately (Supplementary Materials Table 14) as there is significant risk that those presented here do not represent the balance of the literature. Any microarray would include some microglial markers, such as HLA-DR, but studies that did not identify significant differences between AD and control for these markers may be less likely to have mentioned them by name in the title, abstract or article keywords, and would therefore have been missed by the systematic search. Six studies used high-throughput techniques, either microarray11, 123, 124, 125, 126 or proteomics,127 to examine genes, proteins or patterns of gene expression associated with microglia in AD and control in various brain regions, including the entorhinal cortex, hippocampus, postcentral gyrus, superior frontal gyrus, prefrontal cortex, cerebellum, dorsolateral prefrontal cortex, visual cortex and precuneus. All high-throughput studies reported increases in AD relative to control in some of the microglial markers measured.
Nonquantitative comparisons
Fifty-three papers that would otherwise have met the inclusion and exclusion criteria for this review were not included in the full extraction because they were nonquantitative, though results and key methodological details are presented in Supplementary Materials Table 15. Papers were considered nonquantitative if quantitative data for the comparison between AD and control were not presented and/or the methods did not indicate that quantification had occurred. These papers used immunohistochemistry or immunohistochemistry with western blot to measure microglia using various markers, and commented qualitatively on the number of cells, amount of staining/expression or morphology. The most common brain region investigated by the nonquantitative papers was the hippocampus (24/53 studies); however, like in the quantitative papers in the review, many other regions were also investigated, including the entorhinal cortex, temporal cortex, occipital cortex, frontal cortex and anterior cingulate gyrus. Nonquantitative studies used many of the same markers discussed previously, such as HLA,39, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 139, 140, 141, 142, 143, 144 Iba1,77, 145 CD68,142, 146 ferritin,140, 147, 148, 149, 150 CD45,142, 151, 152, 153, 154, 155, 156 RCA-1,138, 157 and various other markers or combinations of markers, considered to be related to microglial activation.158, 159, 160, 161, 162, 163, 164, 165, 166, 167, 168, 169, 170, 171, 172, 173, 174, 175 All but two of these papers,97, 176 measuring CD68 and Iba1, respectively, reported elevations in microglial markers in AD relative to control post-mortem brains. Thus, the null finding rate in the nonquantitative papers is less than a third of the rate of quantitatively assessed markers like MHCII.
Discussion
Most (76/113) of the studies included in this review measured microglia using one of three common markers: MHCII, CD68 and Iba1. Although studies measuring MHCII or CD68 consistently identified increases in AD relative to control in most brain regions studied, 10 of the 20 studies that compared Iba1 identified no difference or a decrease relative to controls. Importantly, 9/10 studies noting an increase in Iba1 in AD relative to controls used expression-based quantification methods (qPCR, western, fluorescence intensity) that indicate only the amount of Iba1 in the sample, whereas most that identified no difference used cell counting, including two studies that used stereological quantification. Iba1 is a pan-microglial marker whose expression increases with microglial activation,58, 77 and hence these results indicate that there are increases in the expression of Iba1, but not the absolute number of microglia in AD. This, along with the increases in MHCII and CD68, which are both markers of activated microglia, suggest that the apparent increases in microglial markers in AD are attributable to increases in activation rather than the absolute number of microglia. This is supported by findings with CD11b that, like Iba1, labels resting and activated microglia, that also had mixed results, and by studies using other activation markers, such as ferritin, IL-1α and CD33, that were consistently increased in AD. However, there is still controversy surrounding what markers are indicative of activation, as well as the type of activation they are associated with,60 and hence more research on the physiological significance of increases in these markers is needed.
The nine studies identified by this review that compared microglial markers between AD and a high pathology control group, the cognitively intact subjects with AD neuropathology, may shed light on whether increased microglial activation is a cause or consequence of the disease. Five of these studies reported higher levels of microglia markers in AD, three using HLA-DR,7, 33, 40 one using CD68,83 and one using both TREM2 and Iba1.61 In contrast, three studies, using CD36, HLA-DR or proteomics, reported no difference between AD and the high-pathology control group despite increases relative to the regular control group,45, 127, 177 whereas one reported more HLA-DR-positive microglia in the high pathology control group than in AD.24 Though there appears to be increased microglia markers in AD relative to high pathology control subjects, more studies using this reference group are needed to help elucidate the role of microglia in AD pathogenesis. Polymorphisms in genes encoding microglial markers HLA-DR, CD33 and TREM2 have been implicated as risk factors for late-onset AD,9, 10, 103 supporting the notion that increased microglial activation is a contributor to AD development, and not merely a response to established AD pathology.
Some of the heterogeneity identified between studies may be attributed to differences in the brain regions examined. More than half the studies that used tissue from the cerebellum identified no difference between control and AD across a range of microglial markers.25, 34, 44, 112, 178 This lack of reactivity in the cerebellum has been remarked upon previously, leading the cerebellum to be proposed as a reference region for positron emission tomography imaging of TSPO.179 Similarly, half the studies examining microglia in the white matter showed either no difference between AD and control25, 44, 52, 69, 94, 109, 139 or higher levels in control.32 Several studies report higher levels of HLA-DR-expressing microglia in the white matter than the gray matter of nondemented cases, suggesting that microglia in this tissue may be constitutively activated.49, 52, 143 The lack of consistent differences in the white matter between patients with AD and controls in the studies in our review could therefore indicate that AD pathology does not stimulate further increases in microglial markers on top of their already activated state, whereas elevations in AD can be observed in the grey matter that does not demonstrate this constitutive elevation.
Heterogeneity can also exist within a brain region. For example, Bachstetter et al.69 counted CD68-positive microglia in the hippocampus CA1, CA2/3, CA4 and the dentate gyrus, and noted increases in the CA1 and dentate gyrus relative to controls, but no difference in CA2/3 or CA4. Similarly, Masliah et al.94 quantified microglia in different layers of the hippocampus and reported higher levels relative to control in the molecular and pyramidal layer, but lower levels in the stratus polymorphous. Thus, studies that examined staining in the whole hippocampus could dilute potential differences between AD and control by examining multiple regions simultaneously, and this may help to explain why nearly a third of studies using hippocampal tissue reported no difference between AD and control. The potential for heterogeneity of results between tissues is particularly important for studies that performed their analyses in a mix of different tissues, rather than examining each region separately.30, 89, 98, 122, 180
Only one of the identified studies reported on the use of anti-inflammatory drugs in their subjects.16 Though this study did not identify significant differences in microglia counts in users and nonusers, nonsteroidal anti-inflammatory drug treatment reduces activated glial cells in a mouse model of AD,181 and the use of nonsteroidal anti-inflammatory drugs is associated with less microglial activation in the brains of elderly patients post-mortem,182 suggesting that unreported nonsteroidal anti-inflammatory drug use could be a potential source of confounding. It is therefore feasible that unreported differences in medication use between AD and controls within and between studies contributed to the variability in the results.
The genotypes of the subjects could also offer a source of heterogeneity. Microglial activation appears to be affected by APOE genotype, with carriers of the ɛ4 risk allele exhibiting greater activation in some48, 56 but not all studies.82 Only 29 of the studies included in this review reported the APOE genotype of their subjects. Among those that reported genotype, there was variability, with some studies excluding those with the ɛ4 allele,68, 172 and others including a majority of AD subjects carrying the risk allele.30, 62, 79
Differences in control group characteristics could also influence the results. Over half the included studies did not report screening controls for neurological or neuropathological abnormalities. In addition, only 11 studies reported excluding subjects with a history of psychiatric disorders. As various neurological and psychiatric diseases have been associated with neuroinflammation in some post-mortem studies,183, 184, 185 this lack of screening could contribute heterogeneity to the results.
The severity of AD pathology in AD cases and controls also varies between studies. Braak stage was the most widely reported pathological score, and is presented in the tables where available. Most studies used AD cases with a Braak score of V–VI, but some used AD cases with scores as low as I and II.18, 45, 53, 64, 67, 123 Similarly for controls, Braak stages less than III were most commonly used, but some studies included controls with Braak stages IV or higher.123, 127 This variation may be important, as some studies identified associations between Braak stage or neurofibrillary tangles and microglia markers, including HLA-DR, Iba1 and CD68.17, 48, 52, 64, 84, 186 Few studies reported on the amount of plaque in the brains included in their cohorts, but differences in plaque load between studies could also influence the results, as microglial markers were found to correlate with plaque loads in several studies.47, 48, 52, 56, 186 These correlations could be functionally important to the progression of AD, as post-mortem analyses from brains of AD patients administered amyloid-β vaccines demonstrate positive correlations between Iba1 and CD68 and markers of neuronal loss and degeneration that are attenuated following immunization and amyloid-β clearance,187 when the number of activated CD68-positive but not total Iba1-positive microglia are reduced.186
Very few studies reported using stereology to complete their cell counts,54, 70, 83, 84, 122 the gold standard for quantifying cells in a tissue without bias. In our review, Serrano-Pozo et al.54 reported that the absolute number of microglia was the same in AD and control samples when stereology was used, but higher in AD when nonstereology-based counting was applied because of an apparent concentration of cells caused by cortical atrophy. The use of nonstereological counting could therefore have introduced some bias into the results. Interestingly, the three stereology studies that counted all microglia, identified by either Iba1 or morphology, reported no differences in total microglial counts in AD relative to control samples,54, 122, 70 whereas increases were noted in the number of cells positive for activation-associated markers MHCII, CD68 and CD33.54, 70, 83, 84 This supports the suggestion that microglial activation is increased in AD without an absolute increase in the number of microglia.
Technical differences could also contribute to variation between studies. For immunohistochemistry, the most commonly used technique in this review, the method and duration of tissue fixation,188, 189 the thickness of the brain sections,189 the use of antigen retrieval,190 the antibody selected77, 188 and variation in many steps in the immunohistochemistry protocol itself can all influence the intensity of staining that could affect the ability of studies to pick up differences between AD and control subjects. Differences in the methods used for measuring gene expression or for quantifying protein by western blot or enzyme-linked immunosorbent assay could similarly contribute to variability in the results. This technical information, with the exception of technique used and post-mortem interval, was not extracted in the tables because the wide variability in methods and reporting made it unfeasible, but it is an important potential contributor to study heterogeneity.
Limitations
Though efforts were made to identify every published paper that compared microglia markers between AD and control post-mortem human brain samples, it is possible that some papers have been missed. The large number of papers returned in the initial search (>20 000) may have increased the risk of misidentifying a paper. In addition, studies that measured a microglial marker, but did not mention it in the title, abstract or keywords would have been missed by the systematic search. Thus, studies that did not examine microglia as a primary objective are more likely to have been omitted which may introduce some bias into the results of this review.
Microglia interact with astrocytes to initiate and drive inflammation within the brain. Like with microglia, markers of astrocytes are reported to be elevated in patients with AD relative to controls,191, 192 where they seem to colocalize with amyloid-β plaques.193, 194 Although our review focused only on microglial markers, it had initially set out to include other inflammatory markers as well, including, astrocytes, cytokines, complement, lipid mediators and other immune cells. Our title and abstract screening uncovered over 700 papers, nearly 297 for astrocytes alone, an unfeasibly large number of papers that led us to focus on microglia for this review. Not including astrocytes and other inflammatory mediators is a limitation of our review because it results in an incomplete picture of neuroinflammation in AD.
Another important limitation of this review is that although it describes the results of the included studies in the context of microglia, it should be noted that nearly all microglia markers are also expressed by other myeloid cells such as infiltrating or perivascular macrophages, neutrophils or T cells (see Supplementary Table 1 for a summary of marker expression in other cell types). Although there are generally few of these cells in the brain parenchyma under normal conditions, there may be increased accumulation and infiltration in AD that could lead to an overestimation of the quantity of microglial markers in diseased relative to control brains.
Conclusion
Microglia markers are increased in various brain regions in patients with AD relative to controls. The balance of the evidence suggests that this increase is more attributable to increased activation than to an increase in absolute cell number, but more research measuring microglial proliferation in post-mortem human brain samples would be useful for clarifying this point.
References
Ma K, Mount HT, McLaurin J . Region-specific distribution of beta-amyloid peptide and cytokine expression in TgCRND8 mice. Neurosci Lett 2011; 492: 5–10.
Souza LC, Filho CB, Goes AT, Fabbro LD, de Gomes MG, Savegnago L et al. Neuroprotective effect of physical exercise in a mouse model of Alzheimer's disease induced by beta-amyloid(1)(-)(4)(0) peptide. Neurotox Res 2013; 24: 148–163.
Hopperton KE, Trepanier MO, Giuliano V, Bazinet RP . Brain omega-3 polyunsaturated fatty acids modulate microglia cell number and morphology in response to intracerebroventricular amyloid-beta 1-40 in mice. J Neuroinflammation 2016; 13: 257.
Vanzani MC, Iacono RF, Caccuri RL, Berria MI . Immunochemical and morphometric features of astrocyte reactivity vs. plaque location in Alzheimer's disease. Medicina 2005; 65: 213–218.
Flanders KC, Lippa CF, Smith TW, Pollen DA, Sporn MB . Altered expression of transforming growth factor-beta in Alzheimer's disease. Neurology 1995; 45: 1561–1569.
Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 1989; 86: 7611–7615.
Lue LF, Brachova L, Civin WH, Rogers J . Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of Alzheimer's disease neurodegeneration. J Neuropathol Exp Neurol 1996; 55: 1083–1088.
Griffin WS . Inflammation and neurodegenerative diseases. Am J Clin Nutr 2006; 83: 470S–474S.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013; 45: 1452–1458.
Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer's disease. Nat Genet 2017; 49: 1373–1384.
Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell 2013; 153: 707–720.
Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A et al. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci 2013; 16: 848–850.
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E et al. TREM2 variants in Alzheimer's disease. N Engl J Med 2013; 368: 117–127.
Lee YB, Nagai A, Kim SU . Cytokines, chemokines, and cytokine receptors in human microglia. J Neurosci Res 2002; 69: 94–103.
Akiyama H, McGeer PL . Brain microglia constitutively express beta-2 integrins. J Neuroimmunol 1990; 30: 81–93.
Halliday GM, Shepherd CE, McCann H, Reid WG, Grayson DA, Broe GA et al. Effect of anti-inflammatory medications on neuropathological findings in Alzheimer disease. Arch Neurol 2000; 57: 831–836.
Hoozemans JJ, van Haastert ES, Veerhuis R, Arendt T, Scheper W, Eikelenboom P et al. Maximal COX-2 and ppRb expression in neurons occurs during early Braak stages prior to the maximal activation of astrocytes and microglia in Alzheimer's disease. J Neuroinflamm 2005; 2: 27.
Kellner A, Matschke J, Bernreuther C, Moch H, Ferrer I, Glatzel M . Autoantibodies against beta-amyloid are common in Alzheimer's disease and help control plaque burden. Ann Neurol 2009; 65: 24–31.
Carpenter AF, Carpenter PW, Markesbery WR . Morphometric analysis of microglia in Alzheimer's disease. J Neuropathol Exp Neurol 1993; 52: 601–608.
Dal Bianco A, Bradl M, Frischer J, Kutzelnigg A, Jellinger K, Lassmann H . Multiple sclerosis and Alzheimer's disease. Ann Neurol 2008; 63: 174–183.
Desai BS, Schneider JA, Li JL, Carvey PM, Hendey B . Evidence of angiogenic vessels in Alzheimer's disease. J Neural Transm 2009; 116: 587–597.
Dhawan G, Floden AM, Combs CK . Amyloid-beta oligomers stimulate microglia through a tyrosine kinase dependent mechanism. Neurobiol Aging 2012; 33: 2247–2261.
Szpak GM, Lechowicz W, Lewandowska E, Bertrand E, Wierzba-Bobrowicz T, Gwiazda E et al. Neurones and microglia in central nervous system immune response to degenerative processes. Part 1: Alzheimer's disease and Lewy body variant of Alzheimer's disease. Quantitative study. Folia Neuropathol 2001; 39: 181–192.
Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ . Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res 2007; 10: 61–74.
Giulian D, Haverkamp LJ, Li J, Karshin WL, Yu J, Tom D et al. Senile plaques stimulate microglia to release a neurotoxin found in Alzheimer brain. Neurochem Int 1995; 27: 119–137.
Gouw AA, Seewann A, Vrenken H, van der Flier WM, Rozemuller JM, Barkhof F et al. Heterogeneity of white matter hyperintensities in Alzheimer's disease: post-mortem quantitative MRI and neuropathology. Brain 2008; 131 (Pt 12): 3286–3298.
Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M et al. Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. J Neurochem 1995; 65: 2146–2156.
Hoozemans JJ, Rozemuller AJ, van Haastert ES, Eikelenboom P, van Gool WA . Neuroinflammation in Alzheimer's disease wanes with age. J Neuroinflamm 2011; 8: 171.
Imamura K, Sawada M, Ozaki N, Naito H, Iwata N, Ishihara R et al. Activation mechanism of brain microglia in patients with diffuse neurofibrillary tangles with calcification: a comparison with Alzheimer disease. Alzheimer Dis Assoc Disord 2001; 15: 45–50.
Itagaki S, McGeer PL, Akiyama H . Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer's disease brain tissue. Neurosci Lett 1988; 91: 259–264.
Jantaratnotai N, Schwab C, Ryu JK, McGeer PL, McLarnon JG . Converging perturbed microvasculature and microglial clusters characterize Alzheimer disease brain. Curr Alzheimer Res 2010; 7: 625–636.
Korvatska O, Leverenz JB, Jayadev S, McMillan P, Kurtz I, Guo X et al. R47H variant of TREM2 associated with Alzheimer disease in a large late-onset family: clinical, genetic, and neuropathological study. JAMA Neurol 2015; 72: 920–927.
Lopes KO, Sparks DL, Streit WJ . Microglial dystrophy in the aged and Alzheimer's disease brain is associated with ferritin immunoreactivity. Glia 2008; 56: 1048–1060.
Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM et al. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer's disease: identification of a cellular activation mechanism. Exp Neurol 2001; 171 (1): 29–45.
Matsuo A, Walker DG, Terai K, McGeer PL . Expression of CD43 in human microglia and its downregulation in Alzheimer's disease. J Neuroimmunol 1996; 71: 81–86.
McGeer PL, Itagaki S, Boyes BE, McGeer EG . Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 1988; 38: 1285–1291.
McGeer PL, Itagaki S, Tago H, McGeer EG . Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett 1987; 79: 195–200.
McGeer PL, McGeer EG, Yasojima K . Alzheimer disease and neuroinflammation. J Neural Transm Suppl 2000; 59: 53–57.
Narayan PJ, Lill C, Faull R, Curtis MA, Dragunow M . Increased acetyl and total histone levels in post-mortem Alzheimer's disease brain. Neurobiol Dis 2015; 74: 281–294.
Parachikova A, Agadjanyan MG, Cribbs DH, Blurton-Jones M, Perreau V, Rogers J et al. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol Aging 2007; 28: 1821–1833.
Rezaie P, Pontikis CC, Hudson L, Cairns NJ, Lantos PL . Expression of cellular prion protein in the frontal and occipital lobe in Alzheimer's disease, diffuse Lewy body disease, and in normal brain: an immunohistochemical study. J Histochem Cytochem 2005; 53: 929–940.
Shepherd CE, Thiel E, McCann H, Harding AJ, Halliday GM . Cortical inflammation in Alzheimer disease but not dementia with Lewy bodies. Arch Neurol 2000; 57: 817–822.
Thal DR, Arendt T, Waldmann G, Holzer M, Zedlick D, Rub U et al. Progression of neurofibrillary changes and PHF-tau in end-stage Alzheimer's disease is different from plaque and cortical microglial pathology. Neurobiol Aging 1998; 19: 517–525.
Van Everbroeck B, Dobbeleir I, De Waele M, De Leenheir E, Lubke U, Martin JJ et al. Extracellular protein deposition correlates with glial activation and oxidative stress in Creutzfeldt-Jakob and Alzheimer's disease. Acta Neuropathol 2004; 108: 194–200.
Vehmas AK, Kawas CH, Stewart WF, Troncoso JC . Immune reactive cells in senile plaques and cognitive decline in Alzheimer's disease. Neurobiol Aging 2003; 24: 321–331.
Wilcock DM, Hurban J, Helman AM, Sudduth TL, McCarty KL, Beckett TL et al. Down syndrome individuals with Alzheimer's disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer's disease. Neurobiol Aging 2015; 36: 2468–2474.
Xiang Z, Haroutunian V, Ho L, Purohit D, Pasinetti GM . Microglia activation in the brain as inflammatory biomarker of Alzheimer's disease neuropathology and clinical dementia. Dis Markers 2006; 22: 95–102.
Egensperger R, Kosel S, von Eitzen U, Graeber MB . Microglial activation in Alzheimer disease: association with APOE genotype. Brain Pathol 1998; 8: 439–447.
Mattiace LA, Davies P, Dickson DW . Detection of HLA-DR on microglia in the human brain is a function of both clinical and technical factors. Am J Pathol 1990; 136: 1101–1114.
Verwer RW, Sluiter AA, Balesar RA, Baayen JC, Noske DP, Dirven CM et al. Mature astrocytes in the adult human neocortex express the early neuronal marker doublecortin. Brain 2007; 130 (Pt 12): 3321–3335.
Wojtera M, Sobow T, Kloszewska I, Liberski PP, Brown DR, Sikorska B . Expression of immunohistochemical markers on microglia in Creutzfeldt-Jakob disease and Alzheimer's disease: morphometric study and review of the literature. Folia Neuropathol 2012; 50: 74–84.
Overmyer M, Helisalmi S, Soininen H, Laakso M, Riekkinen P Sr., Alafuzoff I . Reactive microglia in aging and dementia: an immunohistochemical study of postmortem human brain tissue. Acta Neuropathol 1999; 97: 383–392.
Pugliese M, Rodriguez MJ, Gimeno-Bayon J, Pujadas L, Billett EE, Wells C et al. Alzheimer's disease modifies progenitor cell expression of monoamine oxidase B in the subventricular zone. J Neurosci Res 2010; 88: 2588–2597.
Serrano-Pozo A, Gomez-Isla T, Growdon JH, Frosch MP, Hyman BT . A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am J Pathol 2013; 182: 2332–2344.
Valente T, Gella A, Sole M, Durany N, Unzeta M . Immunohistochemical study of semicarbazide-sensitive amine oxidase/vascular adhesion protein-1 in the hippocampal vasculature: pathological synergy of Alzheimer's disease and diabetes mellitus. J Neurosci Res 2012; 90 (10): 1989–1996.
Minett T, Classey J, Matthews FE, Fahrenhold M, Taga M, Brayne C et al. Microglial immunophenotype in dementia with Alzheimer's pathology. J Neuroinflamm 2016; 13 (1): 135.
Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S . A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun 1996; 224: 855–862.
Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S . Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res 1998; 57: 1–9.
Ohsawa K, Imai Y, Kanazawa H, Sasaki Y, Kohsaka S . Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J Cell Sci 2000; 113 (Pt 17): 3073–3084.
Walker DG, Lue LF . Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther 2015; 7: 56.
Lue LF, Schmitz CT, Serrano G, Sue LI, Beach TG, Walker DG . TREM2 protein expression changes correlate with Alzheimer's disease neurodegenerative pathologies in post-mortem temporal cortices. Brain Pathol 2015; 25: 469–480.
Nielsen HM, Ek D, Avdic U, Orbjorn C, Hansson O et alNetherlands Brain Bank. NG2 cells, a new trail for Alzheimer's disease mechanisms? Acta Neuropathol Commun 2013; 1: 7.
Rangaraju S, Gearing M, Jin LW, Levey A . Potassium channel Kv1.3 is highly expressed by microglia in human Alzheimer's disease. J Alzheimers Dis 2015; 44: 797–808.
Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM . Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer's disease: link to brain reductions in acetylcholine. J Alzheimers Dis 2005; 8: 247–268.
Tang SC, Lathia JD, Selvaraj PK, Jo DG, Mughal MR, Cheng A et al. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp Neurol 2008; 213: 114–121.
Walker DG, Whetzel AM, Serrano G, Sue LI, Beach TG, Lue LF . Association of CD33 polymorphism rs3865444 with Alzheimer's disease pathology and CD33 expression in human cerebral cortex. Neurobiol Aging 2015; 36: 571–582.
Lastres-Becker I, Innamorato NG, Jaworski T, Rabano A, Kugler S, Van Leuven F et al. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 2014; 137 (Pt 1): 78–91.
Satoh J, Kino Y, Asahina N, Takitani M, Miyoshi J, Ishida T et al. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016; 36: 39–49.
Bachstetter AD, Van Eldik LJ, Schmitt FA, Neltner JH, Ighodaro ET, Webster SJ et al. Disease-related microglia heterogeneity in the hippocampus of Alzheimer's disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun 2015; 3: 32.
Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K et al. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013; 78: 631–643.
Magistri M, Velmeshev D, Makhmutova M, Faghihi MA . Transcriptomics profiling of Alzheimer's disease reveal neurovascular defects, altered amyloid-beta homeostasis, and deregulated expression of long noncoding RNAs. J Alzheimers Dis 2015; 48: 647–665.
Davies DS, Ma J, Jegathees T, Goldsbury C . Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer's disease. Brain Pathol 2016; 27: 795–808.
Ekonomou A, Savva GM, Brayne C, Forster G, Francis PT, Johnson M et al. Stage-specific changes in neurogenic and glial markers in Alzheimer's disease. Biol Psychiatry 2015; 77: 711–719.
Lee C, Low CY, Francis PT, Attems J, Wong PT, Lai MK et al. An isoform-specific role of FynT tyrosine kinase in Alzheimer's disease. J Neurochem 2016; 136: 637–650.
Marlatt MW, Bauer J, Aronica E, van Haastert ES, Hoozemans JJ, Joels M et al. Proliferation in the Alzheimer hippocampus is due to microglia, not astroglia, and occurs at sites of amyloid deposition. Neural Plast 2014; 2014: 693851.
Sanchez-Mejias E, Navarro V, Jimenez S, Sanchez-Mico M, Sanchez-Varo R, Nunez-Diaz C et al. Soluble phospho-tau from Alzheimer's disease hippocampus drives microglial degeneration. Acta Neuropathol 2016; 132: 897–916.
Streit WJ, Braak H, Xue QS, Bechmann I . Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol 2009; 118: 475–485.
Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC et al. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer's disease brain and damage the blood-brain barrier. Eur J Clin Invest 2002; 32: 360–371.
Arnold SE, Han LY, Clark CM, Grossman M, Trojanowski JQ . Quantitative neurohistological features of frontotemporal degeneration. Neurobiol Aging 2000; 21: 913–919.
DeLuca GC, Joseph A, George J, Yates RL, Hamard M, Hofer M et al. Olfactory pathology in central nervous system demyelinating diseases. Brain Pathol 2015; 25: 543–551.
Doorn KJ, Goudriaan A, Blits-Huizinga C, Bol JG, Rozemuller AJ, Hoogland PV et al. Increased amoeboid microglial density in the olfactory bulb of Parkinson's and Alzheimer's patients. Brain Pathol 2014; 24: 152–165.
Hultman K, Strickland S, Norris EH . The APOE varepsilon4/varepsilon4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer's disease patients. J Cereb Blood Flow Metab 2013; 33: 1251–1258.
Perez-Nievas BG, Stein TD, Tai HC, Dols-Icardo O, Scotton TC, Barroeta-Espar I et al. Dissecting phenotypic traits linked to human resilience to Alzheimer's pathology. Brain 2013; 136 (Pt 8): 2510–2526.
Serrano-Pozo A, Mielke ML, Gomez-Isla T, Betensky RA, Growdon JH, Frosch MP et al. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer's disease. Am J Pathol 2011; 179: 1373–1384.
Van Everbroeck B, Dewulf E, Pals P, Lubke U, Martin JJ, Cras P . The role of cytokines, astrocytes, microglia and apoptosis in Creutzfeldt-Jakob disease. Neurobiol Aging 2002; 23: 59–64.
Arnold SE, Trojanowski JQ, Gur RE, Blackwell P, Han LY, Choi C . Absence of neurodegeneration and neural injury in the cerebral cortex in a sample of elderly patients with schizophrenia. Arch Gen Psychiatry 1998; 55: 225–232.
Alvarez MI, Rivas L, Lacruz C, Toledano A . Astroglial cell subtypes in the cerebella of normal adults, elderly adults, and patients with Alzheimer's disease: a histological and immunohistochemical comparison. Glia 2015; 63: 287–312.
Falke E, Han LY, Arnold SE . Absence of neurodegeneration in the thalamus and caudate of elderly patients with schizophrenia. Psychiatry Res 2000; 93: 103–110.
Lue LF, Walker DG, Rogers J . Modeling microglial activation in Alzheimer's disease with human postmortem microglial cultures. Neurobiol Aging 2001; 22 (6): 945–956.
Doens D, Fernandez PL . Microglia receptors and their implications in the response to amyloid beta for Alzheimer's disease pathogenesis. J Neuroinflamm 2014; 11: 48.
Shan L, Bossers K, Unmehopa U, Bao AM, Swaab DF . Alterations in the histaminergic system in Alzheimer's disease: a postmortem study. Neurobiol Aging 2012; 33: 2585–2598.
Becher B, Antel JP . Comparison of phenotypic and functional properties of immediately ex vivo and cultured human adult microglia. Glia 1996; 18: 1–10.
Licastro F, Mallory M, Hansen LA, Masliah E . Increased levels of alpha-1-antichymotrypsin in brains of patients with Alzheimer's disease correlate with activated astrocytes and are affected by APOE 4 genotype. J Neuroimmunol 1998; 88: 105–110.
Masliah E, Mallory M, Hansen L, Alford M, Albright T, Terry R et al. Immunoreactivity of CD45, a protein phosphotyrosine phosphatase, in Alzheimer's disease. Acta Neuropathol 1991; 83: 12–20.
Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP . Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflamm 2006; 3: 27.
Kwiatek-Majkusiak J, Dickson DW, Tacik P, Aoki N, Tomasiuk R, Koziorowski D et al. Relationships between typical histopathological hallmarks and the ferritin in the hippocampus from patients with Alzheimer's disease. Acta Neurobiol Exp 2015; 75: 391–398.
Meadowcroft MD, Connor JR, Yang QX . Cortical iron regulation and inflammatory response in Alzheimer's disease and APPSWE/PS1DeltaE9 mice: a histological perspective. Front Neurosci 2015; 9: 255.
Ohgami T, Kitamoto T, Shin RW, Kaneko Y, Ogomori K, Tateishi J . Increased senile plaques without microglia in Alzheimer's disease. Acta Neuropathol 1991; 81: 242–247.
Mochizuki A, Peterson JW, Mufson EJ, Trapp BD . Amyloid load and neural elements in Alzheimer's disease and nondemented individuals with high amyloid plaque density. Exp Neurol 1996; 142: 89–102.
Fukumoto H, Asami-Odaka A, Suzuki N, Iwatsubo T . Association of A beta 40-positive senile plaques with microglial cells in the brains of patients with Alzheimer's disease and in non-demented aged individuals. Neurodegeneration 1996; 5: 13–17.
DiPatre PL, Gelman BB . Microglial cell activation in aging and Alzheimer disease: partial linkage with neurofibrillary tangle burden in the hippocampus. J Neuropathol Exp Neurol 1997; 56: 143–149.
Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF et al. Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet 2008; 83: 623–632.
Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 2011; 43: 429–435.
Malik M, Simpson JF, Parikh I, Wilfred BR, Fardo DW, Nelson PT et al. CD33 Alzheimer's risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci 2013; 33: 13320–13325.
Takahashi K, Rochford CD, Neumann H . Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 2005; 201: 647–657.
Steubl D, Roos M, Hettwer S, Angermann S, Wen M, Schmaderer C et al. Comparison of peritoneal low-molecular-weight-protein-removal in CCPD and CAPD patients based on C-terminal agrin fragment clearance. Kidney Blood Press Res 2016; 41: 175–185.
Roussos P, Katsel P, Fam P, Tan W, Purohit DP, Haroutunian V . The triggering receptor expressed on myeloid cells 2 (TREM2) is associated with enhanced inflammation, neuropathological lesions and increased risk for Alzheimer's dementia. Alzheimers Dement 2015; 11: 1163–1170.
Itoh Y, Yamada M, Suematsu N, Matsushita M, Otomo E . An immunohistochemical study of centenarian brains: a comparison. J Neurol Sci 1998; 157: 73–81.
Paulus W, Bancher C, Jellinger K . Microglial reaction in Pick's disease. Neurosci Lett 1993; 161: 89–92.
Yamada M, Itoh Y, Sodeyama N, Suematsu N, Otomo E, Matsushita M et al. Senile dementia of the neurofibrillary tangle type: a comparison with Alzheimer's disease. Dement Geriatr Cogn Disord 2001; 12: 117–126.
Sheng JG, Jones RA, Zhou XQ, McGinness JM, Van Eldik LJ, Mrak RE et al. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer's disease: potential significance for tau protein phosphorylation. Neurochem Int 2001; 39: 341–348.
Sheng JG, Mrak RE, Griffin WS . Microglial interleukin-1 alpha expression in brain regions in Alzheimer's disease: correlation with neuritic plaque distribution. Neuropathol Appl Neurobiol 1995; 21: 290–301.
Sheng JG, Griffin WS, Royston MC, Mrak RE . Distribution of interleukin-1-immunoreactive microglia in cerebral cortical layers: implications for neuritic plaque formation in Alzheimer's disease. Neuropathol Appl Neurobiol 1998; 24: 278–283.
Mannoji H, Yeger H, Becker LE . A specific histochemical marker (lectin Ricinus communis agglutinin-1) for normal human microglia, and application to routine histopathology. Acta Neuropathol 1986; 71: 341–343.
Mackenzie IR, Hao C, Munoz DG . Role of microglia in senile plaque formation. Neurobiol Aging 1995; 16: 797–804.
Sheffield LG, Marquis JG, Berman NE . Regional distribution of cortical microglia parallels that of neurofibrillary tangles in Alzheimer's disease. Neurosci Lett 2000; 285: 165–168.
Kravitz E, Gaisler-Salomon I, Biegon A . Hippocampal glutamate NMDA receptor loss tracks progression in Alzheimer's disease: quantitative autoradiography in postmortem human brain. PLoS ONE 2013; 8: e81244.
Marutle A, Gillberg PG, Bergfors A, Yu W, Ni R, Nennesmo I et al. (3)H-deprenyl and (3)H-PIB autoradiography show different laminar distributions of astroglia and fibrillar beta-amyloid in Alzheimer brain. J Neuroinflamm 2013; 10: 90.
Knezevic D, Mizrahi R . Molecular imaging of neuroinflammation in Alzheimer's disease and mild cognitive impairment. Prog Neuropsychopharmacol Biol Psychiatry 2017.
Pey P, Pearce RK, Kalaitzakis ME, Griffin WS, Gentleman SM . Phenotypic profile of alternative activation marker CD163 is different in Alzheimer's and Parkinson's disease. Acta Neuropathol Commun 2014; 2: 21.
Shefer VF . Hippocampal pathology as a possible factor in the pathogenesis of senile dementias. Neurosci Behav Physiol 1977; 8: 236–239.
Pelvig DP, Pakkenberg H, Regeur L, Oster S, Pakkenberg B . Neocortical glial cell numbers in Alzheimer's disease. A stereological study. Dement Geriatr Cogn Disord 2003; 16: 212–219.
Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ et al. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflamm 2012; 9: 179.
Durrenberger PF, Fernando FS, Kashefi SN, Bonnert TP, Seilhean D, Nait-Oumesmar B et al. Common mechanisms in neurodegeneration and neuroinflammation: a BrainNet Europe gene expression microarray study. J Neural Transm 2015; 122: 1055–1068.
Li X, Long J, He T, Belshaw R, Scott J . Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer's disease. Sci Rep 2015; 5: 12393.
Podtelezhnikov AA, Tanis KQ, Nebozhyn M, Ray WJ, Stone DJ, Loboda AP . Molecular insights into the pathogenesis of Alzheimer's disease and its relationship to normal aging. PLoS ONE 2011; 6: e29610.
Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L et al. A multi-network approach identifies protein-specific co-expression in asymptomatic and symptomatic Alzheimer's disease. Cell Syst 2017; 4: 60–72 e64.
Su JH, Deng G, Cotman CW . Bax protein expression is increased in Alzheimer's brain: correlations with DNA damage, Bcl-2 expression, and brain pathology. J Neuropathol Exp Neurol 1997; 56: 86–93.
McGeer PL, Akiyama H, Itagaki S, McGeer EG . Immune system response in Alzheimer's disease. Can J Neurol Sci 1989; 16 (4 Suppl): 516–527.
Bayer TA, Buslei R, Havas L, Falkai P . Evidence for activation of microglia in patients with psychiatric illnesses. Neurosci Lett 1999; 271: 126–128.
Togo T, Akiyama H, Kondo H, Ikeda K, Kato M, Iseki E et al. Expression of CD40 in the brain of Alzheimer's disease and other neurological diseases. Brain Res 2000; 885: 117–121.
Bowser R, Reilly S . Expression of FAC1 in activated microglia during Alzheimer's disease. Neurosci Lett 1998; 253: 163–166.
Rogers J, Luber-Narod J, Styren SD, Civin WH . Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer's disease. Neurobiol Aging 1988; 9: 339–349.
Yoshiyama Y, Arai K, Oki T, Hattori T . Expression of invariant chain and pro-cathepsin L in Alzheimer's brain. Neurosci Lett 2000; 290: 125–128.
Wiendl H, Feger U, Mittelbronn M, Jack C, Schreiner B, Stadelmann C et al. Expression of the immune-tolerogenic major histocompatibility molecule HLA-G in multiple sclerosis: implications for CNS immunity. Brain 2005; 128 (Pt 11): 2689–2704.
Christie RH, Chung H, Rebeck GW, Strickland D, Hyman BT . Expression of the very low-density lipoprotein receptor (VLDL-r), an apolipoprotein-E receptor, in the central nervous system and in Alzheimer's disease. J Neuropathol Exp Neurol 1996; 55: 491–498.
Dickson DW, Sinicropi S, Yen SH, Ko LW, Mattiace LA, Bucala R et al. Glycation and microglial reaction in lesions of Alzheimer's disease. Neurobiol Aging 1996; 17: 733–743.
Perlmutter LS, Scott SA, Barron E, Chui HC . MHC class II-positive microglia in human brain: association with Alzheimer lesions. J Neurosci Res 1992; 33: 549–558.
Tooyama I, Kimura H, Akiyama H, McGeer PL . Reactive microglia express class I and class II major histocompatibility complex antigens in Alzheimer's disease. Brain Res 1990; 523: 273–280.
An L, Sato H, Konishi Y, Walker DG, Beach TG, Rogers J et al. Expression and localization of lactotransferrin messenger RNA in the cortex of Alzheimer's disease. Neurosci Lett 2009; 452: 277–280.
Christie RH, Freeman M, Hyman BT . Expression of the macrophage scavenger receptor, a multifunctional lipoprotein receptor, in microglia associated with senile plaques in Alzheimer's disease. Am J Pathol 1996; 148: 399–403.
Sasaki A, Yamaguchi H, Ogawa A, Sugihara S, Nakazato Y . Microglial activation in early stages of amyloid beta protein deposition. Acta Neuropathol 1997; 94: 316–322.
Styren SD, Civin WH, Rogers J . Molecular, cellular, and pathologic characterization of HLA-DR immunoreactivity in normal elderly and Alzheimer's disease brain. Exp Neurol 1990; 110: 93–104.
McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG . Microglia in degenerative neurological disease. Glia 1993; 7: 84–92.
Lopez-Gonzalez I, Schluter A, Aso E, Garcia-Esparcia P, Ansoleaga B, LL F et al. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J Neuropathol Exp Neurol 2015; 74: 319–344.
Wu CK, Thal L, Pizzo D, Hansen L, Masliah E, Geula C . Apoptotic signals within the basal forebrain cholinergic neurons in Alzheimer's disease. Exp Neurol 2005; 195: 484–496.
Jellinger K, Paulus W, Grundke-Iqbal I, Riederer P, Youdim MB . Brain iron and ferritin in Parkinson's and Alzheimer's diseases. J Neural Transm Park Dis Dement Sect 1990; 2: 327–340.
Zeineh MM, Chen Y, Kitzler HH, Hammond R, Vogel H, Rutt BK . Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol Aging 2015; 36: 2483–2500.
van Duijn S, Nabuurs RJ, van Duinen SG, Natte R . Comparison of histological techniques to visualize iron in paraffin-embedded brain tissue of patients with Alzheimer's disease. J Histochem Cytochem 2013; 61: 785–792.
Grundke-Iqbal I, Fleming J, Tung YC, Lassmann H, Iqbal K, Joshi JG . Ferritin is a component of the neuritic (senile) plaque in Alzheimer dementia. Acta Neuropathol 1990; 81: 105–110.
Yamada T, Yoshiyama Y, Sato H, Seiki M, Shinagawa A, Takahashi M . White matter microglia produce membrane-type matrix metalloprotease, an activator of gelatinase A, in human brain tissues. Acta Neuropathol 1995; 90: 421–424.
Akiyama H, Ikeda K, Katoh M, McGeer EG, McGeer PL . Expression of MRP14, 27E10, interferon-alpha and leukocyte common antigen by reactive microglia in postmortem human brain tissue. J Neuroimmunol 1994; 50: 195–201.
Yamada T, Horisberger MA, Kawaguchi N, Moroo I, Toyoda T . Immunohistochemistry using antibodies to alpha-interferon and its induced protein, MxA, in Alzheimer's and Parkinson's disease brain tissues. Neurosci Lett 1994; 181: 61–64.
Yamada T, Yoshiyama Y, Kawaguchi N, Ichinose A, Iwaki T, Hirose S et al. Possible roles of transglutaminases in Alzheimer's disease. Dement Geriatr Cogn Disord 1998; 9: 103–110.
Yamada T, Miyazaki K, Koshikawa N, Takahashi M, Akatsu H, Yamamoto T . Selective localization of gelatinase A, an enzyme degrading beta-amyloid protein, in white matter microglia and in Schwann cells. Acta Neuropathol 1995; 89: 199–203.
Scott SA, Johnson SA, Zarow C, Perlmutter LS . Inability to detect beta-amyloid protein precursor mRNA in Alzheimer plaque-associated microglia. Exp Neurol 1993; 121: 113–118.
Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH . Alzheimer's disease. A double-labeling immunohistochemical study of senile plaques. Am J Pathol 1988; 132: 86–101.
Wirths O, Hillmann A, Pradier L, Hartig W, Bayer TA . Oligomeric pyroglutamate amyloid-beta is present in microglia and a subfraction of vessels in patients with Alzheimer's disease: implications for immunotherapy. J Alzheimers Dis 2013; 35: 741–749.
Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M et al. Occurrence of T cells in the brain of Alzheimer's disease and other neurological diseases. J Neuroimmunol 2002; 124: 83–92.
Yamada T, Yamanaka I . Microglial localization of alpha-interferon receptor in human brain tissues. Neurosci Lett 1995; 189: 73–76.
Akiyama H, Ikeda K, Kondo H, Kato M, McGeer PL . Microglia express the type 2 plasminogen activator inhibitor in the brain of control subjects and patients with Alzheimer's disease. Neurosci Lett 1993; 164: 233–235.
Yamada T, Tsujioka Y, Taguchi J, Takahashi M, Tsuboi Y, Moroo I et al. Melanotransferrin is produced by senile plaque-associated reactive microglia in Alzheimer's disease. Brain Res 1999; 845: 1–5.
Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W et al. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. Brain 2005; 128 (Pt 8): 1778–1789.
Guillemin GJ, Brew BJ, Noonan CE, Takikawa O, Cullen KM . Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer's disease hippocampus. Neuropathol Appl Neurobiol 2005; 31: 395–404.
Akiyama H, Nishimura T, Kondo H, Ikeda K, Hayashi Y, McGeer PL . Expression of the receptor for macrophage colony stimulating factor by brain microglia and its upregulation in brains of patients with Alzheimer's disease and amyotrophic lateral sclerosis. Brain Res 1994; 639: 171–174.
Bryan KJ, Zhu X, Harris PL, Perry G, Castellani RJ, Smith MA et al. Expression of CD74 is increased in neurofibrillary tangles in Alzheimer's disease. Mol Neurodegener 2008; 3: 13.
Minnasch P, Yamamoto Y, Ohkubo I, Nishi K . Demonstration of puromycin-sensitive alanyl aminopeptidase in Alzheimer disease brain. Leg Med 2003; 5 (Suppl 1): S285–S287.
Haga S, Akai K, Ishii T . Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta Neuropathol 1989; 77: 569–575.
Du Yan S, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte WW et al. Amyloid-beta peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci USA 1997; 94: 5296–5301.
Carrano A, Hoozemans JJ, van der Vies SM, Rozemuller AJ, van Horssen J, de Vries HE . Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal 2011; 15: 1167–1178.
Wong A, Luth HJ, Deuther-Conrad W, Dukic-Stefanovic S, Gasic-Milenkovic J, Arendt T et al. Advanced glycation endproducts co-localize with inducible nitric oxide synthase in Alzheimer's disease. Brain Res 2001; 920: 32–40.
Rozemuller AJ, Eikelenboom P, Theeuwes JW, Jansen Steur EN, de Vos RA . Activated microglial cells and complement factors are unrelated to cortical Lewy bodies. Acta Neuropathol 2000; 100: 701–708.
Ryu JK, McLarnon JG . A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer's disease brain. J Cell Mol Med 2009; 13 (9A): 2911–2925.
Xia MQ, Qin SX, Wu LJ, Mackay CR, Hyman BT . Immunohistochemical study of the beta-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer's disease brains. Am J Pathol 1998; 153: 31–37.
Hoozemans JJ, Rozemuller AJ, Janssen I, De Groot CJ, Veerhuis R, Eikelenboom P . Cyclooxygenase expression in microglia and neurons in Alzheimer's disease and control brain. Acta Neuropathol 2001; 101: 2–8.
Arends YM, Duyckaerts C, Rozemuller JM, Eikelenboom P, Hauw JJ . Microglia, amyloid and dementia in Alzheimer disease. A correlative study. Neurobiol Aging 2000; 21: 39–47.
Ricciarelli R, D'Abramo C, Zingg JM, Giliberto L, Markesbery W, Azzi A et al. CD36 overexpression in human brain correlates with beta-amyloid deposition but not with Alzheimer's disease. Free Radic Biol Med 2004; 36: 1018–1024.
Walker DG, Lue LF, Beach TG . Increased expression of the urokinase plasminogen-activator receptor in amyloid beta peptide-treated human brain microglia and in AD brains. Brain Res 2002; 926: 69–79.
Lyoo CH, Ikawa M, Liow JS, Zoghbi SS, Morse CL, Pike VW et al. Cerebellum can serve as a pseudo-reference region in Alzheimer disease to detect neuroinflammation measured with PET radioligand binding to translocator protein. J Nucl Med 2015; 56: 701–706.
Green PS, Mendez AJ, Jacob JS, Crowley JR, Growdon W, Hyman BT et al. Neuronal expression of myeloperoxidase is increased in Alzheimer's disease. J Neurochem 2004; 90: 724–733.
Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C et al. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain 2005; 128 (Pt 6): 1442–1453.
Mackenzie IR, Munoz DG . Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology 1998; 50: 986–990.
Lucas SM, Rothwell NJ, Gibson RM . The role of inflammation in CNS injury and disease. Br J Pharmacol 2006; 147 (Suppl 1): S232–S240.
Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O . Neuroinflammation and psychiatric illness. J Neuroinflamm 2013; 10: 43.
Trepanier MO, Hopperton KE, Mizrahi R, Mechawar N, Bazinet RP . Postmortem evidence of cerebral inflammation in schizophrenia: a systematic review. Mol Psychiatry 2016; 21: 1009–1026.
Zotova E, Bharambe V, Cheaveau M, Morgan W, Holmes C, Harris S et al. Inflammatory components in human Alzheimer's disease and after active amyloid-beta42 immunization. Brain 2013; 136 (Pt 9): 2677–2696.
Paquet C, Amin J, Mouton-Liger F, Nasser M, Love S, Gray F et al. Effect of active Abeta immunotherapy on neurons in human Alzheimer's disease. J Pathol 2015; 235: 721–730.
Lyck L, Dalmau I, Chemnitz J, Finsen B, Schroder HD . . Immunohistochemical markers for quantitative studies of neurons and glia in human neocortex. J Histochem Cytochem 2008; 56: 201–221.
Torres EM, Meldrum A, Kirik D, Dunnett SB . An investigation of the problem of two-layered immunohistochemical staining in paraformaldehyde fixed sections. J Neurosci Methods 2006; 158: 64–74.
Ghatak A, Combs CK . Iba1 immunoreactivity is enhanced following an antigen retrieval treatment with EDTA, pH 6.0. MethodsX 2014; 1: 269–274.
Kashon ML, Ross GW, O'Callaghan JP, Miller DB, Petrovitch H, Burchfiel CM et al. Associations of cortical astrogliosis with cognitive performance and dementia status. J Alzheimers Dis 2004; 6: 595–604.
Muramori F, Kobayashi K, Nakamura I . A quantitative study of neurofibrillary tangles, senile plaques and astrocytes in the hippocampal subdivisions and entorhinal cortex in Alzheimer's disease, normal controls and non-Alzheimer neuropsychiatric diseases. Psychiatry Clin Neurosci 1998; 52: 593–599.
Akiyama H, Mori H, Saido T, Kondo H, Ikeda K, McGeer PL . Occurrence of the diffuse amyloid beta-protein (Abeta) deposits with numerous Abeta-containing glial cells in the cerebral cortex of patients with Alzheimer's disease. Glia 1999; 25: 324–331.
Bouvier DS, Jones EV, Quesseveur G, Davoli MA, Ferreira AT, Quirion R et al. High resolution dissection of reactive glial nets in Alzheimer's disease. Sci Rep 2016; 6: 24544.
Irie-Sasaki J, Sasaki T, Penninger JM . CD45 regulated signaling pathways. Curr Top Med Chem 2003; 3: 783–796.
Kaneko Y, Kitamoto T, Tateishi J, Yamaguchi K . Ferritin immunohistochemistry as a marker for microglia. Acta Neuropathol 1989; 79: 129–136.
Wlodarczyk A, Cedile O, Jensen KN, Jasson A, Mony JT, Khorooshi R et al. Pathologic and protective roles for microglial subsets and bone marrow- and blood-derived myeloid cells in central nervous system inflammation. Front Immunol 2015; 6: 463.
Dinarello CA . Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 2009; 27: 519–550.
Hutchins KD, Dickson DW, Rashbaum WK, Lyman WD . Localization of microglia in the human fetal cervical spinal cord. Brain Res Dev Brain Res 1992; 66: 270–273.
Beckers L, Ory D, Geric I, Declercq L, Koole M, Kassiou M et al. Increased expression of translocator protein (TSPO) marks pro-inflammatory microglia but does not predict neurodegeneration. Mol Imaging Biol 2017.
Bonsack Ft Alleyne CH Jr, Sukumari-Ramesh S . Augmented expression of TSPO after intracerebral hemorrhage: a role in inflammation? J Neuroinflammation 2016; 13: 151.
Acknowledgements
Dr JoAnne McLaurin and Dr Anthony Domenichiello provided insight and resources respectively that aided in the preparation of this review. Dr John Sievenpiper and the staff at the University of Toronto Gerstein Science Information Centre provided assistance in developing the systematic search protocol. This work was supported by a grant from the Canadian Institutes of Health Research (CIHR) to RPB, a CIHR Vanier Canada Graduate Scholarship to KEH, a CIHR Canadian Graduate Scholarship to VG and a National Sciences and Engineering Research Council of Canada Undergraduate Student Research Award to DM. RPB is a Canada Research Chair in Brain Lipid Metabolism.
Author contributions
KEH designed the search with the assistance of MT, reviewed the articles returned by the search for eligibility with the assistance of DM, reviewed all data extraction and wrote the paper. DM, MT and VG assisted with the full-text assessments, data extraction and provided feedback on the paper. RPB oversaw the project, provided feedback on all steps of the search and data extraction and reviewed the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
RPB has received research grants from Bunge Ltd, Arctic Nutrition, the Dairy Farmers of Canada and Nestle Inc, as well as travel support from Mead Johnson and mass spectrometry equipment and support from Sciex. RPB is on the executive of the International Society for the Study of Fatty acids and Lipids and held a meeting on behalf of Fatty Acids and Cell Signaling, both of which rely on corporate sponsorship. RPB has given expert testimony in relation to supplements and the brain. RPB also provides complimentary fatty acid analysis for farmers, food producers and others involved in the food industry, some of whom provide free food samples. The funding body had no role in any step of this paper, or in the decision to publish results. The remaining authors declare no conflicts of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
PowerPoint slides
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Hopperton, K., Mohammad, D., Trépanier, M. et al. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: a systematic review. Mol Psychiatry 23, 177–198 (2018). https://doi.org/10.1038/mp.2017.246
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2017.246
This article is cited by
-
Neuroinflammation is associated with Alzheimer’s disease co-pathology in dementia with Lewy bodies
Acta Neuropathologica Communications (2024)
-
Deterioration of neuroimmune homeostasis in Alzheimer’s Disease patients who survive a COVID-19 infection
Journal of Neuroinflammation (2024)
-
The concept of resilience to Alzheimer’s Disease: current definitions and cellular and molecular mechanisms
Molecular Neurodegeneration (2024)
-
Human brain organoids containing microglia that have arisen innately adapt to a β-amyloid challenge better than those in which microglia are integrated by co-culture
Stem Cell Research & Therapy (2024)
-
Disease and brain region specific immune response profiles in neurodegenerative diseases with pure and mixed protein pathologies
Acta Neuropathologica Communications (2024)
