
Abstract
The diathesis–stress theory for depression states that the effects of stress on the depression risk are dependent on the diathesis or vulnerability, implying multiplicative interactive effects on the liability scale. We used polygenic risk scores for major depressive disorder (MDD) calculated from the results of the most recent analysis from the Psychiatric Genomics Consortium as a direct measure of the vulnerability for depression in a sample of 5221 individuals from 3083 families. In the same we also had measures of stressful life events and social support and a depression symptom score, as well as DSM-IV MDD diagnoses for most individuals. In order to estimate the variance in depression explained by the genetic vulnerability, the stressors and their interactions, we fitted linear mixed models controlling for relatedness for the whole sample as well as stratified by sex. We show a significant interaction of the polygenic risk scores with personal life events (0.12% of variance explained, P-value=0.0076) contributing positively to the risk of depression. Additionally, our results suggest possible differences in the aetiology of depression between women and men. In conclusion, our findings point to an extra risk for individuals with combined vulnerability and high number of reported personal life events beyond what would be expected from the additive contributions of these factors to the liability for depression, supporting the multiplicative diathesis–stress model for this disease.
Similar content being viewed by others

Introduction
A popular explanation for the aetiology of depression is the diathesis–stress model.1,2,3,4,5,6 Initially developed to explain the origins of schizophrenia in the 1960s5,6 and adapted for the study of depression in the 1980s,1,2,3,4 this model states that stress may activate a diathesis or vulnerability, transforming the potential of predisposition into the actuality of psychopathology.7 The model proposes that there is a synergism between the diathesis and stress that yields an effect beyond their combined separate effects into depressive symptomatology and thus, the effects of stress on the depression risk are dependent on the diathesis. Implicit in this theory is that there will be not only additive but multiplicative interactive effects on the liability scale.7
Over 50 years ago Rosenthal6 described the diathesis–stress theories as ‘the ones in which genuine meaning attaches to the commonly repeated statement that heredity and environment interact’. However, he criticised the vague formulations for the predispositions and stressors that these theories propose. This criticism has been highlighted by others like Monroe and Simons,7 who call for more research and more precise measures on the ‘conceptual essence’ of the diathesis–stress premise, that is, ‘the nature of the interaction between elements in the aetiologic process over time’. The diathesis–stress theory and research have been criticised for being ‘unproductive, either theoretically or empirically’.8
The genetically driven sensitivity to environments proposed by the diathesis–stress model can be operationalised as a gene by environment interaction (GxE). GxE studies have commonly focused on single loci in candidate genes, such as the length polymorphism (5HTTLPR) in the serotonin transporter gene (SLC6A4), with mostly inconsistent or negative results.9,10,11,12,13 This approach has limitations related to poor quality genotyping, inconsistent types of interactions, inconsistent grouping of genotypes, selective presentation of results, interactions arising from the scale of measurement and publication bias.9 Moreover, MDD is a polygenic trait, arising from the effect of multiple risk variants, each with small effect sizes.14,15 Therefore, MDD is influenced by many genetic variants of small effect, and it is more likely that affected individuals carry a polygenic burden of risk alleles rather than any single genotype of large effect. However, the progress from a candidate gene to an hypothesis-free genome-wide approach is hampered by the need for extremely large samples due to expected small effect sizes as well as necessarily imperfect assessment of environmental stressors across large cohorts.16,17
Polygenic risk scores (PRS) provide a novel opportunity to test the diathesis–stress model, since PRS can be conceptualised as an indicator of the diathesis and will likely prove a much stronger instrument than any single risk gene. PRS estimation uses Genome-Wide Association Study (GWAS) results to predict the genetic risk of each individual in an independent genotyped sample; PRS are estimated as the sum of risk alleles weighted by their respective independently estimated effect sizes.18 Note that, since GWAS are currently underpowered to detect all common genetic risk variants in complex traits, the variance explained by the PRS is usually lower than the twin heritability.18
The first ones to use PRS for MDD to test for GxE interaction in MDD were Peyrot et al. 17 Using a sample of 1645 participants with a DSM-IV diagnosis for MDD and 340 screened controls from the Netherlands Study of Depression and Anxiety, they showed increased effects of PRS on MDD in the presence of childhood trauma, with evidence for interaction. Musliner et al. 19 studied the association between PRS-MDD, stressful life events (SLEs) and depressive symptoms in a sample of 8761 participants from the Health and Retirement Study in the United States. SLEs were operationalised as a dichotomous variable indicating whether participants had experienced at least one stressful event in the previous 2 years. Depressive symptoms were measured using an 8-item Center for Epidemiological Studies Depression subscale and operationalised as both a dichotomous and a continuous variable. They found that both SLEs and PRS were significantly and independently associated with depressive symptoms, but found no evidence that SLEs moderated the association between PRS-MDD and depressive symptoms. Instead, their results were compatible with an additive model. Most recently, Mullins et al. 20 examined the idea using 1605 cases with recurrent MDD and 1064 controls all with SLE data, and a subset of 240 cases and 272 controls with childhood trauma data from in the RADIANT UK study. Both PRS and SLEs were significant predictors of case/control status but no interactions were found between PRS for MDD and SLEs, in agreement with previous findings by Musliner et al. 19 Significant interactions were found between PRS and childhood trauma but, contrary to Peyrot et al.,17 there was an inverse association with depression status. In summary, these studies do not present consistent results. Studies to date have used the first wave of GWAS data (MDD1) from the Psychiatric Genomics Consortium (PGC) MDD working group (PGC-MDD), based on 9240 cases and 9519 controls,14 and so are likely underpowered.
We report here a direct test of the diathesis–stress model for depression using PRS for MDD and measures of SLEs and social support (SS; lack of SS being considered a stressor); we predict diathesis using an updated version of PGC-MDD GWAS results (N total=159 601, after excluding QIMR data). Given the higher lifetime risk of MDD in women,21 we also tested the hypothesis in sexes separately.
Materials and methods
Phenotypic data were collected as part of a general Health and Lifestyle questionnaire (HLQ) mailed to adult twins enroled in the Australian Twin Registry between 1988 and 1992.22,23,24 It included self-report questions about depression, recent personal or network SLEs (PSLE, NSLE) and SS. The content and details of data collection have been previously described.22,23,24 Data used in this analysis were collected in three waves. The first wave ran between 1988 and 1992 (N=5843) and targeted adult twins (mean age 41.2, s.d.=12.8, range 24–86, 61.0% females).23 The second wave (N=3646, collected between 1990 and 1992) focused on younger twins (mean age 23.2, s.d.=2.2, range 16–31, 65.6% females) and the questionnaire was slightly adapted to cover some of the more common issues of that age group.22,23 Finally, the last wave (N=236) targeted twin pairs whose information was partially missing from the original 1980 survey, using the same questionnaire (collection between 1990 and 1992, mean age 42.0, s.d.=9.9, range 27–73, 58.5% females). This study was approved by the Queensland Institute of Medical Research Human Research Ethics Committee and the storage of the data follows national regulations regarding personal data protection. All of the participants provided informed consent.
Depression scores were calculated by combining the seven depression items from the Delusions-Symptoms-States Inventory (DSSI)25,26 with five depression items from the Symptom CheckList (SCL-90).27 The factor structure of the scale has been reported previously in the younger data set22 and the score has been used in several publications.9,22,23,28,29
The HLQ also assessed PSLE and NSLE, adapted from the List of threatening experiences.30 For PSLE, participants were asked to report adverse events (divorce, marital separation, broken engagement or steady relationship, separation from other loved one or close friend, serious illness or injury, serious accident, burgled or robbed, laid off or sacked from job, other serious difficulties at work, major financial problems, legal troubles or involvement with police, living in unpleasant surroundings) that happened in the last 12 months. In addition, they were asked if they had had serious problems getting along with their close network (spouse, someone living with you, for example, child/elderly parent, other family member, co-twin, a close friend, neighbour or workmate) in the past 12 months. These 19 yes/no items were summed to calculate the PSLE score.
NSLE was calculated from 21 yes/no questions, in which the participants could report death, injury or crisis that their close network (spouse, child, mother/father, co-twin, other brother/sister, other relative, someone else close to them) experienced in the last 12 months.
Perceived social support was measured using the Kessler Perceived Social Support Measure.31 Several publications from our group have made use of these data.9,23,28,32,33,34
We used Item Response Theory (IRT),35,36 which weights the item responses by their difficulty and discrimination, to calculate individuals’ scores of depression, PSLE, NSLE and SS. First, we performed an exploratory non-parametric IRT analysis of the KernSmoothIRT package in R.37 It allows estimation of the probability of endorsing each option of each item as a function of the latent underlying trait (known as Item Response Step Functions: IRSF), without any constraint on the shape of the fitted curve.36 We used it to confirm the monotonicity of IRSF necessary to ensure the property of stochastic ordering on the sum score.38 It further allows choosing the most appropriate parametric IRT models based on the shapes of the non-parametric IRSF.
IRSF plotted in Supplementary Figures 1–4 show, for all scales and items, monotonic IRSF in the normal range of the latent trait continuum (-2 2). Small breaches of monotonicity were observed for extreme values of the latent trait and can be attributed to small numbers of participants that lead to unstable non-parametric kernel estimation (as indicated by widening 95% confidence intervals). Consequently, we estimated the IRT scores using a two-parameter logistic model in WinBUGS v. 1.4.339 that constrains all left asymptotes to be 0 and all right asymptotes to be 1. In such a model, the IRSF only differ in terms of difficulty and discrimination.35,40 Such IRT scores are maximum likelihood estimates of the latent trait and carry the same information as a sum score while presenting more normal distributions, thus reducing the influence of extreme values in later analyses.
Missingness in the depression items was limited to <2% of the respondents and most of the missing answers (88 or 60%) were found in the item ‘recently, I have lost interest in sex or have found not found sex pleasurable’. The 1.6% of the respondents who omitted this item tended to be females (P-value=4.2e−04), 4 months older on average (P-value=9.7e−06), and with a slightly higher DSSI score (+0.1 pts, P-value=3.6e−05). Missingness not at random (that is, potentially dependent on depression level) implies that excluding participants may create a sampling bias. Thus, we chose to impute the missing observations using WinBUGS (described in Wray et al.,34 Rietschel et al. 41) using age, sex and the depression items as predictors. Overall, due the low missingness rate imputation should have little influence on the results.
Lifetime DSM-IV depression diagnoses were obtained in most of the cohort in two telephone interview follow-up studies using the clinical Semi-Structured Assessment for the Genetics of Alcoholism (SSAGA42,43 in 1992–1993 and 1996–2000 (see Supplementary Figure 5 for a summary of the phenotypic data collection). Details of data collection are described elsewhere.29,44,45,46,47 The depression score significantly predicted lifetime DSM-IV MDD status assessed 4–7 years later44,45 (odds ratio=1.96, 95% confidence intervals 1.85–2.08, P-value=3.0e−108, N=8607), which translates to a 6.1-fold increased odds of MDD between participants in the top and bottom deciles of depression IRT scores (Figure 1a), so demonstrating the utility of the score. For our analysis we used the continuous IRT score rather than the binary diagnosis, as continuous models provide greater statistical power than logistic regressions (>99.9% vs 88.0%, N=5221, odds ratio=1.1, beta=0.095, with α=0.05, proportion of cases and s.d. of outcome and predictor measured in our sample) and is available for larger sample size (5179 vs 5221 with IRT score).

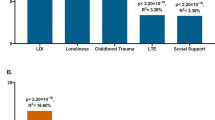
(a) Increased odds of DSM-IV MDD diagnosis per decile of depression IRT score assessed 4–7 years previously. (b) Association between MDD–PRS and depression scores (main effects, one-sided tests, results expressed in % of variance explained). Full sample analyses using the two versions of the polygenic risk scores (PRS) were run in the same target data set with the exact same covariates. Red bars indicate positive correlation with the depression score. PRS were calculated using different P-value thresholds from the Genome-Wide Association Study summary statistics. The most conservative only includes independent loci with genome-wide significant single-nucleotide polymorphism (SNPs) (P-value<5e−8), while the least conservative include the most significant SNP of each haplotype (P-value<1). (c) Association between self-reported stress (personal stressful life events (PSLE) or network stressful life events (NSLE), lack of social support (SS)) and depression IRT score (main effect, one-sided tests, results expressed in % of variance explained). Blue bars indicate negative correlations and red bars indicate positive correlations with the depression score. Dashed bars indicate sex-specific effects. (d) Variance of the depression score explained by the interaction between PRS and PSLE (two-sided tests). Dashed bars indicate sex-specific effects. We focused on the association with the PRS comprising all haplotypes but the other associations are also reported for completeness. Blue bars indicate negative correlations and red bars indicate positive correlations with the depression score. (e) Increase in depression score (fitted values, vertical axis) as a function of PSLE and MDD-PRS. For example, the effect of the PSLE–diathesis interaction is visible when comparing the bottom (minimal PSLE) and top (maximal PSLE) edges of the surface. The difference in slopes indicates that PSLE mediates the effect of the genetic predisposition on the depression score. From right to left, results for the whole sample, females and males. In all analyses, we accounted for familial relatedness using a kinship matrix (a) or a genetic relatedness matrix calculated from SNPs (b–d). For a we used R package ‘hglm’77 to estimate the odds ratios (Student's t-test to test the significance of the fixed effects). For panels b–d, the parameters of the model were estimated using GCTA 1.26.0 (Student's t-test to test the significance of the fixed effects).62 All analyses controlled for age, age2, sex, age × sex and age2 × sex interactions, study, array and the first four genetic principal components in the outcome variable and predictors.60 IRT, Item Response Theory; MDD, major depressive disorder.
DNA collected from blood was genotyped using commercial arrays (Illumina 317 K, 370 K, 610 K, ‘1st generation’, or Core Exome plus Omni-family, ‘2nd generation’.48,49,50) and imputed from a common single-nucleotide polymorphism (SNP) set to the 1000 Genomes (Phase 3 Release 5) reference panel, (http://www.1000genomes.org/)51,52 a strategy that allows genotype data from different arrays to be combined. Observed genotypes were cleaned (by batch) for call rate (⩾95%); minor allele frequency, MAF (⩾1%); Hardy–Weinberg equilibrium (P⩾10−3; PLINK1.953), GenCall score (⩾0.15 per genotype; mean ⩾0.7) and standard Illumina filters, before integrating batches and re-running the quality control and Mendelian checks. We imputed the genotype data via the University of Michigan Imputation Server54 or in-house (chr. X only) using the 1000 genomes Phase 3 Release 5 ‘mixed population’ reference panel), (http://www.1000genomes.org/)51,52 with phasing by SHAPEIT55,56 followed by imputation using minimac3.57 ‘1st generation’ and ‘2nd generation’ were imputed separately due to poor overlap between observed markers. Imputation was based on 277 690 (‘1st generation’) and 240 297 (‘2nd generation’) observed markers; and the two combined after imputation to maximise sample size. This resulted in 9 411 304 SNPs available for analysis, after quality control.
PRS58,59 were calculated from the imputed genotype dosages, using GWAS summary statistics from the most recent PGC-MDD release (9 July 2016), with the exclusion of the contribution of QIMR, for a final sample of 49 524 cases and 110 074 controls (see Supplementary Table 1 for cohort contributions). For comparison, we also calculated the PRS using the first wave GWAS summary statistics published by the PGC-MDD.14 From our data, we excluded SNPs with low imputation quality (r 2<0.6) and MAF below 1%. We selected the most significant independent SNPs using PLINK1.953 in order to correct for signal inflation due to linkage disequilibrium (criteria linkage disequilibrium r 2<0.1 within windows of 10 MBp). We calculated eight different PRS using different P-value thresholding of the GWAS summary statistics (see Supplementary Table 2 for number of SNPs included in each threshold). Histograms of PRS for MDD (1000G imputation, GWAS results from July 2016), together with the histograms of the IRT scores for depression, PSLE, NSLE and SS scores are reported in Supplementary Figure 6.
Our final sample comprised 5221 individuals (from 3083 twin families) of European ancestry with available phenotypic and genetic data (mean age at questionnaire 35.7, s.d.=12.2, range 17–85, 65.6% females). Covariates (age, age2, sex, age × sex and age2 × sex interactions, and the first four genetic principal components) were regressed from the PRS and the stress scores before inclusion in the models to guard against confounding influences on the PRS–stress interactions.60
In order to estimate the variance explained by the PRS, the stressors and their interactions in the depression score, we then fitted linear mixed models, which controlled for relatedness for the whole sample as well as stratified by sex. The parameters of the model were estimated using GCTA 1.26.0 (Student's t-test to test the significance of the fixed effects) that accounts for twin relatedness using a genetic relatedness matrix (GRM). The linear model used is as follows:
Depression=intercept+b × Covariates+c × PRS_z+d × PSLE_z+e × NSLE_z+f × SS_z+g × PRS_z × PSLE_z+h × PRS_z × NSLE_z+i × PRS_z × SS_z+j × G
With b, c, d, e, f, g, h, i the vectors of fixed effects.
Covariates used in this analyses were age, sex, age2, sex × age, sex × age2, GWAS array, wave and first four genetic principal components. Note that sex and its interaction were not included when stratifying the analyses by sex.
PSLE_z, SS_z, NSLE_z and PRS_z are the residuals of the scores after regressing out the covariates listed above.
G is the random effect that models the sample relatedness G~ N(0, GRM), with GRM the N × N matrix of relatedness estimated from SNPs.
We used OpenMx61 to calculate the heritability and correlations (likelihood-ratio test, using a kinship matrix to account for familial relatedness) of the depression score and the stressors, correcting for age, sex, age2, sex × age, sex × age2 and wave. Following the significant genetic correlations estimated from twin models, we investigated how much of the variance in stress scores could be accounted for by the MDD-PRS. We controlled for age, age2, sex (and their interactions), study, imputation batch and four genetic principal components. Model parameters were estimated using GCTA 1.26.062 that accounts from twin relatedness.
Results
PRS for MDD significantly predicted the depression score (maximum variance explained=0.46%. P-value=5.01e−08, Figure 1b, right panel), which represents a substantial improvement compared to PRS predictions based on earlier GWAS14(Figure 1b, left panel, variance explained=0.08%, P-value=0.018), reflecting the increased sample size of the GWAS discovery samples.63,64 The main effects of PSLE, NSLE and lack of SS were also significant, explaining, respectively, 12.9%, 0.3% and 3% of the depression score variance (Figure 1c), with effects in the expected directions. Lack of SS predicted more of the depression score in women than it did in men (between sex differences, P-value=4.7e−03) but there were no other differences between sexes that reached significance.
The significance of main effects allowed testing the significance of the interaction between each stress type (PSLE, NSLE, lack of SS) and the most predictive PRS (using all SNPs: P<1). The interaction with PSLE was significant (0.12% of variance explained, P-value=0.0076) and contributed positively to the risk of depression, predominantly in women (Figure 1d). Overall, the variance explained by PRS main effect plus the interaction was comparable in men (0.73%) and women (0.60%). The interaction was not significant in men while explaining almost as much variance as the main effect in women. However, there was no significant difference when comparing the size of the interaction across sexes (P-value=0.21). For completeness, interactions between each stressor and all PRS are also reported in Supplementary Figure 7.
Discussion
Our finding of a significant diathesis–PSLE interaction points to an extra risk for individuals with combined vulnerability and high number of reported PSLE beyond what would be expected from their additive contributions to liability (Figures 1d and e). In the full sample 0.58% of the depression score variance was explained by the PRS and interaction, of which ~80% corresponds to the main effects and ~20% to the interaction. As the power of the PRS increases with larger GWAS,63 and if these proportions are maintained, the interaction explaining about 20% of the heritability would be typical of the size of GxE estimates for other traits in other species.65 We cannot dismiss the possibility of diathetic interactions with NSLE or SS, as the power of our study is still limited by the PRS instrument63 and our sample size. This is evidenced by our measure of genetic predisposition still only explaining a small fraction of the depression score variance (Figure 1b), in comparison with the twin-based heritability (Supplementary Figure 8 and Sullivan et al. 66), or even the SNP heritability for MDD (h 2 SNP=0.2118). Note that for all psychiatric and complex traits, it is common that the variance explained by PRS corresponds to a fraction of the heritability, especially when only a few variants are known.67 Much larger GWAS samples are required to better differentiate the true SNP signals from the noise and to provide a greater level of prediction.63
We confirmed using a twin analysis of the data set (1110 MZ pairs, 1032 DZ pairs, 961 singletons) that our measure of depression and all three stress measures are moderately heritable (30–40%, P-value<2.1e−25; Supplementary Figure 8), as reported previously.68,69 Additive genetic and unique environment (AE) models showed the best fit to the data and shared environment could not explain the association (P-value<1.7e−03). We also replicated that self-reported measures of stress are genetically correlated with the depression score (Supplementary Figure 9).70,71
PRS for MDD predicted PSLE and SS (P-value<0.001), no significant association was observed with NSLE (Supplementary Figure 10). This is consistent with heritability and genetic correlation results reported from twin models.
On the scale of measurement for depression that we have used, our results support the multiplicative diathesis–stress model for depression proposed in the 1980s. In addition, our results suggest possible differences in the aetiology of depression between women and men, which may have implications for the tailoring of treatments. However, we must caution that the presence and size of interaction are completely dependent on the scale of measurement and more perfectly normal scales for depression and stressors may have produced a different result.65,72 For example, using a simple sum score for depression, with an extreme reverse-J distribution yields an even larger and more significant estimate of interaction (0.39% variance explained; P-value=6.8e−07) whereas using logistic regression to analyse the binary DSM-IV diagnosis, predicated on an underlying normal liability, produces a smaller and only marginally significant estimate (0.06% variance explained; P-value=0.059), although this analysis has much lower power than using a continuous variable.
In addition, the substantial genetic correlation between PSLE and depression hinders attributing the interaction solely to a GxE effect. To investigate this point, we broke down PSLE into events in which the individual may have played an active role (PSLE-active: divorce, separation, having difficulty at work, financial or legal troubles, not getting along with people) as opposed to passive role73,74 (PSLE-passive: illness, accident, being burgled, sacked or living in unpleasant surroundings) and calculated the IRT score for them. This follows previous publications reporting that PSLE-active was more heritable than PSLE-passive,73,74 which we confirmed in our sample (twin h 2 PSLE-Active=0.29, 95% confidence interval 0.24–0.34; h 2 PSLE-Passive=0.11, 0.05–0.17). We tested whether the less heritable PSLE-passive may drive the observed interaction, which may point towards a more likely GxE interaction. However, in the models, active PSLE explained most of the variance explained by the PSLE score (r 2 PSLE- Active=10.5%, P-value=3.2e–123 vs r 2 PSLE-Passive=0.77%, P-value=4.1e−12) and the interaction (r 2 PSLE-Active × PRS=0.085%, P-value=0.030 vs r 2 PSLE-Passive × PRS=0.0084%, P-value=0.46) (results given for the PRS ‘P<1’, consistent with our other analyses, see Supplementary Figure 12 for all details). This approach was unable to confirm the nature of the interaction, since both PSLE-active and -passive are significantly heritable.
To tackle this question more directly, we calculated environmental and genetic factor scores for PSLE (PSLE-E and PSLE-A) via an independent pathway model fitted to the 19 items of the PSLE questionnaire75 (Supplementary Figure 13). Conceptually, this divides the IRT PSLE score, which estimates the (phenotypic) latent trait underlying the participants’ responses in the questionnaire, into its additive genetic and environmental dimensions (Supplementary Figures 13 and 14). We then replaced the PSLE IRT score by each of its components in a mixed model to investigate the source of the interaction: GxG if interaction between PRS and PSLE-A or GxE if interaction between PRS and PSLE-E. As in our previous analyses, we regressed covariates out of the factor scores and also included them in the model. The model including PSLE-E was the best fitting as indicated by lower Akaike information criterion, AIC (AICPSLE-E=2645, AICPSLE-A=2798). Further, the interaction term points towards a greater GxE effect, as the interaction between PRS and PSLE-E explains 0.11% of the variance (P-value=0.0076), although we cannot rule out the presence of an interaction with PSLE-A (r 2=0.076%, P-value=0.037) (results given for the PRS ‘P<1’, see Supplementary Figure 15 for more details). Results showed a similar pattern when we modelled the A and E factors of PLSE-active.
Notwithstanding the above caveats, more work is needed to evaluate different mechanisms of interaction, including a bi-causal relationship between PSLE and depression or molecular interaction (for example, through methylation changes). We are aware of potential confounds of interaction analyses: in addition to sensitivity of the analyses to the properties of the scale76 and unavoidable departures from normality in the outcome and predictors as discussed above (Supplementary Figures 6 and 11), problems may also arise from the stress and depression measures being self-reported in the same questionnaire, and the fact that the stress measures are genetically correlated with the outcome variable. Replication of our findings in independent cohorts, consideration of other variables such as the perceived impact of stressors and improvement of PRS via larger GWAS and larger samples with both depression and risk factors evaluated will allow us further to refine our understanding of the aetiology of depression.
References
Bebbington P. Misery and beyond: the pursuit of disease theories of depression. Int J Soc Psychiatry 1987; 33: 13–20.
Beck AT. Depression: Clinical, Experimental and Theoretical Aspects. Harper & Row: NY, USA, 1967.
McGuffin P, Katz R, Bebbington P. The Camberwell Collaborative Depression Study. III. Depression and adversity in the relatives of depressed probands. Br J Psychiatry 1988; 152: 775–782.
Robins CJ, Block P. Cognitive theories of depression viewed from a diathesis-stress perspective: evaluations of the models of Beck and of Abramson, Seligman, and Teasdale. Cogn Ther Res 1989; 13: 297–313.
Bleuler M. Conception of Schizophrenia within the last fifty years and today [Abridged]. Proc R Soc Med 1963; 56: 945–952.
Rosenthal D. A suggested conceptual framework. In: Rosenthal D(ed). The Genain Quadruplets: A Case Study and Theoretical Analysis of Heredity and Environment in Schizophrenia. Basic Books: NY, USA, 1963, pp 505–511..
Monroe SM, Simons AD. Diathesis-stress theories in the context of life stress research: implications for the depressive disorders. Psychol Bull 1991; 110: 406–425.
Belsky J, Pluess M. Beyond diathesis stress: differential susceptibility to environmental influences. Psychol Bull 2009; 135: 885–908.
Coventry WL, James MR, Eaves LJ, Gordon SD, Gillespie NA, Ryan L et al. Do 5HTTLPR and stress interact in risk for depression and suicidality? Item response analyses of a large sample. Am J Med Genet B Neuropsychiatr Genet 2010; 153B: 757–765.
Haberstick BC, Boardman JD, Wagner B, Smolen A, Hewitt JK, Killeya-Jones LA et al. Depression, stressful life events, and the impact of variation in the serotonin transporter: findings from the National Longitudinal Study of Adolescent to Adult Health (Add Health). PLoS ONE 2016; 11: e0148373.
Karg K, Burmeister M, Shedden K, Sen S. The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch Gen Psychiatry 2011; 68: 444–454.
Munafo MR, Durrant C, Lewis G, Flint J. Gene X environment interactions at the serotonin transporter locus. Biol Psychiatry 2009; 65: 211–219.
Risch N, Herrell R, Lehner T, Liang KY, Eaves L, Hoh J et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA 2009; 301: 2462–2471.
Major Depressive Disorder Working Group of the Psychiatric GC, Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 2013; 18: 497–511.
Wray NR, Pergadia ML, Blackwood DH, Penninx BW, Gordon SD, Nyholt DR et al. Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol Psychiatry 2012; 17: 36–48.
McGuffin P, Rivera M. The interaction between stress and genetic factors in the etiopathogenesis of depression. World Psychiatry 2015; 14: 161–163.
Peyrot WJ, Milaneschi Y, Abdellaoui A, Sullivan PF, Hottenga JJ, Boomsma DI et al. Effect of polygenic risk scores on depression in childhood trauma. Br J Psychiatry 2014; 205: 113–119.
Wray NR, Lee SH, Mehta D, Vinkhuyzen AA, Dudbridge F, Middeldorp CM. Research review: polygenic methods and their application to psychiatric traits. J Child Psychol Psychiatry 2014; 55: 1068–1087.
Musliner KL, Seifuddin F, Judy JA, Pirooznia M, Goes FS, Zandi PP. Polygenic risk, stressful life events and depressive symptoms in older adults: a polygenic score analysis. Psychol Med 2015; 45: 1709–1720.
Mullins N, Power RA, Fisher HL, Hanscombe KB, Euesden J, Iniesta R et al. Polygenic interactions with environmental adversity in the aetiology of major depressive disorder. Psychol Med 2016; 46: 759–770.
Gustavsson A, Svensson M, Jacobi F, Allgulander C, Alonso J, Beghi E et al. Cost of disorders of the brain in Europe 2010. Euro Neuropsychopharmacol 2011; 21: 718–779.
Gillespie N, Kirk KM, Heath AC, Martin NG, Hickie I. Somatic distress as a distinct psychological dimension. Soc Psychiatry Psychiatr Epidemiol 1999; 34: 451–458.
Kirk KM, Birley AJ, Statham DJ, Haddon B, Lake RI, Andrews JG et al. Anxiety and depression in twin and sib pairs extremely discordant and concordant for neuroticism: prodromus to a linkage study. Twin Res 2000; 3: 299.
Treloar SA, Martin NG, Bucholz KK, Madden PAF, Heath AC. Genetic influences on post-natal depressive symptoms: findings from an Australian twin sample. Psychol Med 1999; 29: 645–654.
Bedford A, Foulds GA, Sheffield BF. A new personal disturbance scale (DSSI/sAD). Br J Soc Clin Psychol 1976; 15: 387–394.
Bedford A, Foulds GA. Validation of the delusions‐symptoms‐states inventory. Br J Med Psychol 1977; 50: 163–171.
Derogatis LR, Lipman RS, Covi L. SCL-90: an outpatient psychiatric rating scale—preliminary report. Psychopharmacol Bull 1973; 9: 13–28.
Gillespie NA, Whitfield JB, Williams B, Heath AC, Martin NG. The relationship between stressful life events, the serotonin transporter (5-HTTLPR) genotype and major depression. Psychol Med 2005; 35: 101–111.
Kirk KM, Hickie IB, Martin NG. Fatigue as related to anxiety and depression in a community-based sample of twins aged over 50. Soc Psychiatry Psychiatr Epidemiol 1999; 34: 85–90.
Brugha T, Bebbington P, Tennant C, Hurry J. The list of threatening experiences: a subset of 12 life event categories with considerable long-term contextual threat. Psychol Med 1985; 15: 189–194.
Kessler RC, Kendler KS, Heath A, Neale MC, Eaves LJ. Perceived support and adjustment to stress in a general population sample of female twins. Psychol Med 1994; 24: 317–334.
Coventry WL, Gillespie NA, Heath AC, Martin NG. Perceived social support in a large community sample - age and sex differences. Soc Psychiatry Psychiatr Epidemiol 2004; 39: 625–636.
Coventry WL, Medland SE, Wray NR, Thorsteinsson EB, Heath AC, Byrne B. Phenotypic and discordant-monozygotic analyses of stress and perceived social support as antecedents to or sequelae of risk for depression. Twin Res Hum Genet 2009; 12: 469–488.
Wray NR, Coventry WL, James MR, Montgomery GW, Eaves LJ, Martin NG Use of monozygotic twins to investigate the relationship between 5HTTLPR genotype, depression and stressful life events: an application of item response theory. In: Genetic Effects on Environmental Vulnerability to Disease. John Wiley & Sons Ltd, Chichester, UK, 2008, pp 48–67..
Birnbaum A. Some Latent Trait Models and their Use in Inferring an Examinee's Ability. Statistical Theories of Mental Test Scores. Addison-Wesley: Reading, MA, USA, 1968.
Ramsay JO. Kernel smoothing approaches to nonparametric item characteristic curve estimation. Psychometrika 1991; 56: 611–630.
Mazza A, Punzo A, McGuire B. KernSmoothIRT: An R Package for Kernel Smoothing in Item Response Theory. J Stat Softw 2014; 58: 1–34.
van der Ark LA. Stochastic ordering of the latent trait by the sum score under various polytomous IRT models. Psychometrika 2005; 70: 283–304.
Lunn DJ, Thomas A, Best N, Spiegelhalter D. WinBUGS - a Bayesian modelling framework: concepts, structure, and extensibility. Stat Comput 2000; 10: 325–337.
Mellenbergh GJ. Conceptual notes on models for discrete polytomous item responses. Appl Psychol Meas 1995; 19: 91–100.
Rietschel L, Zhu G, Kirschbaum C, Strohmaier J, Wust S, Rietschel M et al. Perceived stress has genetic influences distinct from neuroticism and depression. Behav Genet 2014; 44: 639–645.
Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger JI Jr. et al. A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. J Stud Alcohol 1994; 55: 149–158.
Hesselbrock M, Easton C, Bucholz KK, Schuckit M, Hesselbrock V. A validity study of the SSAGA—a comparison with the SCAN. Addiction 1999; 94: 1361–1370.
Knopik VS, Heath AC, Madden PA, Bucholz KK, Slutske WS, Nelson EC et al. Genetic effects on alcohol dependence risk: re-evaluating the importance of psychiatric and other heritable risk factors. Psychol Med 2004; 34: 1519–1530.
Bierut LJ, Heath AC, Bucholz KK, Dinwiddie SH, Madden PA, Statham DJ et al. Major depressive disorder in a community-based twin sample: are there different genetic and environmental contributions for men and women? Arch Gen Psychiatry 1999; 56: 557–563.
Heath AC, Madden PA, Bucholz KK, Dinwiddie SH, Slutske WS, Bierut LJ et al. Genetic differences in alcohol sensitivity and the inheritance of alcoholism risk. Psychol Med 1999; 29: 1069–1081.
Heath AC, Whitfield JB, Martin NG, Pergadia ML, Goate AM, Lind PA et al. A quantitative-trait genome-wide association study of alcoholism risk in the community: findings and implications. Biol Psychiatry 2011; 70: 513–518.
Cuellar-Partida G, Springelkamp H, Lucas SE, Yazar S, Hewitt AW, Iglesias AI et al. WNT10A exonic variant increases the risk of keratoconus by decreasing corneal thickness. Hum Mol Genet 2015; 24: 5060–5068.
Medland SE, Nyholt DR, Painter JN, McEvoy BP, McRae AF, Zhu G et al. Common variants in the trichohyalin gene are associated with straight hair in Europeans. Am J Hum Genet 2009; 85: 750–755.
McEvoy BP, Montgomery GW, McRae AF, Ripatti S, Perola M, Spector TD et al. Geographical structure and differential natural selection among North European populations. Genome Res 2009; 19: 804–814.
Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM et al. A global reference for human genetic variation. Nature 2015; 526: 68–74.
Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015; 526: 75–81.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Whitcher B, Schmid V, Thornton A. Working with the DICOM and NIfTI Data Standards in R. J Stat Softw 2011; 44: 1–28.
Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods 2012; 9: 179–181.
Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods 2013; 10: 5–6.
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 2012; 44: 955–959.
Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk of complex disease. Curr Opin Genet Dev 2008; 18: 257–263.
Wray NR, Lee SH, Mehta D, Vinkhuyzen AAE, Dudbridge F, Middeldorp CM. Research review: polygenic methods and their application to psychiatric traits. J Child Psychol Psychiatry 2014; 55: 1068–1087.
Keller MC. Gene x environment interaction studies have not properly controlled for potential confounders: the problem and the (simple) solution. Biol Psychiatry 2014; 75: 18–24.
Boker S, Neale M, Maes H, Wilde M, Spiegel M, Brick T et al. OpenMx: an open source extended structural equation modeling framework. Psychometrika 2011; 76: 306–317.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011; 88: 76–82.
Dudbridge F. Power and predictive accuracy of polygenic risk scores. PLoS Genet 2013; 9: e1003348.
Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014; 511: 421–427.
Eaves LJ, Last K, Martin NG, Jinks JL. A progressive approach to non-additivity and genotype-environmental covariance in the analysis of human differences. Br J Math Stat Psychol 1977; 30: 3.
Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 2000; 157: 1552–1562.
Krapohl E, Euesden J, Zabaneh D, Pingault JB, Rimfeld K, von Stumm S et al. Phenome-wide analysis of genome-wide polygenic scores. Mol Psychiatry 2016; 21: 1188–1193.
Kendler KS. Social support: a genetic-epidemiologic analysis. Am J Psychiatry 1997; 154: 1398–1404.
Kendler KS, Baker JH. Genetic influences on measures of the environment: a systematic review. Psychol Med 2007; 37: 615–626.
Matthews T, Danese A, Wertz J, Odgers CL, Ambler A, Moffitt TE et al. Social isolation, loneliness and depression in young adulthood: a behavioural genetic analysis. Soc Psychiatry Psychiatr Epidemiol 2016; 51: 339–348.
Boardman JD, Alexander KB, Stallings MC. Stressful life events and depression among adolescent twin pairs. Biodemogr Soc Biol 2011; 57: 53–66.
Kang S, Waller NG. Moderated multiple regression, spurious interaction effects, and IRT. Appl Psychol Meas 2005; 29: 87–105.
Plomin R, Lichtenstein P, Pedersen NL, McClearn GE, Nesselroade JR. Genetic influence on life events during the last half of the life span. Psychol Aging 1990; 5: 25–30.
Kendler KS, Karkowski LM, Prescott CA. The assessment of dependence in the study of stressful life events: validation using a twin design. Psychol Med 1999; 29: 1455–1460.
Boomsma DI, Molenaar PC, Orlebeke JF. Estimation of individual genetic and environmental factor scores. Genet Epidemiol 1990; 7: 83–91.
Eaves LJ. Errors of inference in the detection of major gene effects on psychological test scores. Am J Hum Genet 1983; 35: 1179–1189.
Ronnegard L, Shen X, Alam M. hglm: a package for fitting hierarchical generalized linear models. R J 2010; 2: 20–28.
Acknowledgments
Phenotype collection, DNA collection and genotyping were funded by NHMRC (981351) and NIH (R01 AA013326, R01 AA007535, R01 AA010249, R01 AA013321, R37 AA007728) grants to NGM, ACH and PAFM over the past three decades. The Psychiatric Genomics Consortium MDD Working Group depends on the contributions of many parties. LCC was supported by a post-doctoral fellowship from the Fundación Séneca (Seneca Foundation, Regional Agency for Science and Technology, Murcia, Spain, 19151/PD/13) and is supported by a QIMR Berghofer Fellowship. BCD is supported by a UQI scholarship. SEM is supported by a NHMRC research fellowship (1103623). We are grateful to the QIMR participants, data collectors and data managers.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if thematerial is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Colodro-Conde, L., Couvy-Duchesne, B., Zhu, G. et al. A direct test of the diathesis–stress model for depression. Mol Psychiatry 23, 1590–1596 (2018). https://doi.org/10.1038/mp.2017.130
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2017.130
This article is cited by
-
Retrospective ACEs predict complex PTSD symptoms in a large sample of Chinese young adults longitudinally: the moderating role of self-compassion
BMC Psychiatry (2024)
-
Patterns of stressful life events and polygenic scores for five mental disorders and neuroticism among adults with depression
Molecular Psychiatry (2024)
-
Polygenic risk for major depression, attention deficit hyperactivity disorder, neuroticism, and schizophrenia are correlated with experience of intimate partner violence
Translational Psychiatry (2024)
-
Elevated body temperature is associated with depressive symptoms: results from the TemPredict Study
Scientific Reports (2024)
-
Genetics, epigenetics, and neurobiology of childhood-onset depression: an umbrella review
Molecular Psychiatry (2024)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}