
Abstract
Recombinant human erythropoietin (EPO) improves cognitive performance in neuropsychiatric diseases ranging from schizophrenia and multiple sclerosis to major depression and bipolar disease. This consistent EPO effect on cognition is independent of its role in hematopoiesis. The cellular mechanisms of action in brain, however, have remained unclear. Here we studied healthy young mice and observed that 3-week EPO administration was associated with an increased number of pyramidal neurons and oligodendrocytes in the hippocampus of ~20%. Under constant cognitive challenge, neuron numbers remained elevated until >6 months of age. Surprisingly, this increase occurred in absence of altered cell proliferation or apoptosis. After feeding a 15N-leucine diet, we used nanoscopic secondary ion mass spectrometry, and found that in EPO-treated mice, an equivalent number of neurons was defined by elevated 15N-leucine incorporation. In EPO-treated NG2-Cre-ERT2 mice, we confirmed enhanced differentiation of preexisting oligodendrocyte precursors in the absence of elevated DNA synthesis. A corresponding analysis of the neuronal lineage awaits the identification of suitable neuronal markers. In cultured neurospheres, EPO reduced Sox9 and stimulated miR124, associated with advanced neuronal differentiation. We are discussing a resulting working model in which EPO drives the differentiation of non-dividing precursors in both (NG2+) oligodendroglial and neuronal lineages. As endogenous EPO expression is induced by brain injury, such a mechanism of adult neurogenesis may be relevant for central nervous system regeneration.
Similar content being viewed by others

Introduction
Erythropoietin (EPO) and its receptor, EPOR, were originally named because of their pivotal contribution to hematopoiesis, but over the last ~20 years, their important role also in the nervous system became more and more evident.1, 2 EPO and EPOR expression in normal postnatal brain is low3 but distinct in areas like hippocampus and cortex.4, 5 Strong upregulation is observed upon distress, for example, brain injury.2, 6 In healthy rodents and man, recombinant human EPO improves cognition and increases hippocampal long-term potentiation.7, 8 Beneficial effects of EPO on cognitive performance were consistently seen in clinical trials on schizophrenia, multiple sclerosis, major depression or bipolar disease, for example, see refs 9. Importantly, these EPO actions on the brain are essentially independent of hematopoiesis. There was either no increase in hematocrit or no correlation between such increase and cognition in these clinical trials. Moreover, the effect on cognition by far outlasted any transient effect on hematocrit, for example, see refs 9. Even more convincingly, non-hematopoietic EPO analogs14 or the boosted cognition of mice with expression of constitutively active EPOR in pyramidal neurons15 are key arguments for brain effects of EPO in the absence of any blood effects.
Nevertheless, the exact mechanisms of EPO action in brain have still remained widely unclear. Some of the neuroprotective and neuroregenerative effects of EPO in disease models were speculated to be associated with stimulation of neurogenesis, for example, see refs 16, 17, 18, 19, but essentially no data are available to clearly support this hypothesis or to discriminate between EPO effects on neural progenitor proliferation versus differentiation in postnatal brain.17 In the hematopoietic system, EPO is crucial for antiapoptotic effects on erythroid precursors and for their differentiation rather than for proliferation.20 We therefore wondered whether similar mechanisms may also apply for the nervous system.
Adult neurogenesis has been discovered by labeling mitotic cells of the brain with 5’-bromo-deoxyuridine (BrdU) or 3H-thymidine incorporation into nuclear DNA. This led to the identification of restricted neurogenesis areas in the adult brain, mainly dentate gyrus (DG) and subventricular zone (SVZ).21, 22, 23, 24, 25 For oligodendrocytes, the situation is different, as adult precursor cells (OPC) are widely distributed throughout the central nervous system.26 Adult OPC are multipolar cells, morphologically distinct from small and rapidly dividing progenitors in the SVZ and molecularly defined by specific antigens, such as platelet-derived growth factor receptor α (PDGFRα) and the proteoglycan NG2.27, 28, 29, 30
Based on our findings of EPO enhancing cognition and long-term potentiation,7 and the popular concept of neurogenesis leading to more neurons and more neurons to better learning and memory,22, 31, 32, 33 we initiated the present work. We report here the surprising finding that (1) only 3-week administration of EPO to healthy young mice is associated with an approximately 20% increase in the number of mature neurons and oligodendrocytes in hippocampal cornu ammonis (CA1/CA3); (2) under enduring cognitive challenge, these neuron numbers are preserved; (3) this EPO effect involves stimulation of precursor differentiation rather than proliferation or anti-apoptosis. We discuss a resulting working model, in which these observations are—beyond EPO—compatible with a new aspect of postnatal neurogenesis and neuroregeneration.
Materials and methods
Important note: All experiments in this article, including histological counting, were performed by investigators unaware of (i) group assignment and (ii) treatments ('fully blinded’).
In vivo studies
An overview of in vivo studies performed is given in Figure 1a.

Erythropoietin (EPO) effects on neuron number, proliferation and apoptosis in CA1 and CA3. All data are based on bilateral counting. (a) Experimental design of the in vivo experiments (see f for age at treatment in the magnetic resonance imaging (MRI) design). Mice received EPO or placebo intraperitoneally (i.p.) every other day, starting on postnatal day 28. (b) Number of pyramidal neurons in CA1 and CA3 at 1 week after 3-week EPO versus placebo treatment (w4) (analysis was performed in two independent experiments with identical results; n=17 in CA1 for both groups, and n=16 and n=18 in CA3 for placebo and EPO, respectively). (c) Sample cresyl violet staining, illustrating that pyramidal neurons (arrowhead) can be clearly distinguished from other cells (arrow). (d) Number of CTIP2+ pyramidal neurons in CA1 at w4 (n=4 per group). (e) Illustration of the CTIP2 staining in the dorsal hippocampus. The white rectangle indicates the magnified area shown in the lower right corner. (f) MRI-based volumetrical analysis of whole hippocampus (HC) after EPO or placebo (n=6 per group; treatment in this set of male mice was initiated at 11 weeks of age; that is, MRI data were obtained at age 15 weeks). (g) Proliferation determined by 5’-bromo-deoxyuridine (BrdU) incorporation at w4 (placebo n=7 and EPO n=6). (h) Apoptotic cells analyzed with terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (Tunel) staining at w4 (n=10 for both groups). (i) Confocal analysis of BrdU and NeuN double-positive cells at w4 (n=6 for both groups). (j) Confocal picture showing a neuron staining positively for NeuN (green) and BrdU (red). (k and l) Number of Pax6-positive cells at 72h and w1 (n=9 per group). (m) Pax6+ cells (arrows) visualized by 3,3’-diaminobenzidine (DAB) staining. (n) Number of doublecortin (Dcx)-positive cells at w4 (n=9 per group). (o) Sample picture of Dcx+ cells. All bar graphs shown as mean±s.e.m.; all analyses unpaired, two-tailed t-tests; *P<0.05, **P<0.01, *** P<0.001, ****P<0.0001.
Mice
All experiments were approved by and conducted in accordance with the regulations of the local Animal Care and Use Committee (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit—LAVES). Juvenile (28-day-old) male C57BL/6 mice as well as NG2-Cre-ERT2:R26R-td-tomato-mEGFP34, 35 mice on C57BL/6 background were used in all experiments. They were housed in groups of five in standard plastic cages and maintained in a temperature-controlled environment (21±2 °C) on a 12 h light–dark cycle with food and water available ad libitum. Mice were always randomly assigned to treatment groups. Inclusion–exclusion criteria were pre-established. Included were only male mice with same age and similar body weight. Exclusion criteria before experiment start were impaired health or body weight lower than average. No animals had to be excluded during any of the experiments. Criteria to exclude mice from statistical analysis were determined by the Grubbs’ test using GraphPad Software (graphpad.com/quickcalcs). Genotyping of Cre driver and reporter lines have been described.34, 35 Detailed PCR protocols are available on request.
EPO treatment
Male mice were injected intraperitoneally (i.p.) with recombinant human EPO (NeoRecormon, Roche, Welwyn Garden City, UK, 5000 IU kg–1 body weight in 0.01 ml g–1) or placebo (solvent solution, 0.01 ml g–1). Treatment was started at the age of 28 days. Depending on the experiment, EPO was given once (early time point of 6 h) or every other day (later time points of 72 h, 1 week or 3 weeks). BrdU was injected i.p. daily for 3 weeks (5-bromo-2'-deoxyuridin, Sigma-Aldrich, Taufkirchen, Germany, 50 μg g–1 in 0.01ml saline).36 For induction of Cre-activity in NG2-Cre-ERT2 mice, 100 mg kg–1 tamoxifen (in corn oil; Sigma-Aldrich) was injected i.p. at P26 and P27.
Cognitive challenge: touch-screen visual discrimination task
Mice
Male 4-week-old C57BL/6 mice received either EPO (n=10) or placebo (n=11) i.p. every other day over 3 weeks (11 injections in total). After the last injection, the animals were adapted for 1 week to the food deprivation procedure (85% of free feeding body weight) before start of the touch-screen experiment.
Touch-screen-based operant system
Experimental procedures and visual stimuli were based on a previous publication37 with slight modifications. The operant chamber (dimensions: 18.1 cm × 17.6 cm × 18.6 cm), enclosed in a sound and light attenuating box (Med Associates, St Albans, VT, USA), contained a pellet dispenser, which delivered a single 14 mg dustless pellet (TSE Systems, Bad Homburg, Germany) into a food tray (magazine). A touch-sensitive TFT monitor (touch-screen; 18.4 cm × 13.6 cm) was located on the opposite side of the chamber. The touch-screen was divided into two parts, allowing two different visual stimuli (of maximally 7.5 cm × 7.5 cm each) to be presented simultaneously (Controller Software, K-Limbic, Sawbridgeworth, UK).
Touch-screen experimental procedures
Mice were tested on 6 consecutive days per week. Every mouse underwent the following sequence of test phases: (1) acclimation, (2) autoshaping, (3) pre-training 1–3 and (4) visual discrimination. The daily sessions were terminated after 30 min or when the mouse had accomplished the specific learning criterion of the respective test phase.
Acclimation: Pellets were freely available in the magazine (prime reward) with an inter-trial interval of 15 s. A new trial was initiated after collection of the pellet leading to a disruption of an infrared light beam. This test phase was completed after the animal had collected 10 pellets within 30 min.
Autoshaping: The session started with a three pellets prime reward. After the mouse had collected the pellets, two identical stimuli were presented on both sides of the touch-screen for 10 s. The disappearance of the stimuli coincided with a tone, illumination of the pellet tray and the delivery of one pellet. A new trial was initiated after collection of the pellet (inter-trial interval 15 s). Animals that collected 30 pellets within 30 min moved on to the pre-training phase.
Pre-training 1: Again the session started with a three pellets prime reward. After the mouse had collected the pellets, one stimulus was presented pseudo-randomly on the left or right side of the touch-screen. A touch response to the correct side where the stimulus was presented activated the pellet dispenser to release a pellet into the magazine, which was illuminated and signaled by a tone (inter-trial interval 5 s). This test phase was completed after the animal had performed 30 correct responses within 30 min.
Pre-training 2: This test phase was identical to pre-training 1 except that the mouse had to actively initiate a new trial by disrupting the infrared light beam in the magazine a second time (after it had collected the food pellet). Only then the next stimulus could appear.
Pre-training 3: This test phase was identical to pre-training 2 except that an incorrect response to the blank side of the touch-screen where no stimulus was presented resulted in a 5 s timeout with house light turned off and the initiation of a correction trial (maximum of four possibilities to correct the faulty response) with the same stimulus configuration presented. This test phase was completed after the mouse had performed at least 27 correct responses out of 30 trials.
Visual discrimination: This test phase was identical to pre-training 3 except that two different stimuli S+ and S– were presented simultaneously and in a pseudorandom order on the left and right side of the touch-screen. The selection of the stimulus S+ (correct response) led to the disappearance of both stimuli, presentation of a tone, illumination of the food magazine, delivery of a pellet. A new trial could be initiated after the collection of the pellet and an inter-trial interval of 5 s. The selection of the stimulus S– (incorrect response) resulted in a 5 s timeout with house light turned off and the initiation of a correction trial (maximum of four possibilities to correct the faulty response) with the same stimulus configuration presented. The test phase was completed if the mouse accomplished 27 correct responses out of 30 trials or 26 correct responses out of 30 trials on 2 consecutive days, whichever appeared first (correction trials not counted). The experiment was concluded for a given mouse after it had been tested for 61 days including all four test phases.
Hippocampus volumetry using magnetic resonance imaging (MRI)
Male mice at the age of 15 weeks (1 week after 3 weeks of EPO treatment, that is, 4 weeks after EPO treatment start at 11 weeks of age) were anesthetized with 1–1.5% isoflurane in a mixture of oxygen and ambient air and positive ventilated via endotracheal tube. For in vivo volumetry,38 three-dimensional fast low angle shot (FLASH; TR/TE=14.9/3.9 ms) MRI was performed at 9.4T (Bruker Biospin, Rheinstetten, Germany) with an isotropic spatial resolution of 110 μm. Hippocampal volume was determined by manual segmentation using the Amira software (Visage Imaging GmbH, Berlin, Germany).
Histology and immunohistochemistry
Mice were anesthetized by i.p. injection with Avertin (Tribromoethanol, Sigma-Aldrich, St Louis, MN, USA, 0.276 mg g–1) and perfused transcardially with 0.9% saline followed by 4% paraformaldehyde (PFA). Brains were removed, postfixed overnight at 4 °C with 4% PFA and placed in 30% sucrose/phosphate-buffered saline (PBS) for cryoprotection and stored at -80 °C. Whole mouse brains were cut into 30 μm thick coronal sections on a cryostat (Leica, Wetzlar, Germany) and kept in a storage solution (25% ethylene glycol and 25% glycerol in PBS) at –20 °C.
Cresyl violet staining
Every 10th section was mounted on Super Frost microscopic slides (Gerhard Menzel B.V. & Co. KG, Braunschweig, Germany), washed in PBS and immersed for 25 min in dilute cresyl violet stain (0.01%) in acetate buffer (pH 4.5). After dehydration in serial dilutions of ethyl alcohol, sections were coverslipped using DePeX (Serva, Heidelberg, Germany).
Pax6, Dcx, Olig1, PDGFRα, BrdU, Ki67 staining
Every 4th section for BrdU, every 8th for Ki67, every 10th section for Dcx, PDGFRα and Olig1, and every 20th section for Pax6 were washed three times with PBS and mounted on Super Frost microscopic slides. They were dried overnight and rehydrated in PBS. Sections for Pax6, Dcx, Ki67, PDGFRα and Olig1 were microwaved three times for 2 min in citrate buffer. For BrdU staining, sections were pretreated with 1n HCl for 30 min at room temperature (RT). All sections were incubated with 0.5% hydrogen peroxide for 30 min to quench endogenous peroxidases. Afterward, they were permeabilized and blocked with 5% normal horse serum (NHS), 0.5% Triton-X in PBS for 1h at RT. The following primary antibodies (final dilution and source) were used for tissue staining: rabbit anti-Pax6 (1:200, Chemicon, Hampshire, UK), goat anti-Dcx (1:750, Santa Cruz Biotechnologies, Heidelberg, Germany), rabbit anti-PDGFRα (1:2000, Cell Signaling, Danvers, MA, USA), rabbit anti-Olig1 (1:100, Chemicon), mouse anti-BrdU (1:4000, Chemicon) and rabbit anti-Ki67 (1:1000, Novocastra Laboratories, Newcastle Upon Tyne, UK). Antibodies were diluted in 3% NHS, 0.5% Triton-X in PBS. After washing with PBS, sections were incubated with the respective biotinylated secondary antibody for 1.5 h (1:200, Vector Laboratories, Burlingame, CA, USA). The staining was visualized by a peroxidase-labeled avidin-biotin kit (Vector Laboratories) and 3,3’-diaminobenzidine (Sigma-Aldrich). Sections were allowed to dry overnight and coverslipped using DePeX (Serva).
TUNEL staining
To detect cells undergoing apoptosis, an in situ detection kit (DeadEnd Colorimetric TUNEL Kit Promega, Mannheim, Germany) was used. For terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining, every 10th section was washed with PBS and mounted on Super Frost microscopic slides. The staining was performed according to the manufacturer’s protocol.
Immunofluorescence double staining
For BrdU and NeuN, every 20th section was washed with PBS and incubated with 1n HCL for 30 min at 45 °C in a shaking water bath. Afterward, sections were permeabilized and blocked with 5% NHS for 1h at RT. Sections were incubated with mouse anti-NeuN (1:25, Chemicon) in 3% NHS, 0.5% Triton-X in PBS for 48 h at 4 °C. After washing with PBS, sections were incubated with a fluorescently labeled secondary antibody (1:1000 goat anti-mouse Alexa Fluor 488; Invitrogen, Darmstadt, Germany) for 1.5 h at RT. Sections were then washed and incubated with the direct-labeled mouse anti-BrdU Alexa Fluor 594 (1:500, Invitrogen) in 3% NHS, 0.5% Triton-X in PBS for 48 h at 4 °C. After washing in PBS, sections were mounted on Super Frost microscopic slides, dried and coverslipped, using Vectashield HardSet mounting medium containing 4,6-diamidino-2-phenylindole (DAPI; Vector Laboratories). For BrdU and Olig1, every 20th section was washed with PBS and mounted on Super Frost microscopic slides. They were dried overnight and rehydrated in PBS. Sections were pretreated with 1n HCl for 30min at RT. Sections were then boiled in citrate buffer, permeabilized and blocked with 5% NHS for 1h at RT. Sections were incubated with rabbit anti-Olig1 (1:25, Chemicon) and mouse anti-BrdU (1:500, Chemicon) in 3% NHS, 0.5% Triton-X in PBS for 72 h at 4 °C. After washing with PBS, sections were incubated with a fluorescently labeled secondary antibody (1:200 anti-mouse Alexa Fluor 568 Invitrogen,) and anti-rabbit biotinylated (1:200, Vector Laboratories) for 1.5 h at RT. After washing, sections were incubated with streptavidin Alexa 488 (1:200, Invitrogen), washed in PBS and mounted. For green fluorescent protein (GFP) and CC-1, every 10th section was washed with PBS and mounted on Super Frost slides. They were allowed to dry overnight and rehydrated in PBS. The sections were microwaved three times for 2 min in citrate buffer, then permeabilized and blocked with 5% NHS, 0.5% Triton-X in PBS for 1 h at RT. The following primary antibodies (final dilution and source) were used for tissue staining: rabbit anti-GFP (1:500, SYSY, Göttingen, Germany), mouse anti-APC, clone CC-1 (1:100, Merck, Darmstadt, Germany). Antibodies were diluted in 3% NHS, 0.5% Triton-X in PBS. After washing with PBS, sections were incubated with horse anti-mouse biotinylated secondary antibody for 1.5 h (1:200, Vector Laboratories). Staining was visualized by goat anti-rabbit-Cy2 secondary antibody (1:250, Dianova, Hamburg, Germany) and streptavidin conjugated to Alexa633 (1:250, Life Technologies, Darmstadt, Germany). After washing, sections were incubated with DAPI in PBS (0.125 μg ml–1, 406 nm; Sigma-Aldrich), dried overnight and coverslipped by Aqua Poly/Mount (Polysciences, Warrington, PA, USA). For EPOR/GFP double labeling and CTIP2 stainings, sections (for CTIP2 every 10th) were washed with PBS and then permeabilized/blocked with 5% NHS, 0.5% Triton-X in PBS for 1 h at RT on a shaker. As primary antibodies, goat anti-GFP (1:500, Rockland, Limerick, PA, USA), rabbit anti-EPOR6 and guinea pig anti-CTIP2 (both 1:300, SYSY) were used. Antibodies were diluted in 3% NHS, 0.5% Triton-X in PBS and sections incubated with primary antibodies for 72 h at 4 °C on a shaker. After washing with PBS, sections were incubated with fluorescently labeled secondary antibodies (donkey anti-goat Alexa Fluor 488 and donkey anti-rabbit Alexa Fluor 594, both 1:250, Life Technologies, or donkey anti-guinea-pig-Cy3, 1:300, Dianova) for 1.5 h at RT on a shaker. After washing, sections were incubated with DAPI in ddH2O (0.125 μg ml–1, 406 nm; Sigma-Aldrich), dried overnight and coverslipped by Aqua Poly/Mount (Polysciences).
Histological cell counting
Stereological counting of cresyl violet, Dcx, Ki67, PDGFRα, Olig1, BrdU and Tunel-positive cells
Serial coronal sections, spaced at regular intervals for the specific marker under investigation, were taken through the dorsal hippocampus (coordinates from Bregma: −1.34 to −2.54 mm posterior) and the SVZ (coordinates from Bregma: 1.18 to −0.1 mm) for quantitative analysis. Stereological determination of the cell number was carried out using the optical fractionator technique (Stereo Investigator, MicroBrightfield, Magdeburg, Germany). Cells were counted using a light microscope (Olympus BX-50, Tokyo, Japan) modified for stereology with 40x and 100x oil immersion objectives, a computer-driven motorized stage, z axis position encoder (microcator), and a microfire video camera interfaced to a PC with the software Stereo Investigator 6.55 (MicroBrightfield, Inc., Williston, VT, USA). Cell numbers were estimated using the modified optical fractionator technique.39 All stereological analyses for cell counting were performed on both sides of the hippocampus and SVZ. Hippocampus CA2/CA3 region is referred to as CA3 in text and figures. Neurons were counted only in the pyramidal layers of CA1 and CA3 regions (Supplementary Figure 1 and Supplementary Table 1). From area determination of hippocampal subfields, it was estimated that systematic uniform spacing of random sampling site (SURS grid size, x- and y-steps) of 80 × 80 μm would provide >10.000 sampling sites if all sections were analyzed. Accordingly, the x and y dimensions of the counting frame was defined to be 20 × 20 μm, giving an average number of particles counted per optical dissector (with a height of 20 μm and a guard zone of 2 μm from top and bottom) of about 1–2. The estimated total number of neurons in CA1 or CA3 was thus the sum of the counted particles multiplied by the fraction factors 1/ssf, 1/asf and 1/hsf (ssf=section sampling fraction, asf=area sampling fraction, tsf=thickness sampling fraction; Supplementary Figure 1 and Supplementary Table 1). Optimizing the sampling scheme to counting more neurons yielded more precision and a lower coefficient of error.39 For all markers counted, the same inclusion and exclusion criteria were applied. The stereological sampling scheme was considered adequate when coefficient of error was <0.10.40 As this was not the case for Pax6+ cells, because of the relatively low numbers, conventional counting was applied. Pax6+ cells were counted in CA1 and CA3 regions on serial coronal sections through the dorsal hippocampus, using a light microscope (Zeiss Axiostar Plus, Jena, Germany) with 20x and 40x objectives. The counts represent the mean of CA1 or CA3 counts from four sections per animal.
Confocal counting of BrdU+/NeuN+, BrdU+/Olig1+, GFP+/CC-1+ and CTIP2+ cells
Confocal laser scanning microscopy (Leica TCS SP2-AOBS for BrdU+/NeuN+, BrdU+/Olig1+ and Leica TCS SP5-II for GFP+/CC-1+ and CTIP2+) was used to count fluorescently double-labeled cells in anatomically matched sections. Every 10th section obtained from the dorsal part of hippocampus (Bregma –1.34 to –2.54 mm posterior) was analyzed bilaterally in each animal. All BrdU-positive cells in hippocampal formations (DG, CA1 and CA3) were examined for either NeuN or Olig1 co-labeling, the markers for mature neurons and oligodendrocytes respectively. Colocalization was confirmed with a z-stack of the respective cell soma, which was acquired with a line-based sequential scan using 3 μm intervals. The immunofluorescent double-labeled cells were verified using oil immersion 63x objective lens. The volume of hippocampal regions was determined for each section using Cavalieri’s principle.39 Cell density was obtained by dividing the number of double-labeled cells (BrdU+/NeuN+ or BrdU+/Olig1+) per animal by the total volume of hippocampal region in mm3. All GFP+ cells and CTIP2+ in all hippocampus sections were examined for double labeling with CC-1 and DAPI or CTIP2 and DAPI, respectively. Colocalization was confirmed with a z-stack of whole hippocampus sections, which was acquired with a line-based sequential scan, using 0.25-μm intervals and a 20 × glycerol objective. Stacks were further processed by Fiji (http://fiji.sc/Fiji). The final quantification of GFP+/DAPI+, CC-1+/DAPI+, GFP+/CC-1+/DAPI+ and CTIP2+/DAPI+ cells was done using Imaris 7.5.1 (www.bitplane.com).
NanoSIMS experiment
Beginning on postnatal day 28, mice received freely available food pellets containing 1.025% l-Leucine-15N stable isotope (Sigma-Aldrich, Schnelldorf, Germany). On day 28, EPO or placebo treatment was started according to our standard protocol, with injections every other day for 3 weeks. During this treatment, animals received drinking water containing 0.2 mg ml–1 5-ethynyl-2'-deoxyuridine (EdU, Life Technologies, Eugene, OR, USA). EPO/placebo treatment, EdU and l-Leucine-15N feeding were stopped simultaneously. One week later, mice were anesthetized by i.p. injection with Avertin (Tribromoethanol, Sigma-Aldrich, St Louis, MN, USA, 0.276 mg g–1) and perfused transcardially with 0.9% saline followed by 4% PFA. Brains were removed, postfixed overnight at 4 °C with 4% PFA and placed in 30% sucrose/PBS for cryoprotection and stored at −80 °C. Whole mouse brains were cut into 30 μm thick coronal sections on a cryostat (Leica) and kept in a storage solution (25% ethylene glycol and 25% glycerol in PBS) at −20 °C. Slices of the dorsal hippocampus were immunostained for NeuN to identify neuronal cell bodies. Furthermore, EdU detection was accomplished with a Click-iT EdU Alexa Fluorophore 647 Imaging Kit (Life Technologies, Eugene, OR, USA) to label cells proliferating during the EPO/placebo treatment. In brief, after blocking in 5% NHS, sections were incubated with mouse anti-NeuN (1:500, Chemicon) for 48 h at 4 °C. After washing with PBS, sections were incubated with a Cy3-labeled secondary antibody (1:100 goat anti-mouse, Dianova) for 1.5 h at RT. Sections were washed, permeabilized again and incubated with Click-iT reaction mix according to the manufacturer’s instruction. The slices were embedded in LR White resin as previously described,41 and regions from the CA1 pyramidal layer area were excised and processed to 500 nm thick sections using a Leica UC6 microtome (Leica). The sections were put on silicon wafers, and first imaged by fluorescence microscopy, to identify the NeuN-labeled areas. Fiducial markers were imprinted on the sections using a two-photon laser at 100% power output, to enable the identification of the same areas in the nanoscopic secondary ion mass spectrometry (NanoSIMS) instrument. Fluorescence imaging was performed using a TCS STED microscope (Leica Microsystems, Wetzlar, Germany), as described.41 Only areas that did not contain any EdU staining were used, to exclude the analysis of dividing cells. The silicon wafers were then mounted in a NanoSIMS 50L device (Cameca, Gennevilliers, France), and were scanned using Cs+ primary beam. Secondary ions, including 12C14N- and 12C15N-, were detected by mass separation, using a double focalization mass spectrometer (based on 'Mattauch-Herzog' geometry). Areas of 39 by 39 μm were imaged, onto 256 × 256 pixel rasters, using a 10 ms pixel dwell-time. Several planes (10–40) were acquired repeatedly, and were assembled into average images using a self-written routine in Matlab (The Mathworks, Ismaning, Germany). For simplicity, the 12C14N- and 12C15N- ionic species are referred to as 14N and 15N throughout the work.
In vitro studies
Design overviews of in vitro studies are given in Supplementary Figures 4A, C and 5A.
Randomization and exclusion criteria
Samples/dishes per wells were randomly assigned to treatment groups. Exclusion criteria for all experiments were pre-established. Criteria to exclude cultures from statistical analysis were determined by the Grubbs’ test using GraphPad Software (graphpad.com/quickcalcs).
Neural stem cell (NSC) isolation and culture
NSC were isolated from embryonic mice on day 14 (E14) as described previously42 with minor modifications. Whole mouse brains were mechanically dissociated and cultured in NSC medium (Dulbecco’s modified Eagle’s medium/F12 with 15 mM heparin, 2 mM l-glutamine, 2% B27, 0.22% sodium bicarbonate; all from Invitrogen), 1% penicillin/streptomycin (PAA, Cölbe, Germany), 0.6% glucose (Sigma-Aldrich), and supplemented with 20 ng ml–1 human epidermal growth factor and 20 ng ml–1 human basic fibroblast growth factor-2 (both from Peprotech, Hamburg, Germany). NSC were cultured at 37 °C under 5% CO2/95% air and 90% humidity. Medium was replaced every third day. NSC-generated neurospheres were passaged every fifth day according to the following protocol: neurospheres were first incubated in AccuMax (PAA, Hamburg, Germany) for 4 min at 37 °C and mechanically triturated with a fire-polished Pasteur pipette. New NSC cultures were prepared at a density of 40 cells μl–1 in NSC medium and supplemented with 20 ng ml–1 human epidermal growth factor and 20 ng ml–1 human basic fibroblast growth factor-2. Tertiary neurospheres were used for all experiments.
Neurosphere growth assay
Tertiary neurospheres were dissociated into single cells and seeded with a density of 800 cells per well into 96-well suspension culture plates (Greiner bio-one, Frickenhausen, Germany), containing 200 μl of NSC medium supplemented with 5 ng ml–1 human epidermal growth factor and 5 ng ml–1 human basic fibroblast growth factor-2. Cells were immediately treated either with 1IU ml–1 EPO or solvent control, both diluted with PBS, and cultured for 144 h at 37 °C under 5% CO2/95% air and 90% humidity without changing the medium. After 144 h, all wells were analyzed for neurosphere growth: total number of neurospheres per well was counted for each well and every neurosphere was photographed using an Olympus IX70 inverse microscope (Olympus, Münster, Germany) and the AnalySIS software (Soft Imaging System GmbH, Münster, Germany). Sphere diameter was measured using ImageJ (http://imagej.nih.gov/ij/).
NSC cell death assay, TuJ1 and GFAP staining
For the NSC cell death assay, tertiary neurospheres were dissociated into single cells as described above and plated at a density of 25 000 cells on poly-d-lysine (PDL) (Sigma-Aldrich) coated glass coverslips (Paul Marienfeld GmbH & Co. KG, Lauda-Königshofen, Germany) in four-well plates (Nunc/Thermo Scientific, Langenselbold, Germany). NSC were cultured in minimum essential medium, supplemented with 2% B27, 0.22% sodium bicarbonate, 1% sodium pyruvate (all from Invitrogen), 1% penicillin/streptomycin, 2 mM l-glutamine (PAA, Cölbe, Germany) and 0.6% glucose (Sigma-Aldrich). Cultures received 1IU ml–1 EPO or solvent control, both diluted with PBS, for 24 h at 37 °C under 5% CO2/95% air and 90% humidity. After 24 h, NSC cultures were treated with Trypan blue (Sigma-Aldrich) and vital and dead cells were counted for five randomly chosen fields on each coverslip. For each NSC culture and condition, five coverslips were analyzed. For TuJ1 and GFAP staining, NSC were differentiated in minimum essential medium on PDL-coated glass coverslips in 24-well plates (Greiner bio-one) in the presence of 1 IU ml–1 EPO or solvent control. Cells were seeded with a density of 50 000 cells per coverslip. After 96 h, cells were fixed in 4% PFA in PBS (Sigma-Aldrich) and stained with a mouse-anti-TuJ1 (1:500, Sigma-Aldrich) or mouse-anti-GFAP (1:1000, Novocastra, Newcastle Upon Tyne, UK) primary and a goat anti-mouse-Cy3 secondary antibody (1:1000, Dianova). In addition, cell nuclei were counterstained with DAPI. For each NSC culture and staining, photos from eight coverslips (six randomly chosen fields per coverslip) were taken using an Axiophot 2 fluorescence microscope (Zeiss, Jena, Germany) with a Spot Insight camera (Visitron Systems, Puchheim, Germany). From each photo taken, the number of living and dead (DAPI) as well as TuJ1 or GFAP-positive cells were counted.
Knock-down of miR124
Tertiary neurospheres were dissociated into single cells as described above. NSC were transfected with either the FAM-labeled miRCURY LNA miR124 inhibitor or the FAM-labeled miRCURY LNA control oligonucleotide (Exiqon, Skelstedet, Denmark) using the Amaxa mouse neural stem cell nucleofector kit (Lonza, Cologne, Germany) according to the manufacturer's instructions. Both LNA oligonucleotides were used at a concentration of 60 mM. A neurosphere growth assay was performed 96 h after transfection as described above with the following change: 2000 transfected cells per well were plated to compensate for the increased cell death because of the transfection procedure. Cells were treated with 1 IU ml–1 EPO or solvent control (six replicates per condition).
Primary hippocampal neuronal culture E17 (E17-HCC)
Hippocampal cultures from mice at embryonic day 17 (E17) were prepared as described previously.7 The removal of the meninges and dissection of the hippocampi was done in Hank’s balanced salt solution (Invitrogen) supplemented with penicillin/streptomycin (PAA Laboratories GmbH, Pasching, Austria) and HEPES (Invitrogen). The hippocampi were trypsinized and mechanically triturated with a fire-polished Pasteur pipette. Cells were seeded on PDL-coated six-well plates for RNA isolation with a density of 170 000 cells per well. For immunocytochemistry, 25 000 cells were seeded on PDL-coated coverslips. Neurons were cultured in minimum essential medium/B27 medium (Invitrogen) supplemented with sodium bicarbonate, sodium pyruvate, l-glutamine, penicillin, streptomycin and 0.6% glucose. Cultures were incubated at 37 °C under 7.5% CO2/92.5% air and 90% humidity. To analyze the basal EPOR expression, total RNA was isolated 0.3 h and 24h after seeding. In all other experiments, cells were treated with 0.3 IU ml–1 EPO or solvent control 0.3 h after seeding. This time point is declared as time point 0. RNA was isolated at 0.25, 0.5, 1, 3, 6, 12 and 24 h after treatment. The immunocytochemical analysis was performed after 6 h, 12 h, 24 h, and at day 8 (d8).
Immunocytochemistry
After 6, 12 and 24 h in culture, cells grown on coverslips were stained for Dcx or GFAP to analyze the culture composition and double-stained for Dcx and MAP2 to determine the differentiation ratio. Cells were fixed with 4% paraformaldehyde for 20 min, permeabilized and blocked in 0.2% Triton-X-100 with 10% NHS in PBS for 20 min. Cells were incubated with goat anti-Dcx (1:200, Santa Cruz Biotechnologies) or mouse anti-GFAP (1:500, Novocastra, Newcastle Upon Tyne, UK) in 0.02% Triton-X-100 with 1% NHS at 4 °C overnight, followed by the rabbit anti-goat-Cy2 secondary antibody or goat anti-mouse-Cy2 secondary antibody (1:1000, Dianova) at RT. Afterward, coverslips were dried and mounted with fluorescence mounting medium (Vector Laboratories) containing DAPI. For double staining, goat anti-Dcx (1:1000, Santa Cruz Biotechnologies) and primary labeled mouse anti-MAP2 Alexa Fluor 488 antibody (1:2000, Chemicon) were used to colocalize Dcx with MAP2 expression. At d8, neurons were quadruple stained for Dcx, MAP2, SynI and SytI. Cells were first stimulated for 10 s with high K+-base solution (70 mM KCl, 140 mM NaCl, 10 mM HEPES, 10 mM glucose, 4 mM MgCl2, 4 mM CaCl2) in presence of rabbit anti-synaptotagminluminal (1:50, SYSY) and then transferred into low K+-base solution (2.4 mM KCl) in presence of the same antibody for 30 s at 37 °C. After 2-min washing, cells were fixed with 3% paraformaldehyde and 2% acrolein in PBS for 20 min at RT. Cells were permeabilized and blocked in 0.2% Triton-X-100 with 10% NHS in PBS for 20 min and incubated with goat anti-Dcx (1:4000, Santa Cruz Biotechnologies) and mouse anti-synapsin I (1:4000, SYSY) at 4 °C overnight. The next day, cells were incubated with fluorescently labeled secondary antibodies (1:1000 Alexa Fluor 546 donkey anti-mouse, 1:500 Alexa Fluor 594 donkey anti-rabbit, 1:1000 Alexa Fluor 633 donkey anti-goat; Invitrogen) for 1 h at RT. Finally, cells were incubated with primary labeled mouse anti-MAP2 Alexa Fluor 488 antibody (1:2000, Chemicon) at 4 °C overnight. Before mounting with Aqua Poly/Mount (Polysciences), cells were stained with DAPI (0.125 μg ml–1, 406 nm; Sigma-Aldrich).
Fluorescent microscopy counting
For analyzing cell number and cell death, the DAPI-stained cells were counted by using upright light microscope (Axiophot, Zeiss) in two categories. Cells with homogenous oval nuclei were counted representing normal living cells. Cells showing a condensed chromatin formation or fragmented nuclear bodies were considered as apoptotic cells. The number of apoptotic cells is divided by the total number of DAPI-positive cells to obtain the percentage of cell death. To estimate culture composition, four visual fields of Dcx+ or GFAP+ cells were counted and normalized to DAPI.
Confocal microscopy and analysis
Images were obtained using an inverted confocal laser scanning microscope (Leica TCS SP2-AOBS; Mannheim, Germany) equipped with an UV-diode (405 nm), argon (458, 476, 488, 514 nm), DPSS (561 nm) and 2 He–Ne laser (594, 633 nm). Pictures were taken with a 63x Plan-Apo, 1.4NA oil immersion objective.
Double stain
Images for 6, 12 and 24 h were taken with a line average of 4. Acquisition settings were held constant across recordings within each experiment. For the intensity-based comparison of Dcx and MAP2, the free available software Fiji (http://fiji.sc/Fiji) was used. Briefly, the 8-bit pictures were converted into 32-bit pictures, a Gaussian filter with sigma=232 nm was applied, the background was subtracted, the lower threshold was set to 7 and the upper threshold was set to 250. Integrated density of Dcx and MAP2 was measured and ratio taken.
Quadruple stain
Using sequential image recording with fixed emission windows and applying a blind source separation technique, we unmixed the fluorescence contributions of four different fluorescent dyes (Dcx-Alexa Fluor 633, MAP2 Alexa Fluor 488, SytI-Alexa Fluor 594, SynI-Alexa Fluor 546). Twelve-bit images for d8 were taken with a zoom factor 2.5, line average 4 and an accumulation of three scans. Detector gain of 500 was used for all photomultiplier tubes, offset and laser intensity percentages were held constant across images within each experiment. The choice of fluorochromes and emission windows (Figure 6a) was optimized for the subsequent nonnegative tensor factorization decomposition, which further took available lasers of our imaging setup into account. Images were acquired sequentially, whereas emission windows were kept fixed. The images were spectrally decomposed using the nonnegative tensor factorization algorithm described previously43 with minor modifications. The nonnegative tensor factorization algorithm was initialized with spectra as determined by single stained samples. The spectrum of Alexa Fluor 633 (Dcx) was kept fixed during the decomposition. The nonnegative tensor factorization algorithm was coded in Python; the source code is available on request. Further data processing was also done with custom written Python scripts. Using the unmixed images, we determined the total fluorescence intensity of Dcx and MAP2 in the area above a threshold (250 in units of photomultiplier count). To detect synaptic buttons in SynI and SytI stains, we applied a custom-made feature detection algorithm to the images. Images where smoothed using a Gaussian filter with sigma=186nm, Laplace filtered and thresholded to isolate small maxima in fluorescence intensity corresponding to synaptic buttons. To increase the signal to noise ratio, we considered only buttons that have integrated fluorescence intensity >7000. Having detected the putative synaptic buttons, we investigated the morphology of the buttons positive for SynI, SytI, and those buttons positive for both SynI and SytI. Analysis scripts are available on request.
Protein extraction
Tissues
Protein extraction and analysis was done as described previously.44 Mice were decapitated; the hippocampi were taken out, immediately frozen on dry ice and stored at –80°C. For the analysis of myelin and control proteins, samples were homogenized in lysis buffer (50 mM Tris HCl (pH 8.3), 150 mM NaCl, 40 mM NaF, 5 mM EDTA, 5 mM EGTA, 1 mM Na3VO4, 1% Igepal, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate) also containing 1 mM phenylmethysulfonylfluoride, 10 μg ml–1 aprotinin and 10 μg ml–1 leupeptin. The tissue lysates were freeze-thawed four times and homogenized by pulling through a 1 ml syringe 10 times and centrifuged at 12 000 r.p.m. for 45 min. To estimate the phosphorylation of pMAPK/MAPK, total protein was extracted from cultures by using PhosphoSafe (Merck) according to the manufacturer’s instructions.
Neurosphere suspension cultures
Tertiary neurospheres were dissociated into single cells as described before and suspension cultures were prepared with a density of 40 000 cells per suspension culture flask. NSC were cultured 24 h in NSC medium supplemented with 5 ng ml–1 human epidermal growth factor and human basic fibroblast growth factor-2 at 37 °C under 5% CO2/95% air and 90% humidity. After 24 h, 1 IU ml–1 EPO or PBS was added to the suspension culture and total cell protein was isolated 10 min after treatment.
Neurosphere differentiation cultures
Single NSC were plated on six-well plates (Nunc/Thermo Scientific), coated with PDL at a density of 170 000 cells per well in minimum essential medium. NSC were allowed to adhere to the surface for 0.3 h before 1 IU ml–1 EPO or PBS was added and total cell protein was isolated after 10 min.
E17 hippocampal neuronal cultures
Cells were seeded on PDL-coated six-well plates with a density of 170 000 cells per well. Cells were treated with 0.3 IU ml–1 EPO or placebo at 0.3 h or 24 h after seeding and protein was isolated after 10min.
Immunoblot analysis
Supernatant from cells or tissue (10 μl) were used for determination of total protein by the Lowry method. The remaining supernatant was mixed with three volumes of Laemmli buffer (250 mM Tris HCL (pH 8.3), 8% sodium dodecyl sulfate, 40% glycerol, 20% 2-mercaptoethanol, 0.04% pyronin Y) boiled for 5–10min at 95°C and frozen at −80 °C until blotting. In all, 10–20 μg protein were run on NuPAGE 4-12% Bis-Tris Gel (Invitrogen) for 1h at 180 V and transferred to a nitrocellulose membrane for 90 min at 30 V. After blocking with 5% milk in Tween20-Tris-buffered saline at RT for 1h, membranes were incubated with primary antibodies: mouse anti-pMAPK, rabbit anti-MAPK, mouse anti-CNPase (all 1:1000, Sigma-Aldrich), rabbit anti-MBP (1:5000, Dako, Carpinteria, CA, USA) and mouse anti-GAPDH (1:5000, Assay Designs, Ann Arbor, MI, USA), rabbit anti-TOM20 (1:2000, Santa Cruz Biotechnologies), mouse anti-GAD67 (1:5000, SYSY), mouse anti-Grp75 (1:2000, Assay Designs), rabbit anti-TrkB (1:200, Upstate Biotechnology, Lake Placid, NY, USA), rabbit anti-Sox9 (1:2000, Millipore, Billerica, MA, USA), rabbit anti-actin (1:2000, Sigma-Aldrich). Immunoreactive bands were visualized by using secondary antibodies coupled to horseradish peroxidise (1:2000, Sigma-Aldrich) by enhanced chemiluminescence (Immobilon Western, Millipore). Densitometrical analysis of protein bands was performed using ImageJ software. Proteins were either normalized to GAPDH (CNPase, MBP, TOM20, GAD67, Grp75, TrkB) or actin (Sox9).
Isolation of total RNA and quantitative real-time RT-PCR (qPCR)
Tissue or cells were homogenized in Trizol or Quiazol. Total RNA was isolated by using the miRNeasy Mini Kit (Qiagen, Hilden, Germany).
mRNA
First-strand complementary DNA was generated from total RNA using N9 random and Oligo(dT) 18 primers. The relative concentrations of mRNAs of interest in different complementary DNA samples were measured out of three replicates using the threshold cycle method (delta Ct) for each dilution and were normalized to levels of beta-actin RNA. Reactions were performed using SYBR green PCR master mix (ABgene, Foster City, CA, USA) according to the protocol of the manufacturer. Cycling was done for 2 min at 50 °C, followed by denaturation at 95 °C for 10 min. The amplification was carried out by 45 cycles of 95 °C for 15 s and 60 °C for 60 s. The specificity of each primer pair was controlled with a melting curve analysis. For qPCR, we used the following primers: mouse Dcx (NM_001110222.1) fwd: 5′-CCTTGGATGAGAATGAATGC-3′, rev: 5′-TGAGTCAGCTGGAGACTTGC-3′; mouse EPOR (NM_010149.2) fwd: 5′-CCTCATCTCGTTGTTGCTGA-3′, rev: 5′-CAGGCCAGATCTTCTGCTG-3′; mouse GFAP (NM_010277.2) fwd: 5′-ACAGACTTTCTCCAACCTCCAG-3′, rev: 5′-CCTTCTGACACGGATTTGGT-3′; mouse MAP2 (NM_ 001039934) fwd: 5′-AAGCACTGATTGGGAAGCACTC-3′, rev: 5′-CGTCGGCCATCTTTCAGATCTC-3′; mouse ND1 (NM_010894) fwd 5′-CGCAGAAGGCAAGGTGTC-3′, rev 5′-TTTGGTCATGTTTCCACTTCC-3′; mouse S100B (NM_009115) fwd: 5′-AACAACGAGCTCTCTCACTTCC-3′, rev: 5′-CTCCATCACTTTGTCCACCA-3′; mouse Sox9 (NM_011448.3) fwd: 5′-CAAGCGGAGGCCGAAGA-3′, rev: 5′-CAGCTTGCACGTCGGTTT-3′, mouse beta-actin (NM_007393.3) fwd: 5′-CTTCCTCCCTGGAGAAGAGC-3′, rev: 5′-ATGCCACAGGATTCCATACC-3′.
miR124
First-strand complementary DNA synthesis and reactions were generated from total RNA using the TaqMan MicroRNA RT Kit, TaqMan MicroRNA Assay for hsa-miR124, TaqMan MicroRNA Assay for sno-RNA142 as a housekeeper and TaqMan 2x Universal PCR Master Mix (ABgene) according to the protocol of the manufacturer. Cycling was done with 10-min denaturation at 95 °C and amplification for 40 cycles at 95 °C for 15 s and 60 °C for 60 s.
Statistical analyses
Data are expressed as mean±s.e.m. All data sets were normally distributed as ascertained with the Kolmogorov–Smirnov test. Statistical analysis was performed using two-tailed Student’s t-tests for all in vivo and in vitro data. One-tailed Student’s t-tests (stated in the Supplementary Figure legend) were performed whenever experiments were confirmatory or clearly hypothesis driven. Variances were overall similar between compared groups. Touch-screen visual discrimination data were analyzed by means of Kaplan–Meier survival analysis and log ranked Mantel–Cox test. Data plotting and statistical analysis were done using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). Statistical differences were considered to be significant when P<0.05 (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001). Sample size estimates were based on extensive experience with similar experiments; expected effect sizes (Cohen's d see, for example, Cohen45) were according to these previous data strong (d > 1; see for example, Sargin et al.44). Considering all the data, we furthermore performed power analyses to show adequate sample size according to the observed effect sizes of the experiments (software G*Power Version 3.1.7, Düsseldorf, Germany). The adequate power (1–β-error) was defined as ≥ 80% and the α-error as 5%. Overall, adequate sample sizes led to observed effect sizes (1.23–6.90) agreeing with the estimates from previous experiments (strong; d >1). Examples of key experiments: analysis of pyramidal neurons in CA1 in the first cohort of mice (Figure 1b; observed effect size d=1.97). Analysis of pyramidal neurons in the first cohort of mice in CA3 (Figure 1b; observed d=1.23). Analysis of the coefficient of variation of the 15N/14N ratio in the neuronal nuclei in CA1 (Figure 3l; observed d=6.90). Analysis of MBP expression in the hippocampus (Figures 2c and d; observed d=1.45). Analysis of the percentage of CC-1+/GFP+ within the pool of GFP+ cells in the hippocampus of NG2-Cre-ERT2:R26R-td-tomato-mEGFP mice (Figure 2l; observed d=1.86).

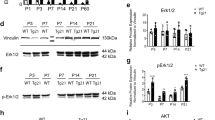
Erythropoietin (EPO) effects on oligodendrocytes and myelination. All data are based on bilateral counting. (a) Number of mature oligodendrocytes with cytoplasmic Olig1 expression in CA1 and CA3 at w4 (placebo n=8 and EPO n=10). (b) Olig1+ cells visualized by 3,3’-diaminobenzidine (DAB) staining. (c) Densitometrical quantification of myelin proteins (housekeeping gene GAPDH) extracted from hippocampal tissue (n=10 for all proteins and groups except n=8 for placebo of MBP and CNPase). (d) Sample western blots. (e) Number of 5’-bromo-deoxyuridine (BrdU) and cytoplasmic Olig1 double-positive cells in CA1 and CA3 at w4 (n=5 for both groups). (f) Illustration of BrdU and Olig1 colocalization. (g) Number of platelet-derived growth factor receptor α+ (PDGFRα+) OPC at 72 h after start of EPO/placebo treatment (n=10 for both groups). (h) PDGFRα+ cells visualized by DAB staining. (i) Number of PDGFRα+ OPC at w4 after start of EPO/placebo treatment (placebo n=8 and EPO n=10). (j) Left hippocampus of NG2-Cre-ERT2:R26R-td-tomato-mEGFP mice at w4 (postnatal day 56), stained for CC-1, green fluorescent protein (GFP) and 4,6-diamidino-2-phenylindole (DAPI); CC, corpus callosum; PL, pyramidal layer. (k) Magnification of the white square area denoted in j, showing a CC-1+/GFP+/DAPI+ cell in all 3 dimensions (xy, xz and yz). (l) CC-1+/GFP+ cells expressed as percentage of total NG2 (GFP+) cells at w4 (n=5 for both groups). (m) CC-1+ cells at w4 (n=5 for both groups). (n) Hippocampus of NG2-Cre-ERT2:R26R-td-tomato-mEGFP mice at postnatal day 31, showing co-labeling for NG2 and EPOR. HC, total hippocampus; All bar graphs shown as mean±s.e.m.; all analyses unpaired, two-tailed t-tests, except for subpanel m one-tailed t-test; *P<0.05, **P<0.01, ***P<0.001.
Results
EPO administration to young mice increases neuron numbers in CA1 and CA3
We applied our EPO treatment protocol known to increase long-term potentiation and to improve memory7 (experimental design; Figure 1a). Stereological quantification of pyramidal neurons bilaterally in CA1/CA3 of 8-week-old mice, that is, 1 week after 3 weeks of EPO (5000 IU kg–1 every other day) or placebo injections, revealed a ~20% increase in mature neurons upon EPO (Figures 1b and c; Supplementary Figure 1, Supplementary Table 1). This increase was determined in two independent experiments on different mouse cohorts. To confirm the augmented pyramidal neuron numbers, we additionally performed anti-CTIP2 staining. CTIP2 is a transcriptional regulatory protein46 that specifically labels pyramidal neurons in CA1. Indeed, the number of CTIP2+ cells in CA1 of the EPO-treated group was ~22% higher compared with the placebo group (Figures 1d and e). Importantly, this increase is also reflected by a larger hippocampal volume in MRI in 15-week-old mice where treatment was started at 11 (instead of 4) weeks of age (Figure 1f). Total brain volume and body weight of EPO-treated mice did not differ from placebo (data not shown).
Higher neuron numbers upon EPO are not explained by increased proliferation or decreased apoptosis
To elucidate the elevated neuron numbers upon EPO in CA1/CA3, we searched for evidence of increased proliferation or reduced apoptosis. Surprisingly, we observed the opposite: BrdU+ cells were diminished (Figure 1g) and apoptotic cells raised (Figure 1h). Double staining with BrdU and NeuN, a marker of mature neurons, showed comparable numbers in EPO and placebo mice (Figures 1i and j). At earlier time points, for example, at 1 week of EPO treatment, neither proliferation nor apoptosis were altered (data not shown). In addition, staining for the cell cycle marker Ki67 in whole hippocampus at 1 week of EPO/placebo injections did not reveal any differences in Ki67+ cells between treatment groups (224±34 versus 221±25; P=0.96), supporting that there is no increased DNA replication in this brain region upon EPO administration.
Reduction of neuronal precursor cells as an early response to EPO
The considerable increase in neurons upon EPO could be due to an accelerated differentiation of neuronal progenitors. In that case, staining for immature markers would yield a respective reduction in number already at early time points. Indeed, cells stained for the transcription factor Pax6 were decreased in CA1/CA3 at 72 h after EPO treatment initiation (Figures 1k–m). Also, the reduction under EPO of doublecortin-positive (Dcx+), that is, still immature neurons, significant in CA3 at 4 weeks, is compatible with accelerated differentiation (Figures 1n and o). In line with this observation, a Dcx mRNA decrease of about 25% was measured by qPCR in hippocampal tissue of EPO versus placebo-treated mice as early as 6 h after the first EPO injection (mean±s.e.m.; 247±13 versus 184±18; original values x105; P=0.012; n=8 per group).
Increase in neuron numbers by EPO is paralleled by increase in oligodendrocytes and myelin proteins
Many hippocampal pyramidal neurons have myelinated axons, and we wondered whether more neurons are associated with more oligodendrocytes. Indeed, numbers of oligodendrocyte lineage cells with cytoplasmic expression of Olig1 were on average >20% increased in CA1/CA3 at week 4 following EPO (Figures 2a and b), as were myelin-associated proteins, Golli33, MBP and CNPase (Figures 2c and d). Control proteins (GAD67, Grp75, Tom20 and TrkB) were all unchanged (data not shown). Double staining for BrdU and Olig1 revealed similar numbers in both treatment groups (Figures 2e and f). Consistent with stimulated differentiation of preexisting OPC by EPO, a transient reduction in PDGFRα+ cells was observed at 72h (Figures 2g and h). This reduction was no longer visible at 4 weeks (Figure 2i).
OPC can differentiate into oligodendrocytes without dividing.26 We repeated the EPO experiment (Figure 1a) in mice, expressing tamoxifen-inducible Cre-ERT2 under control of the endogenous NG2 locus.34 Crossing this line with R26R-td-tomato-mEGFP(mT/mG),35 mice allowed OPC cell fate mapping. In tamoxifen-pretreated mice (two injections on postnatal days 26 and 27, inducing reporter expression in ~60% of hippocampal OPC), EPO increased GFP+/CC-1+ and total CC-1+ oligodendrocytes at week 4 (Figures 2j–m). Supporting a direct EPO effect on OPC, endogenously labeled cells express EPOR (Figure 2n; Supplementary Figures 2A and B). Moreover, cultured OPC produce rising EPOR mRNA levels over time, parallel to myelin gene induction, whereas progenitor-specific transcripts decrease (Supplementary Figure 2C).
Elevated neuronal numbers correlate with elevated 15N-leucine incorporation
Our data on preexisting OPC that differentiate under EPO, led us to search for analogous effects in the neuronal lineage. Unfortunately, specific molecular markers for preexisting neuronal progenitors do not exist at present and thus, there is no corresponding neuronal Cre-ERT2 mouse line available. We therefore searched for alternative ways to explain how so many (~20%) newly differentiated neurons appear upon EPO without prior local cell division. We used multi-isotope imaging mass spectrometry, NanoSIMS,41 to identify pyramidal neurons with higher than average 15N-leucine incorporation. In absence of proliferation markers, this would be indirect evidence for induced protein synthesis, necessarily associated with the differentiation and physical growth of large pyramidal neurons from inconspicuous precursors (Figures 3a–i).

15N-leucine incorporation in CA1 pyramidal neurons evaluated by nanoscopic secondary ion mass spectrometry (NanoSIMS). (a) Overview of the hippocampus showing dense proliferation signals (EdU) in the dentate gyrus. The white square illustrates the EdU signal-free area in the pyramidal layer, analyzed by NanoSIMS. (b and c) Illustration of samples following placebo treatment; (d, e and e’) Illustration of samples following erythropoietin (EPO) treatment; arrows in e’ point to newly generated neuronal cell mass with high15N/14N ratio; stars in e’ denote ‘control’ signal in cytoplasmic regions of older neurons. (f and g) Scatter plots of 15N/14N ratios in pyramidal neurons in both treatment groups. (h and i) Coefficient of variation of 15N/14N ratios in cytoplasm and nuclei of pyramidal neurons (n=3 for both groups). All bar graphs are shown as mean±s.e.m.; **P<0.01 (unpaired two-tailed t-test).
When we compared the 15N/14N ratio in EdU-/NeuN+ pyramidal neurons at week 4 (Figure 1a), this ratio clearly scattered toward higher values upon EPO (Figures 3f and g), indicative of a pronounced increase in newly synthesized protein in a sub-population of pyramidal neurons. Indeed, neurons with higher 15N-leucine-incorporation upon EPO amounted to ~20% in CA1, in agreement with the increased cell number detected by stereology (cresyl violet) and by CTIP2 staining. The coefficient of variation of the15N/14N ratio was augmented in neuronal nuclei and cytoplasm (Figures 3h and i).
Effect of EPO on classical neurogenic regions
Our results do not support newly formed neurons from mitotic (BrdU incorporating) progenitors as a source of the increased numbers in CA1/CA3. However, 1 week after EPO treatment start, also the 'classical' adult neurogenesis areas showed a response. We found an increase under EPO in BrdU+ cells in SVZ and a similar tendency in DG, in both without altered apoptosis (Supplementary Figures 3A and B). The rise in Dcx+ neurons in DG is compatible with stimulated differentiation under EPO also in this niche (Supplementary Figure 3C). At week 4, in contrast, we found a reduction in BrdU+ cells in DG/SVZ, likely as a consequence of temporary depletion of progenitors or negative feedback on neurogenesis (Supplementary Figure 3D). We also noted increased apoptosis (Supplementary Figure 3E), potentially because of abnormal crowding of maturing neurons, comparable numbers of Dcx+ cells at week 4 (Supplementary Figure 3f), and more BrdU/NeuN double-labeled neurons upon EPO (Supplementary Figures 3G–I). These data may suggest that EPO transiently stimulates neuronal precursor proliferation in neurogenesis areas. This stimulation, however, is—according to our working model—probably indirect, that is, a consequence of EPO-induced precursor differentiation (and thus transient depletion) rather than a direct EPO effect, as supported by our in vitro experiments (see below).
Persistence of increased neuron numbers under cognitive challenge
Next, an experiment was designed to explore the old neurologists’ rule, ‘use-it-or-lose-it’. Starting at week 4 (time point 0 in Figure 1a), mice were for 22 weeks either exposed to long-term standard housing without stimulation or to regular cognitive challenge using the touch-screen paradigm. This paradigm, requiring hippocampal functions,47, 48 allows assessment of visual discrimination learning over long time periods. EPO-treated mice needed less time to finish the touch-screen task (Figure 4a). Mice that had completed the task continued with regular touch-screen performance to remain cognitively demanded until 6 months of age. At this time point, EPO-treated mice without challenge revealed only marginally elevated neuron numbers in CA3 (Figure 4b), whereas continuously challenged mice had retained their increased neuron counts in CA1/CA3 (Figure 4c). Independent of EPO or placebo treatment, cognitively challenged mice had higher pyramidal neuron numbers altogether. These data show that enhanced pyramidal neuron numbers induced by EPO in young mice are maintained under conditions of ongoing cognitive challenge but not in its absence: ‘Use-it-or-lose-it’.

Use-it-or-lose-it experiment. (a) Long-term learning curve in the touch-screen visual discrimination task showing significantly earlier task completion of erythropoietin (EPO)-treated mice (placebo n=11 and EPO n=10, log-rank (Mantel–Cox) test). (b and c) Pyramidal neuron counts obtained by stereological counting at 6 months of age (4 months after cessation of EPO/placebo treatment) in mice without (n=10 in case of CA1 for both groups, and n=9 and n=10 in case of CA3 for placebo and EPO groups, respectively) (b) or with (n=11 for placebo, for EPO n=8 and n=9 in CA1 and CA3, respectively) (c) continuous cognitive challenge using touch-screen. All bar graphs shown as mean±s.e.m. (all analyses unpaired one-tailed t-tests).
EPO reduces proliferating neurosphere diameter and number
Based on our in vivo findings, we wondered whether—similar to the hematopoietic system—EPO may drive differentiation rather than directly stimulate proliferation. Dissociated tertiary neurospheres from E14 forebrain cultures (Supplementary Figure 4A) were grown in suspension clusters with FGF-2/EGF.49 Under these conditions, neurospheres sustain mainly symmetrically dividing NSC.50 As prerequisite for testing the EPO influence on these cultures, EPOR mRNA expression was confirmed by qPCR and EPOR function by MAPK activation3 (Supplementary Figure 4B). After 144h of EPO exposure, a decrease in total number and average diameter of neurospheres substantiated an inhibitory EPO effect on NSC proliferation (Figures 5a–c).

Erythropoietin (EPO) effects on proliferation and differentiation of E14 forebrain neurospheres and role of miR124; (a-c) Quaternary E14 forebrain neurospheres cultured under proliferating conditions: histogram showing total number (a) and number by diameter (n=4 per group, paired two-tailed t-test) (b) of quaternary neurospheres at 144 h after EPO/placebo (n=4 per group, paired two-tailed t-test). (c) Phase contrast image of neurospheres. (d-j) Quaternary E14 forebrain neurospheres cultured under differentiating conditions: (d-h) qPCR/western blot results of differentiation markers. EPO-induced changes are expressed as % difference from placebo at the respective time point. Compare Supplementary Table 3. (d) Early neural marker Sox9, shown at mRNA level (0.3 h: n=5, 24 h: n=5, 96 h: n=5, 168 h: n=6, paired two-tailed t-test per time point), as well as protein level (n=3 per group, paired two-tailed t-test per time point) together with a sample western blot. (e) Early neural marker ND1 (0.3 h: n=6, 24 h: n=6, 96 h: n=5, 168 h: n=6, paired two-tailed t-test per time point). (f) Dcx/MAP2 ratio (0.3 h: n=6, 24 h: n=10, 96 h: n=9, 168 h: n=6, paired two-tailed t-test per time point). (g and h) GFAP (0.3 h: n=7, 24 h: n=7, 96 h: n=6, 168 h: n=7, paired two-tailed t-test per time point) and S100B (0.3 h: n=6, 24 h: n=11, 96 h: n=9, 168h: n=6, paired two-tailed t-test per time point). (i and j) miR124 expression and consequence of miR124 knockdown in proliferating neurospheres: (i) miR124 expression in proliferating neurospheres at 6h after addition of EPO/placebo (n=8 per group, paired two-tailed t-test). (j) Neurospheres with diameter >100 μm at 96h after addition of EPO/placebo to neurospheres transfected with miR124 antisense (AS) or scrambled sense (control) oligos (n=4 per group, paired one-tailed t-test). (k) miR124 expression in hippocampus of 28 day-old mice 6h after treatment with EPO/placebo (n=8 per group, unpaired one-tailed t-test). All n-numbers given for the in vitro data are derived from biological replicates, that is, independent cell preparations. All bar graphs are shown as mean±s.e.m.; *P<0.05, **P<0.01, ***P<0.001.
Neurospheres respond to EPO with accelerated differentiation
Next, dissociated tertiary neurospheres were plated in serum-free medium and treated with EPO or placebo (Supplementary Figure 4C). Again, EPOR mRNA expression and functionality were documented (Supplementary Figure 4D). Sox9, ND1, Dcx, MAP2, GFAP and S100B were used as differentiation markers on mRNA level. Compared with placebo (Supplementary Figure 4E; Supplementary Table 2), Sox9 and ND1 strongly and lastingly decreased after EPO (Figures 5d and e; Supplementary Table 3), as did the Dcx/MAP2 ratio, pointing to accelerated neuronal differentiation (Figure 5f; Supplementary Table 3). The decreased Sox9 expression was further confirmed on protein level at the 96 h time point (Figure 5d). GFAP and S100B increased, consistent with enhanced glial cell numbers paralleling the progression of neuronal differentiation (Figures 5g and h; Supplementary Table 3).51 Neurosphere culture composition, cell death and total cell number are presented in Supplementary Figures 4F–H.
Candidate mediator of EPO effects on neural precursors: miR124
Transcription factor Sox9 and microRNA miR124 are critical for the homeostasis of differentiation and proliferation in adult neurogenesis and functionally connected;52, 53 miR124 is increased during differentiation and elevated in type A neuroblasts after transition from amplifying type C cells.54 We asked whether changes in miR124 expression occur also under EPO. Indeed, miR124 was upregulated in NSC 6 h after EPO addition (Figure 5i). Importantly, the EPO-mediated reduction in neurosphere numbers was prevented upon miR124 knockdown (Figures 5b and j). Increased miR124 expression was also found in hippocampus of 28-day-old mice, 6h after a single i.p. injection of EPO (Figure 5k). Thus, EPO may act on neurodifferentiation at least partially via miR124.
E17 hippocampal neurons exposed to EPO show rapid shifts in neural differentiation marker expression
Neurosphere cultures contain mainly early precursor cells, with a small fraction differentiating into neurons (Supplementary Figure 4F). Therefore, we studied E17 hippocampal neuron cultures (E17-HCC) to explore the EPO influence on cells at a more advanced differentiation stage (Supplementary Figure 5A). EPOR mRNA expression and functionality were proven (Supplementary Figure 5B). Comparable to neurospheres, Sox9 mRNA was rapidly upregulated upon EPO and declined thereafter (Supplementary Figures 5C and D; Supplementary Table 5). ND1 (Supplementary Figure 5E) and Dcx/MAP2 decreased as well (Supplementary Figure 5F; Supplementary Table 5). Dcx/MAP2 decrease was confirmed on protein level by immunocytochemistry-based integrated density (Supplementary Figures 5G and H). GFAP and S100B in E17-HCC responded to EPO with short-lived decrease (Supplementary Figures 5I and J; Supplementary Table 5). Culture composition, cell death and total cell number were unchanged (Supplementary Figures 5K–M).
EPO-induced differentiation provokes earlier maturation of neurons
To explore effects on synapse maturation, E17-HCC were treated with EPO/placebo 0.3 h after seeding and analyzed on day 8, when first excitatory synapses are formed.55 As biochemical indicators of synapse maturation/synaptic activity, we used synaptotagmin I (SytI) and synapsin I (SynI). Antibodies against the luminal domain of SytI were used previously to examine the exo-endocytotic recycling of synaptic vesicles and reflect the number of exocytotic events.56 Cells were quadruply stained (Figure 6a).43 Figure 6b illustrates the progressed differentiation at d8, with Dcx expression fading and MAP2 dominating. The EPO effect is again reflected in the decreased Dcx/MAP2 ratio (Figure 6c). We stained for SynI and SytI (Figures 6d and e) and created masks for both markers (Figure 6f). Their colocalization indicates active synapses56 (Figure 6f, white spots) whereas SynI single positive puncta refer to inactive synapses (Figure 6f, red spots). The size of the colocalized area (active area, white spots) was taken as a measure of synaptic activity. Whereas we found no change in the number of active synapses (data not shown), an increase in mean active area per spot was detected (Figure 6g), indicating progressed neuronal maturation under EPO.

Effects of erythropoietin (EPO) on neuronal differentiation and maturation, determined by mean active area of synapses in E17-HCC. Neurons (d8) were quadruply stained for differentiation markers Dcx and MAP2 and activity markers Synaptotagmin I (SytI) and Synapsin I (SynI) (n=8). Staining was analyzed by confocal microscopy. (a) The protocol includes three sequential scans with fixed emission windows (orange) with different excitation wave length. Excitation (dashed line) and emission (solid line) spectra are shown for each fluorescent dye, Alexa Fluor 488 (MAP2, green), Alexa Fluor 546 (SynI, blue), Alexa Fluor 594 (SytI, red) and Alexa Fluor 633 (Dcx, black). 4,6-Diamidino-2-phenylindole (DAPI) fluorescence was determined using an additional excitation at 406 nm. (b) Unmixed confocal picture showing differentiation markers Dcx (red), MAP2 (green) and DAPI (blue). (c) Quantification of the integrated density of Dcx and MAP2 presented as ratio (n=8 per group, paired one-tailed t-test). (d) Unmixed confocal picture showing SytI (green) and SynI (red). (e) Higher magnification of the square mark of picture d, showing single stained dots for SytI (green, arrowhead), single stained dots for SynI (red, star) and colocalized dots (yellow, arrow). (f) Masked images for analyzing the number and area of SytI (green, arrowhead), SynI (red, star) and colocalized (white, arrow) dots. (g) Quantification of the mean overlapping (colocalized) area as mean active area (white spots in f, n=8 per group, paired two-tailed t-test). All n-numbers given are derived from biological replicates, that is, independent cell preparations. All bar graphs are shown as mean±s.e.m.; *P<0.05.
Discussion
We report here the unexpected observation that a 3-week systemic administration of EPO, known to cross the blood–brain barrier,2, 12, 57 induced a ~20% increase in CA1/CA3 neurons and oligodendrocytes in young mice. We ruled out local proliferation of neural precursor cells and antiapoptotic effects of EPO1, 2 as underlying mechanisms. Conversely, we found a decrease in BrdU-positive and an increase in apoptotic cells, compatible with depletion of local precursors and death of a subset of newly generated (supernumerary) neurons. However, the concept of ‘preexisting neuronal precursors’ is at variance with the conventional view of adult neurogenesis, as opposed to preexisting oligodendrocyte precursors, which are widely accepted.26 We are certainly aware of open experimental questions that preclude final conclusions at this stage, but we will discuss our data in a ‘working model’ to help guiding future research. This working model is novel and, if confirmed by other labs, the underlying concept of adult neurogenesis without entering cell cycle may be reminiscent of adult OPC that can also trans-differentiate without proliferation. Both could be of major significance for rapid regenerative processes in the brain.
For proof-of-concept that the elevated number of oligodendrocytes upon EPO is derived from preexisting OPC, we used mice expressing an inducible oligodendrocyte lineage marker, the tamoxifen-inducible Cre-ERT2 gene58 under control of the endogenous NG2 promoter.34 Indeed, we found increased numbers of labeled oligodendrocytes at 4 weeks after initiation of EPO treatment. Unfortunately, specific molecular markers for genetically targeting analogous neuronal precursor stages remain to be defined and Cre-ERT2 tools to be developed. We therefore used advanced mass spectroscopy, NanoSIMS,41 to support the possibility that ~20% of CA1/CA3 neurons (as detected by stereology and CTIP2 staining) were newly generated. Intriguingly, we found exactly this percentage of neurons in CA1 with highly elevated 15N-leucine incorporation upon EPO in absence of proliferation markers. This finding is indeed consistent with enhanced protein synthesis underlying the growth of newly differentiated pyramidal neurons from preexisting postmitotic local precursors. Future studies will have to address whether similar results can be obtained in other central nervous system regions.
Complementing the pivotal work of many groups on adult neurogenesis in distinct regions of the brain, such as DG and SVZ, for example, see refs 21, 52, 59, our findings have led us to a working model, in which we discuss the postnatal delivery of new functional neurons and oligodendrocytes in the hippocampus from a pool of local precursors, outside the 'classical' neurogenic regions. In this working model, migration of precursors from DG/SVZ to the hippocampus, as induced in disease states, for example, see refs 60, may ultimately replenish this pool. Interestingly, using a nestin-GFP reporter mouse, a population of bipolar cells was identified in CA1 that did not incorporate BrdU and was negative for GFAP, S100B and Dcx. These cells were traced back to the corpus callosum alveus, a region dorsal of CA1/CA3, where neuronal stem cells have also been extracted from.63, 64
In connection with disease models, EPO treatment was associated with increased numbers of BrdU+ cells in DG and SVZ.16, 17 This fact and also the timing when BrdU+ cells were found enhanced are in good agreement with our data. Even the observation of a decrease in BrdU+ cells in neurogenic areas at week 4 as described here was similarly noted before, but understood as lacking long-term effects of EPO on adult neurogenesis.16 The conclusions drawn by others that EPO stimulates neuroproliferation, however, are not supported here. In contrast, we interpret our findings not (or only to a limited degree) as direct effects of EPO on neuroproliferation but rather as feedback signaling of differentiating neurons to proliferating progenitors. For example, the mere depletion of neuroblasts (by EPO stimulated differentiation) could release the reported GABA brake on stem cell proliferation.65, 66
In neural stem cells (E14-NSC) and hippocampal cultures (E17-HCC), we found evidence for EPO-induced differentiation/maturation rather than proliferation. These findings are a major supportive argument for the validity of our working model, and as such a central part of the present work. Although the cultures differ in their differentiation state, EPO apparently targets the same transcription factors, Sox9 and ND1. Similar findings on differentiation of secondary NSC were reported previously,17 whereas discrepant results were seen using unphysiologically high doses, for example, Chen et al.67 Importantly, miR124, known to regulate Sox952, 53 and drive neuronal differentiation at the expense of proliferation, is shown here to be stimulated by EPO and mediate its inhibitory effect on neurosphere number/diameter. Clearly, the increase in miR124 upon EPO in vivo and in vitro is small but consistent. Notably, the growing literature on microRNAs in the brain indicates that even relatively small changes in their levels can have substantial impact on cellular function.68, 69
The presently lacking information on further brain areas, for example, cortex or cerebellum, female gender, other species or older age at EPO treatment still limits the generalizability of our findings. Nevertheless, our MRI data showing hippocampal enlargement were obtained from 15-week-old male mice, that is, from animals where EPO administration had started at 11 (instead of 4) weeks of age. We note that similar volume increases of hippocampal CA1/CA3 subfields were reported recently by high-resolution MRI following EPO treatment in human patients.11 This clinical observation is in perfect agreement with our findings in healthy young mice and may point to the relevance of EPO for neurogenesis also in advanced adulthood and across species.
Exploring how EPO acts on cognition, we presumably 'tapped' into a physiological system of neuronal (re)generation from local precursors that can be pharmacologically manipulated with recombinant human EPO. One important explanation why this novel type of postnatal neurogenesis has escaped detection in the past is the astonishing finding that it occurs in absence of mitotic cell division. Thus, it cannot be captured by incorporation of BrdU, tritiated thymidine, or retroviral labeling strategies, classical tools in the field.23 The second reason for detecting this type of neurogenesis by serendipity is that we have enhanced the underlying transition of resident precursor cells to mature neurons and oligodendrocytes by EPO. If this type of postnatal neurogenesis and gliogenesis is likewise present in untreated mice, it may also occur at a low rate for continuous on-demand delivery of neurons, which cannot easily be demonstrated by absolute cell counts. First support for this hypothesis is the increase in pyramidal CA1/CA3 neurons upon long-term cognitive challenge, with and without EPO.
In conclusion, our data provide a working model for the cellular basis of so far unexplained but consistent clinical findings that EPO improves cognition in mouse and man. Specifically, we note the possibility that neurons can be generated rapidly, that is, without entering S-phase, and also outside the 'classical’ neurogenesis areas, that is, from inconspicuous local precursors that may have been overlooked. A conceptually similar scenario was proven here for oligodendrocytes, demonstrating that their adult precursors (NG2 cells)27, 29, 30 can more efficiently differentiate under EPO without proliferation. Functionality of the newly generated neurons is suggested by enhanced long-term potentiation in Schaffer collateral CA1 synapses7 and by faster learning in the touch-screen setup as shown here, and of newly generated oligodendrocytes by enhanced myelin protein expression. Given these findings, the upregulation of EPO and EPOR in the injured brain is intriguing.2, 6 But independent of its role in central nervous system regeneration, our observations add a new layer of complexity to postnatal neurogenesis.
References
Brines M, Cerami A . Emerging biological roles for erythropoietin in the nervous system. Nat Rev Neurosci 2005; 6: 484–494.
Sirén AL, Fasshauer T, Bartels C, Ehrenreich H . Therapeutic potential of erythropoietin and its structural or functional variants in the nervous system. Neurotherapeutics 2009; 6: 108–127.
Ehrenreich H, Hasselblatt M, Knerlich F, von Ahsen N, Jacob S, Sperling S et al. A hematopoietic growth factor, thrombopoietin, has a proapoptotic role in the brain. Proc Natl Acad Sci USA 2005; 102: 862–867.
Digicaylioglu M, Bichet S, Marti HH, Wenger RH, Rivas LA, Bauer C et al. Localization of specific erythropoietin binding sites in defined areas of the mouse brain. Proc Natl Acad Sci USA 1995; 92: 3717–3720.
Marti HH, Wenger RH, Rivas LA, Straumann U, Digicaylioglu M, Henn V et al. Erythropoietin gene expression in human, monkey and murine brain. Eur J Neurosci 1996; 8: 666–676.
Ott C, Martens H, Hassouna I, Oliveira B, Erck C, Zafeiriou MP et al. Widespread expression of erythropoietin receptor in brain and its induction by injury. Mol Med 2015 (in press).
Adamcio B, Sargin D, Stradomska A, Medrihan L, Gertler C, Theis F et al. Erythropoietin enhances hippocampal long-term potentiation and memory. BMC Biol 2008; 6: 37.
Miskowiak K, O'Sullivan U, Harmer CJ . Erythropoietin enhances hippocampal response during memory retrieval in humans. J Neurosci 2007; 27: 2788–2792.
Ehrenreich H, Fischer B, Norra C, Schellenberger F, Stender N, Stiefel M et al. Exploring recombinant human erythropoietin in chronic progressive multiple sclerosis. Brain 2007; 130: 2577–2588.
Wustenberg T, Begemann M, Bartels C, Gefeller O, Stawicki S, Hinze-Selch D et al. Recombinant human erythropoietin delays loss of gray matter in chronic schizophrenia. Mol Psychiatry 2011; 16: 21.
Miskowiak KW, Vinberg M, Macoveanu J, Ehrenreich H, Koster N, Inkster B et al. Effects of erythropoietin on hippocampal volume and memory in mood disorders. Biol Psychiatry 2014; 78: 270–277.
Ehrenreich H, Degner D, Meller J, Brines M, Behe M, Hasselblatt M et al. Erythropoietin: a candidate compound for neuroprotection in schizophrenia. Mol Psychiatry 2004; 9: 42–54.
Ehrenreich H, Hinze-Selch D, Stawicki S, Aust C, Knolle-Veentjer S, Wilms S et al. Improvement of cognitive functions in chronic schizophrenic patients by recombinant human erythropoietin. Mol Psychiatry 2007; 12: 206–220.
Leist M, Ghezzi P, Grasso G, Bianchi R, Villa P, Fratelli M et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science 2004; 305: 239–242.
Sargin D, El-Kordi A, Agarwal A, Muller M, Wojcik SM, Hassouna I et al. Expression of constitutively active erythropoietin receptor in pyramidal neurons of cortex and hippocampus boosts higher cognitive functions in mice. BMC Biol 2011; 9: 27.
Ransome MI, Turnley AM . Systemically delivered erythropoietin transiently enhances adult hippocampal neurogenesis. J Neurochem 2007; 102: 1953–1965.
Shingo T, Sorokan ST, Shimazaki T, Weiss S . Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J Neurosci 2001; 21: 9733–9743.
Tsai PT, Ohab JJ, Kertesz N, Groszer M, Matter C, Gao J et al. A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. J Neurosci 2006; 26: 1269–1274.
Leconte C, Bihel E, Lepelletier FX, Bouet V, Saulnier R, Petit E et al. Comparison of the effects of erythropoietin and its carbamylated derivative on behaviour and hippocampal neurogenesis in mice. Neuropharmacology 2011; 60: 354–364.
Hattangadi SM, Wong P, Zhang L, Flygare J, Lodish HF . From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood 2011; 118: 6258–6268.
Altman J, Das GD . Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol 1965; 124: 319–335.
Aimone JB, Li Y, Lee SW, Clemenson GD, Deng W, Gage FH . Regulation and function of adult neurogenesis: from genes to cognition. Physiol Rev 2014; 94: 991–1026.
Gould E . How widespread is adult neurogenesis in mammals? Nat Rev Neurosci 2007; 8: 481–488.
Kaplan MS, Hinds JW . Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science 1977; 197: 1092–1094.
Rakic P . Adult neurogenesis in mammals: an identity crisis. J Neurosci 2002; 22: 614–618.
Hughes EG, Kang SH, Fukaya M, Bergles DE . Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat Neurosci 2013; 16: 668–676.
Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE . NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 2010; 68: 668–681.
Morrens J, Van Den Broeck W, Kempermann G . Glial cells in adult neurogenesis. Glia 2012; 60: 159–174.
Nishiyama A, Komitova M, Suzuki R, Zhu X . Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci 2009; 10: 9–22.
Richardson WD, Young KM, Tripathi RB, McKenzie I . NG2-glia as multipotent neural stem cells: fact or fantasy? Neuron 2011; 70: 661–673.
Leuner B, Gould E, Shors TJ . Is there a link between adult neurogenesis and learning? Hippocampus 2006; 16: 216–224.
Deng W, Aimone JB, Gage FH . New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci 2010; 11: 339–350.
Burke SN, Barnes CA . Neural plasticity in the ageing brain. Nat Rev Neurosci 2006; 7: 30–40.
Huang W, Zhao N, Bai X, Karram K, Trotter J, Goebbels S et al. Novel NG2-CreERT2 knock-in mice demonstrate heterogeneous differentiation potential of NG2 glia during development. Glia 2014; 62: 896–913.
Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L . A global double-fluorescent Cre reporter mouse. Genesis 2007; 45: 593–605.
Breunig JJ, Arellano JI, Macklis JD, Rakic P . Everything that glitters isn't gold: a critical review of postnatal neural precursor analyses. Cell Stem Cell 2007; 1: 612–627.
Izquierdo A, Wiedholz LM, Millstein RA, Yang RJ, Bussey TJ, Saksida LM et al. Genetic and dopaminergic modulation of reversal learning in a touchscreen-based operant procedure for mice. Behav Brain Res 2006; 171: 181–188.
Hagemeyer N, Boretius S, Ott C, Von Streitberg A, Welpinghus H, Sperling S et al. Erythropoietin attenuates neurological and histological consequences of toxic demyelination in mice. Mol Med 2012; 18: 628–635.
Keuker JI, Vollmann-Honsdorf GK, Fuchs E . How to use the optical fractionator: an example based on the estimation of neurons in the hippocampal CA1 and CA3 regions of tree shrews. Brain Res Protoc 2001; 7: 211–221.
West MJ, Gundersen HJ . Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol 1990; 296: 1–22.
Saka SK, Vogts A, Krohnert K, Hillion F, Rizzoli SO, Wessels JT . Correlated optical and isotopic nanoscopy. Nat Commun 2014; 5: 3664.
Ahlenius H, Kokaia Z . Isolation and generation of neurosphere cultures from embryonic and adult mouse brain. Methods Mol Biol 2010; 633: 241–252.
Neher RA, Mitkovski M, Kirchhoff F, Neher E, Theis FJ, Zeug A . Blind source separation techniques for the decomposition of multiply labeled fluorescence images. Biophys J 2009; 96: 3791–3800.
Sargin D, Hassouna I, Sperling S, Sirén AL, Ehrenreich H . Uncoupling of neurodegeneration and gliosis in a murine model of juvenile cortical lesion. Glia 2009; 57: 693–702.
Cohen J . Statistical Power Analysis for the Behavioral Sciences, 2 (edn). Lawrence Erlbaum Associates: Hillsdale, 1988.
Leid M, Ishmael JE, Avram D, Shepherd D, Fraulob V, Dolle P . CTIP1 and CTIP2 are differentially expressed during mouse embryogenesis. Gene Expr Patterns 2004; 4: 733–739.
Fidalgo C, Conejo NM, Gonzalez-Pardo H, Arias JL . Functional interaction between the dorsal hippocampus and the striatum in visual discrimination learning. J Neurosci Res 2012; 90: 715–720.
Lee AC, Yeung LK, Barense MD . The hippocampus and visual perception. Front Hum Neurosci 2012; 6: 91.
Reynolds BA, Rietze RL . Neural stem cells and neurospheres—re-evaluating the relationship. Nat Methods 2005; 2: 333–336.
Conti L, Pollard SM, Gorba T, Reitano E, Toselli M, Biella G et al. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol 2005; 3: e283.
Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods 2015; 12: 671–678.
Cheng LC, Pastrana E, Tavazoie M, Doetsch F . miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci 2009; 12: 399–408.
Scott CE, Wynn SL, Sesay A, Cruz C, Cheung M, Gomez Gaviro MV et al. SOX9 induces and maintains neural stem cells. Nat Neurosci 2010; 13: 1181–1189.
Papagiannakopoulos T, Kosik KS . microRNA-124: micromanager of neurogenesis. Cell Stem Cell 2009; 4: 375–376.
Grabrucker A, Vaida B, Bockmann J, Boeckers TM . Synaptogenesis of hippocampal neurons in primary cell culture. Cell Tissue Res 2009; 338: 333–341.
Matteoli M, Takei K, Perin MS, Sudhof TC, De Camilli P . Exo-endocytotic recycling of synaptic vesicles in developing processes of cultured hippocampal neurons. J Cell Biol 1992; 117: 849–861.
Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C et al. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA 2000; 97: 10526–10531.
Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P et al. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res 1999; 27: 4324–4327.
Encinas JM, Michurina TV, Peunova N, Park JH, Tordo J, Peterson DA et al. Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell 2011; 8: 566–579.
Cayre M, Canoll P, Goldman JE . Cell migration in the normal and pathological postnatal mammalian brain. Prog Neurobiol 2009; 88: 41–63.
Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N et al. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell 2002; 110: 429–441.
Schmidt W, Reymann KG . Proliferating cells differentiate into neurons in the hippocampal CA1 region of gerbils after global cerebral ischemia. Neurosci Lett 2002; 334: 153–156.
Kronenberg G, Wang LP, Geraerts M, Babu H, Synowitz M, Vicens P et al. Local origin and activity-dependent generation of nestin-expressing protoplasmic astrocytes in CA1. Brain Struct Funct 2007; 212: 19–35.
Seaberg RM, van der Kooy D . Adult rodent neurogenic regions: the ventricular subependyma contains neural stem cells, but the dentate gyrus contains restricted progenitors. J Neurosci 2002; 22: 1784–1793.
Liu X, Wang Q, Haydar TF, Bordey A . Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat Neurosci 2005; 8: 1179–1187.
Kriegstein AR . GABA puts the brake on stem cells. Nat Neurosci 2005; 8: 1132–1133.
Chen ZY, Asavaritikrai P, Prchal JT, Noguchi CT . Endogenous erythropoietin signaling is required for normal neural progenitor cell proliferation. J Biol Chem 2007; 282: 25875–25883.
Banzhaf-Strathmann J, Benito E, May S, Arzberger T, Tahirovic S, Kretzschmar H et al. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer's disease. EMBO J 2014; 33: 1667–1680.
Zovoilis A, Agbemenyah HY, Agis-Balboa RC, Stilling RM, Edbauer D, Rao P et al. microRNA-34c is a novel target to treat dementias. EMBO J 2011; 30: 4299–4308.
Acknowledgements
This study was supported by the Max Planck Society, the DFG Research Center for Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB), a DFG grant to TD (Da 208/12-1), and the Interdisciplinary Center for Clinical Research (IZKF), University of Würzburg (TP D103; TP N229). The research stay of Maria Florencia Rossetti was supported by the Fundación Bunge y Born, Argentina. We thank Barbara Gado for expert technical assistance in maintenance and preparation of cell lysates of NSC cultures. The authors are indebted to Michael Frotscher, MD, PhD, ZMNH—University of Hamburg, to Gerd Kempermann, MD, CRT-Technical University of Dresden, and to Irmgard Amrein, PhD, University of Zürich, Switzerland, for critical reading of the manuscript and highly appreciated feedback and advice.
Author contributions
HE developed the study concept and design, supported in respective parts by A-LS, K-AN, EN, SOR and TD. IH and CO performed all stereological and ex vivo confocal analysis, partly guided by MM. SS conducted histological staining and, together with NH, western blot analyses. DW, MFR and ED conducted and analyzed the touch-screen trial. CO, IH, LW, ICV, KK and CH were responsible for the NanoSIMS experiment, guided and supervised by SOR. LW, with the help of KH, performed essentially all the work on E17 hippocampal cultures as well as all biochemical analyses of E14-NSC cultures. RAN, MM, AZ and EN were involved in experimental design and analysis of quadruple-stained neuronal cultures. NO performed culture work and bioassays on E14-NSC. LF together with NO stained and counted NSC cultures. SB performed MRI analysis. RAN, TD, MD, AZ, SG, K-AN and EN gave input to data analysis and interpretation. HE and K-AN wrote the manuscript. HE and A-LS had full access to all data in the study and take responsibility for the integrity of the data and the accuracy of analyses. All authors have seen and approved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
HE has submitted/holds user patents for EPO in stroke, schizophrenia and MS. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Hassouna, I., Ott, C., Wüstefeld, L. et al. Revisiting adult neurogenesis and the role of erythropoietin for neuronal and oligodendroglial differentiation in the hippocampus. Mol Psychiatry 21, 1752–1767 (2016). https://doi.org/10.1038/mp.2015.212
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2015.212
This article is cited by
-
Stable isotope labeling and ultra-high-resolution NanoSIMS imaging reveal alpha-synuclein-induced changes in neuronal metabolism in vivo
Acta Neuropathologica Communications (2023)
-
Erythropoietin regulates developmental myelination in the brain stimulating postnatal oligodendrocyte maturation
Scientific Reports (2023)
-
Introducing the brain erythropoietin circle to explain adaptive brain hardware upgrade and improved performance
Molecular Psychiatry (2022)
-
Hippocampal neurons respond to brain activity with functional hypoxia
Molecular Psychiatry (2021)
-
Erythropoietin promotes hippocampal mitochondrial function and enhances cognition in mice
Communications Biology (2021)
