
Abstract
Inflammation and altered glutamate metabolism are two pathways implicated in the pathophysiology of depression. Interestingly, these pathways may be linked given that administration of inflammatory cytokines such as interferon-α to otherwise non-depressed controls increased glutamate in the basal ganglia and dorsal anterior cingulate cortex (dACC) as measured by magnetic resonance spectroscopy (MRS). Whether increased inflammation is associated with increased glutamate among patients with major depression is unknown. Accordingly, we conducted a cross-sectional study of 50 medication-free, depressed outpatients using single-voxel MRS, to measure absolute glutamate concentrations in basal ganglia and dACC. Multivoxel chemical shift imaging (CSI) was used to explore creatine-normalized measures of other metabolites in basal ganglia. Plasma and cerebrospinal fluid (CSF) inflammatory markers were assessed along with anhedonia and psychomotor speed. Increased log plasma C-reactive protein (CRP) was significantly associated with increased log left basal ganglia glutamate controlling for age, sex, race, body mass index, smoking status and depression severity. In turn, log left basal ganglia glutamate was associated with anhedonia and psychomotor slowing measured by the finger-tapping test, simple reaction time task and the Digit Symbol Substitution Task. Plasma CRP was not associated with dACC glutamate. Plasma and CSF CRP were also associated with CSI measures of basal ganglia glutamate and the glial marker myoinositol. These data indicate that increased inflammation in major depression may lead to increased glutamate in the basal ganglia in association with glial dysfunction and suggest that therapeutic strategies targeting glutamate may be preferentially effective in depressed patients with increased inflammation as measured by CRP.
Similar content being viewed by others
Introduction
Two evolving theories regarding the development of mood disorders involve excessive activation of inflammatory pathways and alterations in glutamate metabolism.1, 2, 3 For example, increased inflammatory markers have been reliably found in the peripheral blood and cerebrospinal fluid (CSF) of patients with unipolar and bipolar depression.1, 4, 5 Moreover, administration of inflammatory stimuli including interferon (IFN)-α, typhoid vaccination or endotoxin have all been shown to lead to behavioral changes that characterize these disorders including depressed mood, anhedonia and psychomotor slowing.6, 7, 8, 9, 10, 11 Finally, inhibition of inflammatory cytokines has been shown to reduce depressed mood in several patient populations.12, 13, 14
Alterations in glutamate metabolism have also been implicated in mood disorders.3 Indeed, a number of studies using magnetic resonance spectroscopy (MRS) have found alterations in glutamate and associated metabolite levels in multiple brain regions of patients with depression, bipolar depression in particular.15, 16, 17, 18, 19, 20, 21, 22, 23 Probably the most dramatic evidence of the role of glutamate in psychopathology of mood disorders is derived from the profound and rapid response of treatment-resistant depressed patients to ketamine, an antagonist of the glutamate N-methyl-D-aspartate receptor.24, 25, 26
Regarding mechanisms that may link alterations in inflammation and glutamate metabolism in mood disorders, inflammatory cytokines have been shown to decrease the expression of glutamate transporters on relevant glial elements (for example, astrocytes) and increase astrocytic glutamate release through reverse efflux.27, 28, 29, 30, 31 Glutamate released by astrocytes has preferential access to extrasynaptic N-methyl-D-aspartate receptors, which have been shown to decrease brain-derived neurotrophic factor and increase excitotoxicity.1, 32 In addition, our previous data have demonstrated that administration of the inflammatory cytokine IFN-α increases the glutamate to creatine (Glu/Cr) ratio using MRS in the left basal ganglia and dorsal anterior cingulate cortex (dACC),33 both of which have been implicated as targets of peripherally administered inflammatory stimuli.6, 7, 9, 10, 11 Increased glutamate in these brain regions in turn correlated with depressive symptoms including anhedonia and fatigue.33, 34 Interestingly, in a recent report, pretreatment with ketamine blocked the development of depressive-like behavior in mice as a result of administration of lipopolysaccharide, a potent activator of the inflammatory response.35 Surprisingly, ketamine had no effect on lipopolysaccharide-induced inflammatory activation in the brain.
Another pathway that may contribute to increased glutamate in the context of inflammation is the impact of inflammatory cytokines on indoleamine 2,3 dioxygenase, which breaks down tryptophan into kynurenine.31, 36 Kynurenine is converted into quinolinic acid by activated microglia and macrophages in the brain.36 Quinolinic acid not only directly binds the N-methyl-D-aspartate receptor but has been shown to stimulate the release and block the reuptake of glutamate by astrocytes.37 Increases in quinolinic acid have been found to correlate with interleukin (IL)-6 in suicide attempters38 and the ratio of kynurenic acid (a putative neuroprotective metabolite of kynurenine) to quinolinic acid was found to be lower in patients with depression and correlated negatively with anhedonia in depressed subjects.39 In sum, inflammation has several points of intersection with glutamate metabolism in the brain and thus may be associated with increased glutamate in patients with major depressive disorder and increased inflammation.
We therefore endeavored to examine the hypothesis that increased inflammation in patients with major depression would be associated with increased glutamate in the left basal ganglia and dACC, and that these increases would be associated with behavioral changes including anhedonia and psychomotor slowing as previously described in IFN-α-treated patients.33 To test this hypotheses, we used MRS, a noninvasive neuroimaging technique that allows in vivo quantification of metabolites including glutamate in localized brain regions.17 Furthermore, we used the concentration of the acute phase protein, C-reactive protein (CRP) in the plasma as the primary biomarker of inflammation. Plasma CRP concentrations have been shown to be reliably elevated in depressed patients and have been associated with the development of depression,40, 41 as well as with the antidepressant response to the tumor necrosis factor antagonist infliximab.12
Materials and methods
Study subjects
Fifty subjects aged 21–65 years diagnosed with major depressive disorder or bipolar disorder, depressed type using Structured Clinical Interview for DSM-IV,42 were enrolled. Exclusion criteria included any unstable medical condition or evidence of active infection (as determined by physical examination and laboratory testing), a history of schizophrenia as determined by Structured Clinical Interview for DSM-IV, active psychotic symptoms of any type, substance abuse/dependence within the past 6 months (determined by Structured Clinical Interview for DSM-IV and urine drug testing), active suicidal ideation reflected by a score of ⩾3 on item 3 of the 17-item Hamilton Scale of Depression43 and/or a score <28 on the Mini-Mental State Examination.44 All patients were free of psychotropic medications for at least 4 weeks (8 weeks for fluoxetine) and had not taken any medications known to affect the immune system (for example, glucocorticoids, statins, angiotensin-2 inhibitors and non-steroidal anti-inflammatory agents excluding aspirin 81 mg per day) within the past 6 months (2 weeks for non-steroidal anti-inflammatory agents). Medications for other medical conditions were allowed as dictated by the patients’ treating physicians. All participants signed informed consent and the study was approved a priori by the Institutional Review Board of Emory University. Subjects were recruited from a parent study on phenotyping depressed patients with increased inflammation (ClinicalTrials.gov NCT01426997).
Study procedures
Study procedures occurred in the same order over 3 days. MRS scans were conducted on Day 1 between 1400 and 1600 h. On Day 2, blood sampling was conducted between 0800 and 1000 h (to minimize circadian variations), neuropsychiatric assessments between 1000 and 1200 h and neurocognitive testing between 1400 and 1600 h. Lumbar puncture was conducted on Day 3 between 1200 and 1600 h.
Behavioral assessments
Depression severity was measured using the Hamilton Scale of Depression and Inventory of Depressive Symptoms–Self Rating.43, 45 Age of onset of depression (years), duration of current episode (months), number of episodes and family history (yes/no) were collected using structured interview.12 Treatment history was assessed using the Antidepressant Treatment History Questionnaire, providing number of antidepressant trials in the current episode.46 Anhedonia was assessed using a subscale of the Inventory of Depressive Symptoms–Self Rating consisting of three items (reduced mood reactivity, reduction in general interest and reduced capacity for pleasure or enjoyment). This subscale has acceptable face and construct validity in measuring hedonic capacity and correlates with other more nuanced assessments of anhedonia.47
Neurocognitive assessments
Psychomotor performance was measured by the finger-tapping test, a task of pure motor function that requires patients to tap with the index finger of the dominant hand as fast as possible for 10 s intervals;48 the Reaction Time Task of the Cambridge Neuropsychological Test Automated Battery, which includes simple and five-choice reaction time segments and provides distinction between reaction time and movement latencies;49, 50 and the Digit Symbol Substitution Test of Wechsler Adult Intelligence Scale that measures psychomotor processing speed.51
Scanning protocol
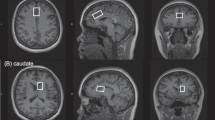
Scanning was performed on a Siemens 3-Tesla Magnetom Trio System (Siemens Medical Solutions USA, Malvern, PA, USA; VB17, 12-channel head coil).33, 34 Briefly, for image guidance and prescription of voxels of interest, axial T1 images were obtained using three-dimensional magnetization-prepared rapid gradient-echo with settings of time to repetition (TR)=2300 millseconds (ms), time to echo (TE)=3.02 ms, time following inversion pulse (TI)=1100 ms, flip angle=8o and voxel size 1 × 1 × 1 mm3. MRS was acquired using point-resolved spectroscopy with the following parameters: TR=3000 ms, TE=30 ms, sampling size=1024 complex spectral points and 128 averages. One voxel sized 17 × 30 × 17 mm3 located on the left basal ganglia and one voxel sized 20 × 30 × 10 mm3 located on the dACC (Brodmann’s Area 24) were used to obtain single-voxel 1H-MRS as previously reported.33, 34
MRS postprocessing
Postprocessing to obtain absolute glutamate concentrations was accomplished using LCModel package-Version 6-2-2b.52 The water-suppressed time-domain data were analyzed between 0.2 and 4.0 p.p.m. using the basis set provided by the vendor (http://s-provencher.com/pages/lcm-applications.shtml). Concentrations and ratios with Cramer–Rao lower bounds >20% were excluded from analysis. Single-voxel MRS-based metabolite values from the left basal ganglia of sixpatients and dACC region of five patients had to be excluded due to motion-related inferior quality spectra (Cramer–Rao lower bounds >20). T1-weighted images were segmented into gray matter, white matter and CSF compartments using FreeSurfer (https://surfer.nmr.mgh.harvard.edu/fswiki) on the whole brain T1 images. A volume of interest was generated on the T1 images, which matched the location and size of MRS voxel. Volumes of gray matter, white matter and CSF segments in this volume of interest were then generated using FreeSurfer. The absolute glutamate concentrations generated by the LCModel was corrected for CSF using the formula: C=Co × 1/(1−fCSF), where C is the corrected metabolite concentrations, Co is the metabolite concentrations generated by LCModel output and fCSF is the fraction of the CSF volume.53, 54 Investigators blinded to group assignment conducted all image analyses.
Multivoxel chemical shift imaging
To complement single-voxel assessments, multi-voxel metabolite data were acquired using chemical shift imaging (CSI) in exploratory analyses. First, the axial brain image was placed on a grid measuring 164.8 × 164.8 mm subdivided into 16 × 16 voxels. Within this large grid, a rectangular region comprising 8 × 10 voxels, which included the primary regions of interest (subcortical regions), measuring 82.4 × 103mm was selected for CSI data acquisition. Individual voxels were localized during the scan and individual spectra were acquired from each voxel. The primary regions of interest in this analysis were the basal ganglia regions, and therefore a 2 × 3 voxel subregion spanning parts of the right and left caudate, putamen and pallidum, and the intervening white matter (internal capsule) were chosen for study. The scanning parameters included a double spin-echo point-resolved spectroscopy sequence (TR=1590 ms, TE=30 ms, 1024 complex spectral points and 6 averages). Mean data from 6 acquisitions of all 80 voxels (10.3 × 10.3 × 15 mm3 each) were averaged post hoc during the postprocessing using the LCModel. The total acquisition time was as follows: water-suppressed metabolite scan: 11 min 13 s and non-water suppressed scan: 4 min 3 s. Metabolite by creatine+phosphocreatine (Cr) ratios were used for these analyses, because of the methodological challenges inherent to calculating absolute metabolite values using CSI data including B0 inhomogeneity and tissue variations within the large number of CSI voxel spectra.55
Immune assessments
Blood was obtained in EDTA tubes through an indwelling catheter after 30 min of rest and immediately centrifuged (1000 g for 15 min at 4 °C). Plasma was collected and stored at −80 °C until batched assay. CSF samples were aliquoted into 1 ml vials and immediately frozen at −80 °C until batched assay. CRP was measured by the immunoturbidometric method, using the Beckman AU 480 chemistry analyzer and the Ultra WR CRP reagent kit (Sekisui Diagnostics, LLC, Lexington, MA, USA).12 Concentrations of IL-1β, IL-6 and tumor necrosis factor, and their soluble receptors were assessed in duplicate using multiplex bead-based assays (R&D Systems, Minneapolis, MN, USA) and analyzed on a MAGPIX CCD imaging system (Luminex Corporation, Austin, TX, USA). Mean inter- and intra-assay coefficients of variation were reliably ⩽10%.
Statistical analysis
Linear regression analyses were used to determine the association between independent and dependent variables, controlling for relevant covariates. The primary analyses examined the relationship between plasma and CSF CRP, as well as other inflammatory markers and absolute glutamate concentrations in the left basal ganglia and dACC as determined by single-voxel MRS. The relationship between absolute glutamate concentrations and anhedonia and psychomotor speed were also considered in the primary analyses. The association of CRP and other inflammatory markers with CSI metabolite ratios were considered in exploratory analyses. Age, sex, body mass index, smoking status (yes/no) and depression severity as measured by the Hamilton Scale of Depression were included as covariates in all primary analyses. These covariates have all been shown to be related to inflammation in previous reports and in our own studies as well.34, 56, 57, 58 In exploratory analyses, body mass index was not included in the models because of its strong correlation with both the plasma and CSF CRP (r=0.64, P<0.001 and r=0.57, P<0.001, respectively). To examine stepwise relationships between inflammation and absolute glutamate concentrations, depressed patients were also stratified into low (CRP<1 mg l−1), medium (CRP=1–3 mg l−1) and high (CRP>3 mg l−1) inflammation groups based on recommended guidelines.59 To examine the main effects of inflammatory status (low, medium and high) on absolute glutamate, one-way analysis of variance (ANOVA) was used followed by post-hoc testing using Bonferroni correction. One-way ANOVA and Fisher’s exact tests were used to determine differences in sociodemographic and clinical variables among the inflammation groups. For analysis of CSI data, to control for multiple comparisons, a non-biased omnibus multivariate ANOVA was used to examine the relationship between CRP and other inflammatory markers, and all relevant metabolites (normalized to Cr) including n-acetyl aspartate (NAA/Cr), myo-inositol (mI/Cr), choline (Cho/Cr) and glutamate (Glu/Cr) obtained concurrently from the left and right basal ganglia regions. Before this analysis, it was confirmed that Cr concentrations were not significantly correlated with CRP. In the case of a significant overall multivariate ANOVA, a false discovery rate (FDR) of <0.05 was employed to indentify individual metabolites that were significantly associated with inflammatory markers.60 Non-normally distributed data were natural log-transformed for all analyses. Statistical computations were performed using IBM-SPSS (International Business Machines, New York, NY, USA). Power assessments were measured using G*Power.61
Results
Patient characteristics
The sociodemographic and clinical characteristics of the study sample are indicated in Table 1. Table 2 shows absolute concentrations of glutamate in the regions of interest and concentrations of cytokines in the plasma and CSF.
Relationship between plasma CRP and absolute glutamate concentrations as measured by single-voxel MRS
Log CRP was the only significant predictor of log left basal ganglia glutamate after exclusion of covariates (β=0.36, t=2.57, P=0.01, Cohen’s f2=0.33, power=0.95; Figure 1). One-way ANOVA also revealed a significant main effect of CRP group on log left basal ganglia glutamate (F[2,43]=4.42, P=0.02, d=0.90, power=0.80), with the high CRP group (>3 mg l−1, n=14) exhibiting significantly greater log glutamate concentrations than the low CRP group (<1 mg l−1, n=22; mean difference=0.062, SE=0.021, Pcorr=0.02, d=1.15, power=0.95; Figure 1). This relationship remained significant after controlling for covariates including body mass index, which was the only covariate which differed among CRP groups (high CRP group: 38.7±7.1; medium CRP group: 32.6±7.2; low CRP group: 26.5±4.3; F[2,49]=17.7, P<0.001). Although log plasma CRP was highly correlated with log CSF CRP (r=0.87, P<0.001, d=3.53), log CSF CRP did not predict log left basal ganglia glutamate concentrations (β=0.29, t=1.8, P=0.085, Cohen’s f2=0.09, power=0.49), albeit the analysis was underpowered. No significant associations were found between log plasma or log CSF CRP and log absolute glutamate concentrations in the dACC using linear regression or ANOVA (P=NS). Therefore, the dACC was not considered in further analyses.

Association between plasma C-reactive protein (CRP) and the left basal ganglia glutamate in depressed patients. Plasma CRP was positively associated with the left basal ganglia absolute glutamate in both a linear (a) and step-wise manner (b), in medication-free patients with major depressive disorder. (a) Increased log plasma CRP correlated and log left basal ganglia absolute glutamate concentrations in depressed subjects after controlling for age, sex, race, smoking status, body mass index and 17-item Hamilton Depression Rating Scale scores (β=0.36, t=2.57, P=0.014). (b) Group-wise comparison of log left basal ganglia absolute glutamate concentrations among patients grouped by CRP revealed a significant main effect of group (F[2,43]=4.42, P=0.018). *P<0.025.
Relationship between other plasma and CSF inflammatory markers and absolute glutamate concentrations in the left basal ganglia as determined by single-voxel MRS
Log plasma concentrations of the chemokine monocyte chemoattractant factor-1 was the only inflammatory marker that was significantly correlated with the left basal ganglia glutamate controlling for covariates (β=0.33, t=2.39, P=0.022).
Relationship of the left basal ganglia absolute glutamate concentrations with anhedonia and psychomotor speed
After controlling for covariates, the left basal ganglia glutamate was positively associated with anhedonia subscale scores (β=0.42, t=3.03, P=0.004, Cohen’s f2=0.21, power=0.82, predictors=8). To correct for multiple comparisons between glutamate and the measures of psychomotor speed, simple correlation coefficients between log left basal glutamate and neurocognitive assessments were calculated using an FDR <0.05. Three measures, that is, mean finger-tapping frequency in the dominant hand (r=−0.42, p(FDRcorr)=−0.032), simple reaction time (ms; r=0.32, p(FDRcorr)=0.048) and Digit Symbol Substitution Test performance (r=−0.36, p(FDRcorr)=0.048), survived FDR correction and thus were considered in subsequent analyses. Log left basal ganglia glutamate positively predicted a simple reaction time (β=0.32, t=2.18, P=0.035, Cohen’s f2=0.11, power=0.66, predictors=8; Figure 2) and negatively predicted mean frequency of taps on the finger-tapping test and performance on the Digit Symbol Substitution Test (β=−0.38, t=2.95, P=0.005, Cohen’s f2=0.32, power=0.90 and β=−0.36, t=2.54, P=0.01, Cohen’s f2=0.15, power=0.74, predictors=8, respectively; Figure 2).

Correlations between the left basal ganglia glutamate and anhedonia and psychomotor speed in depressed patients. (a) Logged values of absolute glutamate concentrations in the left basal ganglia were correlated with the IDS-SR–Anhedonia subscale (β=0.42, t=3.03, P=0.004). (b) Log absolute basal ganglia glutamate concentrations were correlated with finger-tapping frequency on the FTT (β=−0.40, t=−2.96, P=0.005). (c) Log absolute basal ganglia glutamate concentrations were correlated with log simple reaction time assessed using the CANTAB (β=0.35, t=2.44, P=0.019). (d) Log absolute basal ganglia glutamate concentrations were correlated with performance on the DSST (β=−0.36, t=−2.57, P=0.01). Statistical values controlling for covariates including age, sex, race, body mass index, smoking status, Hamilton Scale of Depression scores and plasma CRP are indicated in parentheses. CANTAB, Cambridge automated neuropsychological test battery; DSST, digit symbol substitution test; FTT, finger-tapping test; IDS-SR, inventory for depressive symptoms-self rated.
Relationship between plasma CRP and other metabolites in basal ganglia as measured by CSI
We conducted a non-biased exploratory analysis of all metabolites acquired simultaneously from both basal ganglia using CSI. An omnibus analysis using multivariate ANOVA was employed to test the association between log plasma CRP concentrations and log-transformed values of MRS metabolites after adjusting for covariates (see Subjects and methods). The overall model was significant (Roy’s largest root (F[8,35]=2.66, P=0.02, partial η2=0.38, power=0.86). Three metabolites, that is, the left basal ganglia Glu/Cr (F[6,48]=3.22, p(FDRcorr)=0.03, partial η2=0.35 power=0.90), the left basal ganglia mI/Cr (F[7,48]=3.4, p(FDRcorr)<0.02, partial η2=0.37, power=0.93) and the right basal ganglia mI/Cr (F[7,49]=4.36, p(FDRcorr)<0.008, partial η2=0.43, power=0.97), remained significant after correcting for FDR (<0.05) and were considered in subsequent analyses. Log left basal ganglia Glu/Cr was significantly associated with both log plasma (β=0.32, t=2.55, P=0.01) and log CSF CRP (β=0.29, t=2.07, P=0.04), validating the association of glutamate with plasma CRP using single-voxel MRS. Log left basal ganglia mI/Cr was also significantly associated with both log plasma (β=0.29, t=2.37, P=0.02) and log CSF CRP (β=0.32, t=2.40, P=0.02) as was log right basal ganglia mI/Cr (log plasma CRP: β=0.42, t=3.68, P=0.001 and log CSF CRP: β=0.40, t=3.22, P=0.003).
Relationship between other inflammatory markers and glutamate and myoinositol as measured by CSI
FDR-corrected independent regression analyses were run to explore the association between metabolite ratios obtained using CSI and plasma, and CSF immune markers other than CRP. Both log CSF IL-1β and log plasma IL-1receptor antagonist predicted log left basal ganglia Glu/Cr (β=0.31, t=2.27, P=0.03 and β=0.35, t=2.80, P=0.008, respectively). Similarly, log plasma IL-1receptor antagonist positively predicted log mI/Cr in both the left and right basal ganglia (β=0.24, t=2.04, P=0.05 and β=0.23, P=2.05, P=0.05, respectively), whereas log plasma tumor necrosis factor receptor type 2 predicted log right basal ganglia mI/Cr (β=0.30, t=2.71, P=0.01). Log plasma concentrations of the anti-inflammatory cytokine IL-10 were negatively associated with log right basal ganglia mI/Cr (β=−0.29, t=−2.53, P=0.02). Finally, in univariate analyses, log left and right basal ganglia mI/Cr was significantly associated with the left and right total basal ganglia volume (r=−0.45, N=50, P=0.001 and r=−0.38, N=50, P<0.01, respectively) as measured by Freesurfer software including the caudate, putamen and pallidum. After controlling for relevant covariates, these associations were no longer significant. No relationship was found between log left or right basal ganglia Glu/Cr and the left or right total basal ganglia volume.
Discussion
The data support a relationship between inflammation and glutamate metabolism in patients with major depression as manifested by a linear and stepwise association between plasma CRP and absolute concentrations of glutamate in the left basal ganglia as measured by MRS. These findings are consistent with previous work by our group demonstrating that administration of IFN-α is associated with increased left basal ganglia glutamate in otherwise non-depressed subjects with hepatitis C.33, 34 Consistent with our findings with IFN-α, we also found that changes in glutamate concentrations were in turn associated with anhedonia and psychomotor slowing.33, 34 Taken together, these data support the notion that inflammation may not only serve as a biomarker of altered glutamate metabolism in depression and other psychiatric disorders, but also may represent a pathophysiologic pathway by which inflammation has an impact on the brain to influence behavior.
We have previously demonstrated that administration of IFN-α increased glutamate in the left basal ganglia and the dACC.33, 34 However, in our previous study, we measured the Glu/Cr ratio. In the current study, we measured absolute glutamate concentrations, making it difficult to compare changes in glutamate as a function of inflammation across studies. Nevertheless, using our CSI measures of Glu/Cr in the left basal ganglia from the current study, we were able to compare results with those from IFN-α-treated patients. Interestingly, our low inflammation depressed group exhibited a Glu/Cr ratio almost indistinguishable from that seen in our control subjects in the IFN-α study (1.06 s.d. 0.09 versus 0.95 s.d. 0.23, t=0.82, P=0.41). Moreover, the absolute magnitude of the difference between the high and low inflammation depressed groups was roughly half of that observed following IFN-α, a powerfully potent inflammatory stimulus (0.08 s.e. 0.03 versus 0.17 s.e. 0.08, t=5.73, P=0.001). The comparison between these studies and their results suggests that there is good consistency in our measures of glutamate across study populations and across time, and that the impact of increased inflammation appears to be somewhat dose dependent regardless of whether the source is exogenous or endogenous. Moreover, as noted above, our findings in both studies, examining a host of metabolites, were largely significant for glutamate. These findings suggest that the effects of inflammation may be relatively exclusive to glutamate, albeit in the current study we also found evidence of increased myoinositol. In both studies we also found associations between inflammation-related changes in glutamate to be related to behaviors suggestive of basal ganglia dysfunction. These data are compatible with a growing literature, suggesting that a primary target of cytokines in the brain is the basal ganglia.6, 7, 9, 10, 11 Finally, in both studies we found the left-lateralized effects of inflammation consistent with the left-lateralized microstructural alterations in the striatal region of IFN-α-treated patients as measured by quantitative magnetization transfer imaging.62
Consistent with the regulation of extracellular glutamate concentrations by astrocytes,63 we also found that myoinositol in the basal ganglia was associated with increased inflammatory markers. Myoinositol is believed to be a glial marker of astrocytes64, 65 and its positive relationship with inflammation may be evidence of inflammation-induced astrocytic activation or dysfunction.66 Indeed, recent data have provided evidence of astrocytic dysfunction in postmortem samples of depressed subjects.67 Thus, further studies examining the relationship among glutamate, myoinositol and inflammation in depression are warranted. In addition, the association between plasma monocyte chemoattractant factor-1 and glutamate lends support to preclinical evidence that invading macrophages recruited into the brain during activated inflammatory states (including depression) might also contribute to increased glutamate.68
A recent meta-analysis of glutamate concentrations in cortical regions of depressed patients versus controls revealed considerable heterogeneity in the findings.22 Given our results, inflammation may serve as an important between-subjects factor that may account for a significant amount of the variance in glutamate concentrations, especially as it relates to the basal ganglia. Thus, these data indicate that inflammatory biomarkers may identify patients with altered glutamate metabolism who might be ideal candidates for glutamate-targeted therapies alone or in combination with anti-inflammatory treatments.
There are several strengths and limitations of the study. All patients were medication free and the sample was diverse regarding sex and race. In addition, study procedures were standardized to limit circadian variations and ordering effects. However, the study did not contain a healthy control group and whether glutamate concentrations in patients with high inflammation are greater than healthy controls cannot be determined. Nevertheless, as noted above, our low inflammation group had glutamate concentrations similar to the control group in our previous study on IFN-α. It should also be noted that MRS measurements of glutamate do not differentiate intracellular versus extracellular glutamate, albeit the majority of glutamate is intracellular. However, a relationship between MRS-assessed glutamate and a transcranial magnetic stimulation-based measure of global cortical excitability has been documented, suggesting that MRS measurements of glutamate reflect neural glutamatergic activity.69 Relative to our assessments of behavior, we used a subscale of the Inventory of Depressive Symptoms–Self Rating to measure anhedonia. Although this subscale has shown good correlations with other scales of anhedonia,70 the study would have benefitted from more objective measures of motivation including tasks of effort-based motivation, which we and others have shown to be altered by inflammatory stimuli.71, 72 In addition, as we recruited exclusively medication-free subjects for this study, our sample was less likely to include patients who had multiple antidepressant trials or were treatment resistant. Finally, we did not find any changes in dACC glutamate in depressed patients as a function of inflammation. The lack of changes in dACC glutamate is in contradistinction to our previous findings with IFN-α. One explanation for this discrepancy is that IFN-α-induced increases in dACC glutamate were associated with IFN-α-induced development of depression. However, in the current study, all patients were depressed, thereby obviating any indirect effects of inflammation on the development of depression, which may have in turn contributed to the effects of IFN-α on dACC glutamate.
References
Miller AH, Maletic V, Raison CL . Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 2009; 65: 732–741.
Sanacora G, Banasr M . From pathophysiology to novel antidepressant drugs: glial contributions to the pathology and treatment of mood disorders. Biol Psychiatry 2013; 73: 1172–1179.
Sanacora G, Treccani G, Popoli M . Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 2012; 62: 63–77.
Stertz L, Magalhaes PV, Kapczinski F . Is bipolar disorder an inflammatory condition? The relevance of microglial activation. Curr Opin Psychiatry 2013; 26: 19–26.
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK et al. A meta-analysis of cytokines in major depression. Biol Psychiatry 2010; 67: 446–457.
Capuron L, Pagnoni G, Drake D, Woolwine B, Spivey J, Crowe R et al. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry 2012; 69: 1044–1053.
Brydon L, Harrison NA, Walker C, Steptoe A, Critchley HD . Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol Psychiatry 2008; 63: 1022–1029.
Raison CL, Borisov AS, Woolwine BJ, Massung B, Vogt G, Miller AH . Interferon-alpha effects on diurnal hypothalamic-pituitary-adrenal axis activity: relationship with proinflammatory cytokines and behavior. Mol Psychiatry 2010; 15: 535–547.
Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR . Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry 2010; 68: 748–754.
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD . Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry 2009; 66: 407–414.
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Dolan RJ et al. Neural origins of human sickness in interoceptive responses to inflammation. Biol Psychiatry 2009; 66: 415–422.
Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry 2013; 70: 31–41.
Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet 2006; 367: 29–35.
Monk JP, Phillips G, Waite R, Kuhn J, Schaaf LJ, Otterson GA et al. Assessment of tumor necrosis factor alpha blockade as an intervention to improve tolerability of dose-intensive chemotherapy in cancer patients. J Clin Oncol 2006; 24: 1852–1859.
Frye MA, Watzl J, Banakar S, O'Neill J, Mintz J, Davanzo P et al. Increased anterior cingulate/medial prefrontal cortical glutamate and creatine in bipolar depression. Neuropsychopharmacology 2007; 32: 2490–2499.
Xu J, Dydak U, Harezlak J, Nixon J, Dzemidzic M, Gunn AD et al. Neurochemical abnormalities in unmedicated bipolar depression and mania: a 2D 1H MRS investigation. Psychiatry Res 2013; 213: 235–241.
Yüksel C, Öngür D . Magnetic resonance spectroscopy studies of glutamate-related abnormalities in mood disorders. Biol Psychiatry 2010; 68: 785–794.
Yoon SJ, Lyoo IK, Haws C, Kim TS, Cohen BM, Renshaw PF . Decreased glutamate/glutamine levels may mediate cytidine's efficacy in treating bipolar depression: a longitudinal proton magnetic resonance spectroscopy study. Neuropsychopharmacology 2009; 34: 1810–1818.
Hashimoto K, Sawa A, Iyo M . Increased levels of glutamate in brains from patients with mood disorders. Biol Psychiatry 2007; 62: 1310–1316.
Strawn JR, Chu WJ, Whitsel RM, Weber WA, Norris MM, Adler CM et al. A pilot study of anterior cingulate cortex neurochemistry in adolescents with generalized anxiety disorder. Neuropsychobiology 2013; 67: 224–229.
Castillo M, Kwock L, Courvoisie H, Hooper SR . Proton MR spectroscopy in children with bipolar affective disorder: preliminary observations. AJNR Am J Neuroradiol 2000; 21: 832–838.
Arnone D, Mumuni AN, Jauhar S, Condon B, Cavanagh J . Indirect evidence of selective glial involvement in glutamate-based mechanisms of mood regulation in depression: Meta-analysis of absolute prefrontal neuro-metabolic concentrations. Eur Neuropsychopharmacol 2015; 25: 1109–1117.
Ajilore O, Haroon E, Kumaran S, Darwin C, Binesh N, Mintz J et al. Measurement of brain metabolites in patients with type 2 diabetes and major depression using proton magnetic resonance spectroscopy. Neuropsychopharmacology 2007; 32: 1224–1231.
Duman RS, Li N, Liu RJ, Duric V, Aghajanian G . Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology 2012; 62: 35–41.
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 2000; 47: 351–354.
aan het Rot M, Collins KA, Murrough JW, Perez AM, Reich DL, Charney DS et al. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry 2010; 67: 139–145.
Tilleux S, Hermans E . Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res 2007; 85: 2059–2070.
Ida T, Hara M, Nakamura Y, Kozaki S, Tsunoda S, Ihara H . Cytokine-induced enhancement of calcium-dependent glutamate release from astrocytes mediated by nitric oxide. Neurosci Lett 2008; 432: 232–236.
Matute C, Domercq M, Sanchez-Gomez MV . Glutamate-mediated glial injury: mechanisms and clinical importance. Glia 2006; 53: 212–224.
Malarkey E, Parpura V . Mechanisms of glutamate release from astrocytes. Neurochem Int 2008; 52: 142–154.
Dantzer R, Walker A . Is there a role for glutamate-mediated excitotoxicity in inflammation-induced depression? J Neural Transm (Vienna) 2014; 121: 925–932.
Hardingham GE, Bading H . Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 2010; 11: 682–696.
Haroon E, Woolwine BJ, Chen X, Pace TW, Parekh S, Spivey JR et al. IFN-alpha-induced cortical and subcortical glutamate changes assessed by magnetic resonance spectroscopy. Neuropsychopharmacology 2014; 39: 1777–1785.
Haroon E, Felger JC, Woolwine BJ, Chen X, Parekh S, Spivey JR et al. Age-related increases in basal ganglia glutamate are associated with TNF, reduced motivation and decreased psychomotor speed during IFN-alpha treatment: preliminary findings. Brain Behav Immun 2015; 46: 17–22.
Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B et al. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6 J mice. Neuropsychopharmacology 2013; 38: 1609–1616.
Schwarcz R . The kynurenine pathway of tryptophan degradation as a drug target. Curr Opin Pharmacol 2004; 4: 12–17.
Tavares RG, Tasca CI, Santos CE, Alves LB, Porciuncula LO, Emanuelli T et al. Quinolinic acid stimulates synaptosomal glutamate release and inhibits glutamate uptake into astrocytes. Neurochem Int 2002; 40: 621–627.
Erhardt S, Lim CK, Linderholm KR, Janelidze S, Lindqvist D, Samuelsson M et al. Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology 2013; 38: 743–752.
Savitz J, Drevets WC, Smith CM, Victor TA, Wurfel BE, Bellgowan PS et al. Putative neuroprotective and neurotoxic kynurenine pathway metabolites are associated with hippocampal and amygdalar volumes in subjects with major depressive disorder. Neuropsychopharmacology 2015; 40: 463–471.
Gimeno D, Kivimaki M, Brunner EJ, Elovainio M, De Vogli R, Steptoe A et al. Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychol Med 2009; 39: 413–423.
Howren MB, Lamkin DM, Suls J . Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med 2009; 71: 171–186.
First MB, Spitzer RL, Gibbon M, JBW Williams . Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version. (SCID-IV). Biometrics Research, New York State Psychiatric Institute: New York, 2002.
Hamilton M . A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23: 56–62.
Folstein MF, Folstein SE, McHugh PR . “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12: 189–198.
Trivedi MH, Rush AJ, Ibrahim HM, Carmody TJ, Biggs MM, Suppes T et al. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med 2004; 34: 73–82.
Chandler GM, Iosifescu DV, Pollack MH, Targum SD, Fava M . RESEARCH: validation of the Massachusetts General Hospital Antidepressant Treatment History Questionnaire (ATRQ). CNS Neurosci Ther 2010; 16: 322–325.
Ameli R, Luckenbaugh DA, Gould NF, Holmes MK, Lally N, Ballard ED et al. SHAPS-C: the Snaith-Hamilton pleasure scale modified for clinician administration. PeerJ 2014; 2: e429.
Collyer CE, Broadbent HA, Church RM . Categorical time production: evidence for discrete timing in motor control. Percept Psychophys 1992; 51: 134–144.
Majer M, Welberg LA, Capuron L, Pagnoni G, Raison CL, Miller AH . IFN-alpha-induced motor slowing is associated with increased depression and fatigue in patients with chronic hepatitis C. Brain Behav Immun 2008; 22: 870–880.
Sahakian BJ, Owen AM . Computerized assessment in neuropsychiatry using CANTAB: discussion paper. J R Soc Med 1992; 85: 399–402.
Wechsler D, Coalson D, Raiford S . Wechsler Adult Intelligence Test Fourth Edition Technical and Interpretative Manual Pearson: San Antonio, 2008.
Provencher SW . Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magnetic Resonance in Medicine 1993; 30: 672–679.
Gussew A, Erdtel M, Hiepe P, Rzanny R, Reichenbach JR . Absolute quantitation of brain metabolites with respect to heterogeneous tissue compositions in (1)H-MR spectroscopic volumes. Magma (New York, NY) 2012; 25: 321–333.
Hall MH, Jensen JE, Du F, Smoller JW, O'Connor L, Spencer KM et al. Frontal P3 event-related potential is related to brain glutamine/glutamate ratio measured in vivo. Neuroimage 2015; 111: 186–191.
Posse S, Otazo R, Dager SR, Alger J . MR spectroscopic imaging: principles and recent advances. J Magn Reson Imaging 2013; 37: 1301–1325.
O'Connor MF, Irwin MR . Links between behavioral factors and inflammation. Clin Pharmacol Ther 2010; 87: 479–482.
Valkanova V, Ebmeier KP, Allan CL . CRP, IL-6 and depression: a systematic review and meta-analysis of longitudinal studies. J Affect Disord 2013; 150: 736–744.
Liu Y, Al-Sayegh H, Jabrah R, Wang W, Yan F, Zhang J . Association between C-reactive protein and depression: modulated by gender and mediated by body weight. Psychiatry Res 2014; 219: 103–108.
Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO 3rd, Criqui M et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003; 107: 499–511.
Hochberg Y, Benjamini Y . More powerful procedures for multiple significance testing. Stat Med 1990; 9: 811–818.
Faul F, Erdfelder E, Buchner A, Lang A . Statistical power analyses using G*Power 3.1: tests for correlation and regression analyses. Behav Res Methods 2009; 41: 1149–1160.
Dowell NG, Cooper EA, Tibble J, Voon V, Critchley HD, Cercignani M et al. Acute changes in striatal microstructure predict the development of interferon-alpha induced fatigue. Biol Psychiatry 2015; pii: S0006-3223: 00464–00473.
Schousboe A, Westergaard N, Sonnewald U, Petersen SB, Huang R, Peng L et al. Glutamate and glutamine metabolism and compartmentation in astrocytes. Dev Neurosci 1993; 15: 359–366.
Brand A, Richter-Landsberg C, Leibfritz D, Multinuclear NMR . studies on the energy metabolism of glial and neuronal cells. Dev Neurosci 1993; 15: 289–298.
Chang L, Munsaka SM, Kraft-Terry S, Ernst T . Magnetic resonance spectroscopy to assess neuroinflammation and neuropathic pain. J Neuroimmune Pharmacol 2013; 8: 576–593.
Muller N, Schwarz MJ . The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry 2007; 12: 988–1000.
Nagy C, Suderman M, Yang J, Szyf M, Mechawar N, Ernst C et al. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol Psychiatry 2014; 20: 320–328.
Piani D, Frei K, Do KQ, Cuenod M, Fontana A . Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci Lett 1991; 133: 159–162.
Stagg CJ, Bestmann S, Constantinescu AO, Moreno LM, Allman C, Mekle R et al. Relationship between physiological measures of excitability and levels of glutamate and GABA in the human motor cortex. J Physiol 2011; 589: 5845–5855.
Iwata M, Ota KT, Duman RS . The inflammasome: pathways linking psychological stress, depression, and systemic illnesses. Brain Behav Immun 2013; 31: 105–114.
Felger JC, Mun J, Kimmel HL, Nye JA, Drake DF, Hernandez CR et al. Chronic interferon-alpha decreases dopamine 2 receptor binding and striatal dopamine release in association with anhedonia-like behavior in nonhuman primates. Neuropsychopharmacology 2013; 38: 2179–2187.
Vichaya EG, Hunt SC, Dantzer R . Lipopolysaccharide reduces incentive motivation while boosting preference for high reward in mice. Neuropsychopharmacology 2014; 39: 2884–2890.
Acknowledgements
This work was funded in part by grants from the National Institute of Mental Health K23MH091254 (EH), KL2TR000455 (JCF), Young Investigator Award from The Brain & Behavior Research Foundation (JCF and EH) and R01MH08760 (AHM). We acknowledge the support of the BITC-CSI core of the School of Medicine at Emory University, ACTSI-CRN funded by National Center for Advancing Translational Sciences-CTSA Award (UL1TR000454) and Training Support from Advanced Research Institute for Geriatric Mental Health funded by the National Institute of Mental Heath R25MH068502 (EH).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
PowerPoint slides
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Haroon, E., Fleischer, C., Felger, J. et al. Conceptual convergence: increased inflammation is associated with increased basal ganglia glutamate in patients with major depression. Mol Psychiatry 21, 1351–1357 (2016). https://doi.org/10.1038/mp.2015.206
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2015.206
This article is cited by
-
Involvement of kynurenine pathway between inflammation and glutamate in the underlying etiopathology of CUMS-induced depression mouse model
BMC Neuroscience (2022)
-
Metabolomic biomarkers related to non-suicidal self-injury in patients with bipolar disorder
BMC Psychiatry (2022)
-
The role of the immune system in posttraumatic stress disorder
Translational Psychiatry (2022)
-
Immune targets for therapeutic development in depression: towards precision medicine
Nature Reviews Drug Discovery (2022)
-
The kynurenine pathway and bipolar disorder: intersection of the monoaminergic and glutamatergic systems and immune response
Molecular Psychiatry (2021)
