
Abstract
Central nervous glycogen synthase kinase 3β (GSK3β) is implicated in a number of neuropsychiatric diseases, such as bipolar disorder, depression, schizophrenia, fragile X syndrome or anxiety disorder. Many drugs employed to treat these conditions inhibit GSK3β either directly or indirectly. We studied how conditional knockout of GSK3β affected structural synaptic plasticity. Deletion of the GSK3β gene in a subset of cortical and hippocampal neurons in adult mice led to reduced spine density. In vivo imaging revealed that this was caused by a loss of persistent spines, whereas stabilization of newly formed spines was reduced. In electrophysiological recordings, these structural alterations correlated with a considerable drop in the frequency and amplitude of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor-dependent miniature excitatory postsynaptic currents. Expression of constitutively active β-catenin caused reduction in spine density and electrophysiological alterations similar to GSK3β knockout, suggesting that the effects of GSK3β knockout were mediated by the accumulation of β-catenin. In summary, changes of dendritic spines, both in quantity and in morphology, are correlates of experience-dependent synaptic plasticity; thus, these results may help explain the mechanism of action of psychotropic drugs inhibiting GSK3β.
Similar content being viewed by others


Introduction
Wnt signaling regulates several important facets of central nervous system development, including the proliferation of neuronal precursors, differentiation and integration of juvenile neurons into developing networks and neuronal polarity.1,2 In the adult nervous system, the same molecular mechanisms also fine-tune synaptic plasticity,3,4 and thereby influence cognitive functions and behavior. One of the main effectors in Wnt signaling is β-catenin, which promotes the transcription of Wnt target genes. The enzyme glycogen synthase kinase 3β (GSK3β) is a potent inhibitor of Wnt signaling as it phosphorylates β-catenin, leading to its degradation. Inhibition of GSK3β activity itself by various enzymes is a critical step upon activation of the Wnt signaling cascade and its downstream target genes.5 In addition to β-catenin, GSK3β phosphorylates several other substrates, including microtubule-associated protein MAP 1B and tau protein, which are implicated in synaptogenesis and stabilization of neurites.6 Notably, many psychotropic drugs with diverse mechanisms of action, such as antipsychotics, selective serotonin uptake inhibitors and the mood stabilizer lithium, are known to inhibit GSK3β indirectly, or—as in the case of lithium—directly, which contributes to their psychotropic effects.7 At the level of glutamatergic synapses, inhibition of GSK3β suppresses the induction of NMDA receptor-dependent long-term depression,8,9 a recognized physiological correlate of synaptic plasticity and memory.10 However, it is unknown whether inhibition of GSK3β also has consequences at the level of structural synaptic plasticity, which typically is associated with alterations in the number and turnover rate of dendritic spines. Structural changes of dendritic spines, which correspond to glutamatergic postsynapses, are prominent during experience-dependent learning, where increases in spine turnover correlate with the ability to learn new tasks.11 Decreased turnover of spines, on the other hand, is associated with neuropsychiatric diseases. A prominent example is fragile X syndrome, a disorder characterized by mental retardation and an increased number of misshaped dendritic spines.12 Neurodegenerative diseases, such as Alzheimer’s disease, in contrast are associated with a reduced number of dendritic spines,12 whereas psychiatric diseases such as schizophrenia or depression are associated with alterations in either direction, occurring in specific regions of the brain.13
To study how the loss of GSK3β affects structural synaptic plasticity in the adult central nervous system, we generated mice with inducible knockout of GSK3β in a fluorescently labeled subset of neurons and analyzed spine numbers and dynamics as well as miniature postsynaptic currents.
Materials and methods
Transgenic mice
SlickV mice, which coexpress a drug-inducible form of Cre recombinase and the fluorescent protein YFP,14 were crossed with floxed GSK3β animals homozygous for GSK3βLoxP/LoxP15 to yield SlickV × GSK3βLoxP/LoxP mice, in which GSK3β knockout can be induced in a fluorescently labelled subset of neurons upon tamoxifen administration (cf. Supplementary Methods; Figure 1a). This was confirmed immunohistochemically in all mice analyzed (n=24). Two-month-old SlickV × GSK3βLoxP/LoxP mice were treated with tamoxifen for 5 days, which resulted in almost complete loss of GSK3β immunoreactivity in YFP-positive neurons (Figure 1d). SlickV × GSK3βLoxP/LoxP mice were used in the experiments, whereas SlickV × GSK3β+/+ animals, which were offsprings of the same founder animals, but not littermates, served as controls. Spine densities in SlickV × GSK3βLoxP/LoxP mice were indistinguishable from those in SlickV × GSK3β+/+ mice before tamoxifen administration (P>0.7, t-test, n=4; Figure 2b, ‘control’).

Knockout of GSK3β and β-catenin in a sparse subset of hippocampal and cortical neurons. (a) Breeding scheme to obtain inducible GSK3β knockout. SlickV mice co-express tamoxifen-inducible cre recombinase (CreERT2) with YFP under the Thy-1 promoter. These were crossed with GSK3βLoxP/LoxP mice, in which GSK3β is flanked by LoxP sites. Administration of tamoxifen irreversibly excises GSK3β in YFP-expressing neurons. (b) Breeding scheme to obtain inducible constitutive activation of β-catenin. SlickV mice were crossed with Catnb+/lox(ex3) mice, in which exon 3 of the Catnb gene is flanked by LoxP sites. Administration of tamoxifen irreversibly excises exon 3 in YFP-expressing neurons. (c) Sagittal section through the brain of a SlickV mouse, YFP stain. Note the sparse labelling in the cortex and CA1 region. I–VI, cortical layers I–VI. CC, corpus callosum. CA1, CA3, CA4 hippocampal fields CA1, CA3 and CA4, respectively. DG, dentate gyrus. Dashed boxes show typical fields, where dendritic spines were quantified (AP, apical and BA, basal dendrites; SO, stratum oriens; SR, stratum radiatum). Scale bar, 400 μm. (d) Immunohistochemical stain against GSK3β reveals selective loss in YFP-expressing hippocampal neurons (arrows). Single confocal section; SP, stratum pyramidale; SR, stratum radiatum. Scale bar, 20 μm.

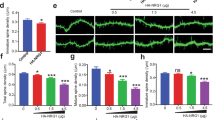
Cell autonomous GSK3β knockout in neurons alters dendritic spine density and morphology. (a) Apical dendrites of CA1 neurons after induction of GSK3β knockout by tamoxifen administration. Scale bar, 2 μm. (b) Spine densities of CA1 neurons in untreated (control) and tamoxifen-treated animals. (n=10 dendrites per animal; N=4 animals for control; N=12–16 after tamoxifen). ***P<0.001 (t-test). (c) Spine morphologies after tamoxifen administration. *P<0.05 (X2 test, n=3). (d) Cumulative histogram of spine head volumes after induction of GSK3β knockout (P<0.001, n>900 spines; Mann–Whitney test).
The effect of stabilized β-catenin was studied by crossing Catnblox(ex3) mice16 with Slick-V mice, to yield SlickV × Catnb+/▵ex3 mice, which express stabilized and therefore constitutively active β-catenin (Figure 1b). Stabilization of β-catenin was immunohistochemically confirmed in all mice analyzed (n=4). SlickV × Catnb+/+ littermates served as controls and spine densities in this line did not differ significantly from values in SlickV × GSK3βLoxP/LoxP animals (P>0.7, t-test, n=4; not shown). Tamoxifen was administered to 2-month-old animals for 5 days, as with SlickV × GSK3βLoxP/LoxP animals.
Mice were group-housed under pathogen-free conditions, except for those that received cranial window implants, which were singly housed after the surgery. All mice were kept at a 12/12-h light–dark cycle with ad libitum access to food and water. All animal procedures were performed in accordance with a protocol approved by the Ludwig Maximilian University of Munich and the Government of Upper Bavaria.
Immunohistochemistry
Animals were killed by transcardiac perfusion with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (w/v) in deep ketamine/xylazine anesthesia. The brains were removed and post-fixed in PBS containing 4% paraformaldehyde over night before cutting 60-μm-thick coronal sections on a vibratome (VT 1000 S from Leica, Wetzlar, Germany). Floating sections were permeabilized with 2% Triton X-100 in PBS over night at room temperature. For anti-GSK3β and β-catenin staining, sections were blocked with I-Block reagent (2 mg ml−1; Applied Biosystems, Life Technologies GmbH, Darmstadt, Germany). Primary antibodies (GSK3β, H-76, rabbit polyclonal, Santa Cruz Biotechnology, Heidelberg, Germany; β-catenin ab6302, rabbit polyclonal, Abcam, Cambridge, UK) were incubated overnight at room temperature in a 1:100 dilution, according to the manufacturer’s recommendations. Slices were washed 3 × 10 min with PBS and then incubated with the secondary antibodies (1:200; goat anti-rabbit Alexa 647, Invitrogen, Life Technologies GmbH) for 5 h. After 3 × 10-min washing in PBS, sections were incubated for 30 mins with anti-YFP Alexa 488 antibodies (Invitrogen, Life Technologies GmbH). Sections were finally washed for 5 × 10 min with PBS before mounting them on glass coverslips with Dako fluorescence conserving media (Dako, Hamburg, Germany).
Image acquisition and analysis
Settings for confocal and in vivo multiphoton microscopy are detailed in the Supplementary Methods. All images were deconvolved before further analysis (AutoQuant, Version x2.0.1, Media Cybernetics, Rockville, MD, USA). Figures show maximum intensity projected images (except where specified otherwise), whereas the analysis was performed in three dimensions. Spines were counted in z-stacks by manually scrolling through the images. Dendrite lengths were measured in the Zeiss LSM510 Software (Carl Zeiss AG, Oberkochen, Germany). Spine densities refer to the amount of spines per dendrite length from which they protrude. Spine morphology was determined by manually tracing dendrites in the Imaris software (Bitplane, Zürich, Switzerland) and then classified in Matlab based on morphological criteria.17 Imaris also reported spine head volumes.
Electrophysiological recordings
Hippocampal slices were prepared from mice 8–9 weeks of age. Slice preparation is detailed in the Supplementary Methods. The recording solution contained (mM): NaCl, 125; KCl, 2.5; NaH2PO4, 1.25; NaHCO3, 25; CaCl2, 2; MgCl2, 2; glucose, 25. Slices were kept at room temperature until use under continuous bubbling (95% O2, 5% CO2). Recordings were made from CA1 pyramidal neurons. Patch pipettes were pulled from borosilicate glass (HEKA Elektronik GmbH, Lambrecht/Pfalz, Germany) using a P87 puller (Sutter Instruments, HEKA Elektronik GmbH) and filled with an internal solution containing (in mM): 140 KCl, 1 CaCl2, 10 EGTA, 2 MgCl2, 4 Na2ATP, 0.5 Na2GTP, 10 HEPES, pH 7.3. The bath input resistance was 6–8 M. The membrane potential was corrected for a liquid junction potential of +8 mV. Miniature EPSCs and IPSCs (mEPSC, and mIPSC, respectively) were recorded in voltage clamp mode from a holding potential of −65 mV after the blockade of the action potential-dependent synaptic activity with tetrodotoxin (0.5 μM). NMDA (L-AP5, 50 μM) and GABAA (picrotoxin, 200 μM) receptor-channel blockers were used to isolate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor-mediated mEPSCs, whereas glycine/GABAergic mIPSCs were recorded in the presence of NMDA and AMPA receptor blockers (L-AP5, 50 μM and CNQX, 50 μM). Off-line analysis was performed in Mini Analysis (Synaptosoft, Decatur, GA, USA). Raw current traces were smoothed (5+ binomial filter). The threshold defining synaptic events was 2.5–3 times the SD of the noise. Synaptic currents were detected automatically and were frequently re-examined by eye to exclude false-positive results. Curves were fitted and graphs were generated in IgorPro (WaveMetrics, Lake Oswego, OR, USA, 6.22A).
Statistical analysis
Statistics were calculated in Prism v 5.04 (GraphPad Software, San Diego, CA, USA). Spine densities between two groups were compared using unpaired, two-sided t-tests, assuming normal distributions according to the central limit theorem, as spine densities were calculated as the means of means for every mouse. For t-tests, the variance between groups was tested (F-test) and not found to be significantly different. For multiple comparisons, one-way analysis of variance (ANOVA) with Bonferroni’s post hoc test were used. For in vivo time series data, two-way ANOVA was used to test the effects of genotypes. Curve fit parameters were compared using F-tests. The numbers of differently shaped spines were compared using a X2 test. Electrophysiology data were compared using non-parametric Mann–Whitney tests on frequencies and amplitudes calculated as the means of the means for each cell. About 30–50 excitatory or 50–70 inhibitory events were recorded per cell. To compare the cumulative histograms of these data, probability density functions were generated from all pooled events per condition and P-values were calculated using Kolmogorov–Smirnov tests.
Variance is reported as s.d. Error bars are not displayed when the s.d. is smaller than the size of the symbols.
Analyses of data shown in Figures 2b, 3, 5c and d and Supplementary Figure 1 were carried out with the investigator blinded to the genotypes. In the remaining experiments, no blinding was performed.

GSK3β knockout alters spine turnover and stabilization in vivo. GSK3β knockout was induced starting on day 0, after the recording. (a) In vivo two photon recordings of apical tufts of layer 5 neurons in the somatosensory cortex. Arrows denote persistent spines (present for ⩾4 days, white), lost and gained spines (green and pink, respectively). Scale bar, 5 μm. (b–d) Spine densities of all spines (b), persistent spines (present⩾4 days) (c) and transient spines (present <4 days) (d). (e) Daily turnover ratio of all spines. (f) Survival of all spines that were present on day −4. (g) Survival of all spines gained on day 10. Data from n=3–18 dendrites per animal; N=4 animals per group. (f, g) Lines show fits of monoexponential decay functions. TWA, two-way analysis of variance (ANOVA).
Results
We used a mouse model co-expressing inducible Cre recombinase and the fluorophore YFP (SlickV)14 to specifically delete GSK3β (Figure 1a), or exon 3 of the β-catenin gene (Figure 1b) in a small subset of neurons (Figure 1c). GSK3β knockout was induced in 2-month-old animals by application of tamoxifen, which caused almost complete loss of GSK3β immunoreactivity in YFP-expressing neurons (Figure 1d). As the fraction of these neurons is very low, they are not expected to provide a substantive fraction of presynaptic input into any given neuron (Figure 1c and Young et al.14). Therefore, any observed effects are assumed to be caused by the loss of cell autonomous GSK3β.
Spine densities were quantified in apical dendrites of the CA1 region of the hippocampus in transgenic animals treated with tamoxifen, whereas tamoxifen-treated GSK3β wild-type animals served as controls (Figure 2a). Untreated animals of both genotypes served as further controls (Figure 2b). A strong reduction in spine densities was observed after knockout induction (from 2.01 ±0.06 to 1.52±0.10 μm−1, P<0.001, n=12–16, t-test, Figures 2a and b). Similar reductions in spine densities were observed in other brain areas (Supplementary Figure 1a). Pharmacological inhibition of GSK3 α and β with AR-A014418 (30 μmol kg−1, intraperitoneally, daily18) for 3 weeks reduced spine density from 1.92±0.03 to 1.59±0.11 μm−1 (n=4, P<0.01, t-test; Supplementary Figure 1b). In GSK3β knockout, however, AR-A014418 had no further effect on spine density (Supplementary Figure 1b), suggesting that the observed effect on spines was maximal and solely caused by the GSK3β isoform.
Morphologically, three basic types of dendritic spines are distinguished: stubby, mushroom- shaped and thin spines,19 which reflect different functional states.20 In neurons with GSK3β knockout, we detected an increase in the number of stubby spines and a reduction in both thin and mushroom spines (Figure 2c; P<0.001, X2 test, n=3), whereas the overall spine head volume was reduced from 0.12±0.14 to 0.09±0.10 μm3 (Figure 2d; P<0.001, n>900 spines; Mann–Whitney test).
A fraction of dendritic spines is constantly being lost and replaced, and alterations in this dynamic turnover are part of physiological processes such as learning and memory.11 To understand how dynamic spine turnover was altered upon deletion of GSK3β, we performed in vivo imaging of cortical layer 5 neurons before and during knockout of GSK3β (Figure 3a). GSK3β knockout caused a significant reduction in spine density over the observation period of 25 days (P<0.001, n=4, two-way ANOVA; Figure 3b). This reduction was mediated by the loss of persistent spines—that is, spines which had existed for at least 4 days (P<0.0001, n=4, two-way ANOVA; Figure 3c). Conversely, the number of transient spines were increased P<0.0001, n=4, two-way ANOVA; Figure 3d), which was also reflected by the increased daily turnover ratio of spines21 (P<0.0001, n=4, two-way ANOVA; Figure 3e). The spine loss was attributed to a shortened life time of all spines, expressed as the time constant of an exponential decay function (tau), which was reduced from 36.8 d to 13.8 d (P<0.001, F-test; Figure 3f). We further analyzed the fate of all spines that were gained on day 10 and found that these, too, showed significantly reduced life times, from tau=17.0 d to 4.16 d (P<0.05, F-test; Figure 4g). These data suggest that the lack of cell autonomous GSK3β causes the loss of persistent spines and, at the same time, reduces the ability of newly formed spines to stabilize.

Excitatory synaptic neurotranstransmission is impaired in GSK3β knockout. (a, b) GSK3β knockout neurons were identified by their YFP fluorescence, whereas adjacent non-fluorescent neurons served as controls. SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum; SL, stratum lacunosum-moleculare. Scale bars: (a), 50 μm; (b) 10 μm. The arrow in b marks the patch pipette. (c) Miniature excitatory postsynaptic currents (mEPSCs). (d) Miniature inhibitory postsynaptic currents (mIPSCs). (e) Cumulative probability densities of mEPSC frequency and (f) amplitude of GSK-3+/+ (solid line) and GSK-3 LoxP/LoxP (dashed line) neurons from tamoxifen-treated mice. Quantification of miniature inhibitory postsynaptic current (mIPSC) frequencies (g) and amplitudes (h) of the same genotypes indicated in e, f: GSK-3+/+ (solid line) and GSK-3LoxP/LoxP (dashed line). Insets present the mean and s.d. of mEPSC and mIPSC frequency and amplitude, respectively. Data are collected from n=7/9 neurons of N=5 animals per group. *P<0.05 (Mann–Whitney test); n.s., not significant.
To appreciate the potential functional consequences of the cell-autonomous deficiency of GSK3, we carried out voltage-clamp recordings of AMPA-receptor-mediated mEPSC in hippocampal CA1 pyramidal neurons in slices from tamoxifen-treated SlickV × GSK3βLoxP/LoxP mice (n=7 versus n=9; from N=5 mice; Figures 4a and b). We recorded mEPSCs and mIPSCs in fluorescent GSK3βLoxP/LoxP neurons and non-fluorescent GSK3β+/+ neurons of tamoxifen-treated animals (Figures 4c and d). These experiments revealed a significant decrease in mEPSC frequency (5.9±2.1 versus 3.2±0.8 Hz; P<0.05; Mann–Whitney test; Figure 4e) and amplitude (35.4±9.3 versus 26.5±1.2 pA; P<0.05; Mann–Whitney test; Figure 4f) in GSK3βLoxP/LoxP neurons, with neither the rise nor the decay time constants of mEPSCs being altered (not shown). These findings were corroborated by comparing data from the respective cumulative probability density functions of mEPSC frequencies (P<0.001, Figure 4e) and amplitudes (P<0.001, Figure 4f) using Kolmogorov–Smirnov tests. Whereas the decrease in mEPSC frequency, in light of reduced spine density in GSK3βLoxP/LoxP, was expected, the reduction in the amplitude of mEPSCs was surprising. To define whether the latter was caused by reduced quanta size or decreased fraction of multiquantal mEPSCs, we compared the distribution of quanta amplitude and coefficient of variation (Supplementary Figure 2). We found both, a decrease in large amplitude events as well as coefficient of variation of mEPSCs amplitude, indicative of a reduction in the fraction of multiquantal events,22, 23, 24 possibly due to preferential loss of mushroom spines that receive axon terminals with multiple active zones.
To find out whether the reduction in the frequency and amplitude of synaptic currents are specific functional manifestations of the spine loss of pyramidal neurons, we assessed mIPSCs in GSK3βLoxP/LoxP pyramidal neurons of tamoxifen-treated animals. There was no significant change in the mean frequency of mIPSCs (6.8±1.5 versus 7.1±3.2 Hz; P>0.05, Mann–Whitney test; n=12/7; from N=5 mice; Figure 4h) nor in the cumulative probability density function (P>0.05, Kolmogorov–Smirnov test; Figure 4h). The mean amplitudes of mIPSCs in GSK3βLoxP/LoxP pyramidal neurons were also not significantly altered (37.8±5.1 versus 45.5±11.4 pA; P>0.05, Mann–Whitney test; Figure 4g), whereas the cumulative probability density function showed significant differences, perhaps because of the high number of events analyzed (P<0.05, Kolmogorov–Smirnov test; Figure 4h). The kinetics of mIPSCs remained unaltered (not shown). Overall, these observations imply that the structural and functional changes associated with cell-autonomous deficiency of GSK3βLoxP/LoxP are limited to glutamatergic synaptic inputs. Taken together, these results strongly suggest that the cell-autonomous knockout of GSK3β impairs the stability of dendritic spines and attenuates the excitatory synaptic transmission in pyramidal neurons of the adult brain, through post-synaptic alterations.
Among the numerous substrates of GSK3β,6 β-catenin is a major signal transducer in the Wnt signalling pathway and has been directly implicated in synaptic plasticity.25 Phosphorylation of β-catenin by GSK3β causes rapid degradation of β-catenin, whereas inhibition of GSK3β leads to β-catenin accumulation. Accordingly, we observed increased cytoplasmic and nuclear levels of β-catenin in GSK3β knockout neurons (Figure 5a and Supplementary Figure 3). To test whether this had an effect on spine density, we crossed Catnblox(ex3) mice,16 in which exon 3 of β-catenin is flanked by loxP sites, with SlickV mice, resulting in SlickV × Catnb+/lox(ex3) animals (Figure 1b). Exon 3 encodes for the residues that are phosphorylated by GSK3β, and their deletion stabilizes the β-catenin protein without affecting its function.16 We quantified spine densities before and 1 week after induction of β-catenin stabilization and found a significant reduction in spine density in Catnblox(ex3) neurons of tamoxifen-treated animals, from 2.0±0.04 μm−1 in controls to 1.58±0.15 μm−1 (P<0.01, n=4, t-test; Figures 5b and c). Furthermore, we tested whether GSK3 inhibition had an additional effect in Catnblox(ex3) neurons. We found that spine densities in vehicle-treated Catnblox(ex3) neurons were indistinguishable from those from mice, which had been treated with AR-A014418 (1.61±0.06 and 1.60±0.05 μm−1, respectively; P>0.5, n=4, t-test; Figure 5d). This suggests that GSK3β knockout and constitutively active β-catenin reduce spine densities through the same downstream mechanism, which was saturated in both models. Voltage-clamp analysis of the AMPA-receptor-mediated mEPSCs in YFP-positive Catnblox(ex3) neurons of the hippocampal CA1 area in slices from tamoxifen-treated mice (n=4; Figure 5e) revealed significant decrease in both frequency (5.9±1.6 versus 4.1±1.2 Hz; P<0.05, Mann–Whitney test; Figure 5f) and amplitude (31.2±4.4 versus 26.2±0.7 pA; P<0.05, Mann–Whitney test; n=7/7 neurons; Figure 5g) of synaptic events as compared with those of Catnb+/+ controls. Analysis of the respective cumulative probability density functions also revealed signficant differences between two groups (frequency: P<0.001; amplitude: P<0.001; Kolmogorov–Smirnov test). Neither the rise nor decay time constants of mEPSCs were altered (not shown). Taken together, these findings strongly suggest β-catenin as a major downstream effector of GSK3β in the regulation of spine dynamics and synaptic function.

GSK3β knockout leads to the accumulation of β-catenin, which reduces dendritic spine densities. (a) Maximum intensity projection of 21-μm-deep confocal sections of the CA1 region, one week after induction of GSK3β knockout. YFP (green) identifies neurons with GSK3β knockout. Immunohistochemical staining against β-catenin (magenta) shows the accumulation of β-catenin. Scale bar, 10 μm. (b) Apical dendrites of CA1 neurons after the induction of β-catenin stabilization with tamoxifen. Scale bar, 2 μm. (c) Spine densities of CA1 neurons in untreated (control) and tamoxifen-treated animals. *P<0.05; t-test, n=10 dendrites per animal, N=3–4 mice. (d) Administration of the GSK3 blocker AR-A014418 has no effect in Catnblox(ex3) neurons. Wild-type controls are duplicated from Supplementary Figure 1 for comparison. **P<0.01 (t-test, n=10 dendrites per animal, N=4 mice). (e) Miniature excitatory postsynaptic currents (mEPSCs) from Catnb+/+ and Catnblox(ex3) pyramidal neurons of tamoxifen-treated mice with cumulative probability densities of mEPSC frequency (f) and amplitude (g), respectively. Catnb+/+ (solid line) and Catnblox(ex3) (dashed line). Insets present the mean and s.d. of mEPSC and miniature inhibitory postsynaptic current (mIPSC) frequency and amplitude, respectively. Data are collected from n=7/7 neurons of N=4 animals per group *P<0.05; Mann–Whitney test.
Discussion
Both density and dynamic turnover of spines are structural correlates of experience-dependent plasticity. For instance, sensory deprivation increased spine densities in several experimental paradigms, whereas enrichment increased spine turnover.11 Moreover, learning is characterized by a transient increase in spine numbers, resulting in stabilization of some newly formed spines, whereas some formerly present spines are eliminated.11 Decreased activity of GSK3β may impose a state of increased plasticity to neurons, characterized by destabilization and increased turnover of spines (Figure 3). This notion agrees well with the finding that induction of long-term potentiation, a functional correlate of memory formation, caused inhibition of GSK3β.8,9 Furthermore, GSK3 inhibition alleviated behavioural symptoms in animal models and in patients with fragile X syndrome, which is characterized by an increased density of spines.26 Thus, our recent findings offer insight into the mechanism of action of GSK3 inhibition in neuropsychiatric diseases.
References
Budnik V, Salinas PC . Wnt signaling during synaptic development and plasticity. Curr Opin Neurobiol 2011; 21: 151–159.
Salinas PC, Zou Y . Wnt signaling in neural circuit assembly. Annu Rev Neurosci 2008; 31: 339–358.
Salinas PC, Hall AC . Lithium and synaptic plasticity. Bipolar Disord 1999; 1: 87–90.
Salcedo-Tello P, Ortiz-Matamoros A, Arias C . GSK3 Function in the brain during development, neuronal plasticity, and neurodegeneration. Int J Alzheimers Dis 2011; 2011: 189728.
Sutherland C, Leighton IA, Cohen P . Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J 1993; 296: 15–19.
Sutherland C . What are the bona fide GSK3 Substrates? Int J Alzheimers Dis 2011; 2011: 505607.
Beaulieu J-M, Gainetdinov RR, Caron MG . Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol 2009; 49: 327–347.
Dewachter I, Ris L, Jaworski T, Seymour CM, Kremer A, Borghgraef P et al. GSK3beta, a centre-staged kinase in neuropsychiatric disorders, modulates long term memory by inhibitory phosphorylation at serine-9. Neurobiol Dis 2009; 35: 193–200.
Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J et al. LTP Inhibits LTD in the hippocampus via regulation of GSK3[beta]. Neuron 2007; 53: 703–717.
Collingridge GL, Peineau S, Howland JG, Wang YT . Long-term depression in the CNS. Nat Rev Neurosci 2010; 11: 459–473.
Fu M, Zuo Y . Experience-dependent structural plasticity in the cortex. Trends Neurosci 2011; 34: 177–187.
Fiala JC, Spacek J, Harris KM . Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Rev 2002; 39: 29–54.
Penzes P, Cahill ME, Jones KA, VanLeeuwen J-E, Woolfrey KM . Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 2011; 14: 285–293.
Young P, Qiu L, Wang D, Zhao S, Gross J, Feng G . Single-neuron labeling with inducible Cre-mediated knockout in transgenic mice. Nat Neurosci 2008; 11: 721–728.
Jaworski T, Dewachter I, Lechat B, Gees M, Kremer A, Demedts D et al. GSK-3α/β kinases and amyloid production in vivo. Nature 2011; 480: E4–E5.
Harada N, Tamai Y, Ishikawa T-o, Sauer B, Takaku K, Oshima M et al. Intestinal polyposis in mice with a dominant stable mutation of the [beta]-β-catenin gene. EMBO J 1999; 18: 5931–5942.
Harris K, Jensen F, Tsao B . Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation [published erratum appears in J Neurosci 1992 Aug;12(8):following table of contents]. Journal Neurosci 1992; 12: 2685–2705.
Gould TD, Einat H, Bhat R, Manji HK . AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int J Neuropsychopharmacol 2004; 7: 387–390.
Peters A, Kaiserman-Abramof IR . The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am J Anat 1970; 127: 321–355.
Hayashi Y, Majewska AK . Dendritic spine geometry: functional implication and regulation. Neuron 2005; 46: 529–532.
Holtmaat AJGD, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW et al. Transient and persistent dendritic spines in the neocortex in vivo. Neuron 2005; 45: 279–291.
Auger C, Kondo S, Marty A . Multivesicular release at single functional synaptic sites in cerebellar stellate and basket cells. J Neurosci 1998; 18: 4532–4547.
Lisman JE, Raghavachari S, Tsien RW . The sequence of events that underlie quantal transmission at central glutamatergic synapses. Nat Rev Neurosci 2007; 8: 597–609.
Raghavachari S, Lisman JE . Properties of quantal transmission at CA1 synapses. J Neurophysiol 2004; 92: 2456–2467.
Maguschak KA, Ressler KJ . The dynamic role of beta-β-catenin in synaptic plasticity. Neuropharmacology 2012; 62: 78–88.
Mines MA, Jope RS . Glycogen synthase kinase-3: a promising therapeutic target for fragile X syndrome. Front Mol Neurosci 2011; 4: 35.
Acknowledgements
This project was funded by the EU 7th framework program (Neuro.GSK3), the German Center for Neurodegenerative Diseases (DZNE), the Deutsche Forschungsgemeinschaft (DFG) and the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung; 13N12778). Catnblox(ex3) mice were kindly provided by Makoto M Taketo (Kyoto University). We thank the technical expertise and assistance of Sonja Steinbach, Patrizia Bonert, Julia Vlcek and Pitt Liebmann.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Ochs, S., Dorostkar, M., Aramuni, G. et al. Loss of neuronal GSK3β reduces dendritic spine stability and attenuates excitatory synaptic transmission via β-catenin. Mol Psychiatry 20, 482–489 (2015). https://doi.org/10.1038/mp.2014.55
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2014.55
This article is cited by
-
Wnt signalling pathways as mediators of neuroprotective mechanisms: therapeutic implications in stroke
Molecular Biology Reports (2024)
-
Lithium rescues dendritic abnormalities in Ank3 deficiency models through the synergic effects of GSK3β and cyclic AMP signaling pathways
Neuropsychopharmacology (2023)
-
Aerobic exercise training improves memory function through modulation of brain-derived neurotrophic factor and synaptic proteins in the hippocampus and prefrontal cortex of type 2 diabetic rats
Journal of Diabetes & Metabolic Disorders (2023)
-
Light at night: effect on the daily clock, learning, memory, cognition, and expression of transcripts in different brain regions of rat
Photochemical & Photobiological Sciences (2023)
-
Understanding Glycogen Synthase Kinase-3: A Novel Avenue for Alzheimer’s Disease
Molecular Neurobiology (2023)
