
Abstract
A growing body of research demonstrates that individuals diagnosed with major depressive disorder (MDD) are characterized by shortened telomere length, which has been posited to underlie the association between depression and increased instances of medical illness. The temporal nature of the relation between MDD and shortened telomere length, however, is not clear. Importantly, both MDD and telomere length have been associated independently with high levels of stress, implicating dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis and anomalous levels of cortisol secretion in this relation. Despite these associations, no study has assessed telomere length or its relation with HPA-axis activity in individuals at risk for depression, before the onset of disorder. In the present study, we assessed cortisol levels in response to a laboratory stressor and telomere length in 97 healthy young daughters of mothers either with recurrent episodes of depression (i.e., daughters at familial risk for depression) or with no history of psychopathology. We found that daughters of depressed mothers had shorter telomeres than did daughters of never-depressed mothers and, further, that shorter telomeres were associated with greater cortisol reactivity to stress. This study is the first to demonstrate that children at familial risk of developing MDD are characterized by accelerated biological aging, operationalized as shortened telomere length, before they had experienced an onset of depression; this may predispose them to develop not only MDD but also other age-related medical illnesses. It is critical, therefore, that we attempt to identify and distinguish genetic and environmental mechanisms that contribute to telomere shortening.
Similar content being viewed by others

Introduction
A growing body of research demonstrates that individuals diagnosed with major depressive disorder (MDD) are characterized by shortened telomere length, which has been posited to underlie the association between depression and increased rates of medical illness, including cardiovascular disease, diabetes, metabolic syndrome, osteoporosis and dementia (see Wolkowitz et al.1 for a review). Indeed, a number of studies have documented shorter telomere length in depressed than in nondepressed individuals (e.g., refs 2,3,4,5,6,7).
Telomeres are specialized nucleoprotein structures that cap the ends of linear chromosomes; they consist of double-stranded, repetitive TTAGGG DNA repeats and associated proteins known as the shelterin complex.8,9 Telomere structure is critical for maintaining chromosomal stability by protecting against premature replication termination. Telomeres may shorten as a result of degradation by oxidative stress or other genotoxic stimuli, or by multiple cycles of mitosis in the absence of sufficient telomerase activity. When telomeres shorten to a critical length, the telomere ends are unprotected and initiate classic DNA damage responses, which may lead to apoptosis or to genomic instability. Telomeres shorten with each cell division and, therefore, are believed to be a marker of cellular aging. Although variability in telomere length has been found to be attributable in large part to chronological age,10 telomere shortening has also been associated with the presence of stressors, such as childhood adversity and the stress of caring for an ill family member.11, 12, 13 Interestingly, Wolkowitz et al.14 found an inverse relation between telomere length and lifetime exposure to depression, a finding that suggests that shortened telomere length is a consequence of the stressfulness associated with experiencing depressive episodes. It is also possible, however, that exposure to stressors in childhood is associated with shortened telomeres, which may then contribute to the onset of depressive episodes. Indeed, Verhoeven et al.15 recently found that both currently and remitted depressed individuals had shorter telomeres than did healthy controls, suggesting either that MDD episodes leave a lasting imprint on telomere length or that shortened telomere length antedates the onset of depression. To address this question, it is critical to assess telomere length in individuals who are at high risk of developing MDD but who have not yet experienced a depressive episode.
In this context, numerous studies have shown that stress has a central role in MDD. Researchers have documented a consistent association between stressful life events and the onset of depression.16,17 Moreover, a growing body of research is demonstrating dysregulated hypothalamic-pituitary-adrenal (HPA)-axis responses to stress in individuals diagnosed with MDD (e.g., Chopra et al.18 and Weinstein et al.19). In fact, dysregulation of the HPA axis, reflected in anomalous levels of cortisol secretion, has been posited to be a core neurobiological abnormality in depression.20,21
Although this depression-associated dysregulation of HPA-axis functioning may be a consequence of the disorder, recent evidence suggests that abnormalities in the HPA-axis system precede and even contribute to the onset of MDD. Thus, theorists have posited that dysregulation of the HPA axis contributes to the increase risk for MDD documented in children of depressed parents.22,23 Indeed, beginning as early as infancy, offspring of depressed mothers, who are at a three- to fivefold increase in the risk of developing a depressive episode by virtue of having a depressed parent,24 exhibit higher levels of cortisol in response to a psychosocial stressor than do infants of mothers with no history of depression.25 Similar adverse effects of parental depression on children’s HPA-axis functioning have also been documented in older offspring. For example, Mackrell et al. 26 found that maternal and paternal depression predicted greater cortisol reactivity to stress in 9-year-old children at a 2-year follow-up assessment. Similarly, Lupien et al.27 recently reported that 10-year-old children who were exposed to maternal depression since birth exhibited higher levels of glucocorticoids than did their non-exposed peers. Moreover, longitudinal studies have found that higher levels of salivary cortisol in high-risk children predict the subsequent onset of depressive symptoms and MDD.28,29
Taken together, these studies indicate that depressed individuals and children of depressed parents are characterized by high levels of cortisol secretion in response to stress and that this dysregulation of the HPA axis may place offspring of depressed parents at increased risk for MDD. Investigators have just begun to examine explicitly the association between HPA-axis functioning and telomere length (e.g., refs 30, 31,32,33). Indeed, only one study has examined the relation between HPA-axis activity and telomere length in depressed individuals. In that study, Wikgren et al.7 not only found shorter telomeres in depressed individuals than in nondepressed controls but also observed an association between shorter telomeres and hypocortisolism in both the depressed and nondepressed groups (as measured by low postdexamethasone cortisol and high percentage of cortisol reduction after the dexamethasone suppression test).
No study has yet examined telomere length, or the relation between telomere length and HPA-axis activity, in individuals at risk for depression. Studying such a population is critical in assessing whether shortened telomere length is a pre-existing condition or risk factor for developing depression, or, alternatively, is a response to, or concomitant of, major depressive episodes. The current study was designed to examine the relation between these two central markers of stress in a sample of children at risk of developing MDD. We focused in this study on daughters of depressed mothers, given the high intergenerational transmission of risk for depression.24 We assessed both cortisol levels in response to a laboratory stressor and telomere length in never-disordered daughters of mothers with recurrent episodes of depression (girls at familial risk for depression, or ‘high-risk’ girls) and age-matched daughters of mothers with no history of Axis I disorders (‘low-risk’ girls). We expected that the daughters of depressed mothers would have shorter telomeres than would their low-risk peers. Further, based on data showing that cortisol diminishes the activity of telomerase, the major enzyme responsible for preserving telomere length,34 we also predicted that, across the low- and high-risk groups, shorter telomeres would be associated with greater cortisol reactivity to stress.
Materials and Methods
Participants
Participants were 97 girls aged 10–14 years with no current or past Axis I disorder. Forty-seven girls had mothers with no current or past Axis I disorder (low risk (CTL)), and 50 girls had mothers with a history of recurrent episodes of depression during their daughter’s lifetime (high risk (RSK)). Participants were recruited through the Department of Psychiatry and Behavioral Sciences at Stanford University and advertisements were posted online and throughout the community. Interested individuals were screened for initial inclusion and exclusion criteria via a telephone interview. Participants who were likely to be eligible for the CTL or RSK group were invited to the laboratory for more extensive screening.
Clinical assessment
Interviews
Diagnoses were determined via in-person structured clinical interviews. The Kiddie Schedule for Affective Disorders and Schizophrenia35 was administered to daughters and mothers (about their daughters); both informants had to report an absence of current or past Axis I disorder in the daughter. The Structured Clinical Interview for DSM-IV36 was administered to mothers; mothers who reported recurrent episodes of MDD in the daughter’s lifetime were included in the RSK group, and mothers who reported an absence of current or past Axis I disorder were included in the CTL group. An independent rater who was blind to group membership randomly evaluated 10% of the Structured Clinical Interview for DSM-IV and Kiddie Schedule for Affective Disorders and Schizophrenia interviews. In all cases, diagnosis of recurrent episodes of depression in RSK mothers, absence of an Axis I disorder in CTL mothers, and absence of an Axis I disorder in daughters matched the diagnosis made by the original interviewer (κ=1.00). Eligible participants returned to the laboratory within 1 week to provide a saliva sample for telomere measurement and complete the stress task.
Questionnaires
Daughters completed the 10-item version of the Children’s Depression Inventory (CDI-S37). The CDI is a self-report measure of depressive symptomatology for children aged 8–17 years. Pubertal status was assessed via self-report Tanner staging.38 Ratings were made on a five-point scale, with Tanner stage I representing an absence of secondary sexual characteristics and Tanner stage 5 representing physiologic sexual maturity.
Telomere assessment
Genomic DNA was purified from 500 μl of saliva collected in the Oragene DNA Kit (DNA Genotek, Kanata, ON, Canada) with the DNA Agencourt DNAdvance Kit (cat. no. A48705; Beckman Coulter Genomics, Brea, CA, USA) according to the manufacturer’s instruction. Although telomere length is most commonly assessed in leukocytes, Mitchell et al.39 recently reported a significant positive correlation of 0.72 (P=0.002) between leukocyte telomere length and telomere length measured in saliva, and Daniali et al.40 found significant positive correlations between telomere length measured in leukocytes, skeletal muscle, skin and subcutaneous fat). DNA was quantified by Quant-iT PicoGreen dsDNA Assay Kit (cat. no. P7589; Life Techonologies, Grand Island, NY, USA) and run on 0.8% agarose gels to check the integrity. DNA samples were stored at −80 °C; any samples that were degraded were excluded from telomere length analysis.
Telomere length measurement assay was adapted from the method originally published by Cawthon.41,42 The cycling profile for T (telomeric) PCR consisted of: denature at 96 °C for 1 min; denature at 96 °C for 1 s, anneal/extend at 54 °C for 60 s, with fluorescence data collection, 30 cycles. The cycling profile for S (single-copy gene) PCR consisted of: denature at 96 °C for 1 min, denature at 95 °C for 15 s, anneal at 58 °C for 1 s, extend at 72 °C for 20 s, 8 cycles, followed by denature at 96 °C for 1 s, anneal at 58 °C for 1 s, extend at 72 °C for 20 s, hold at 83 °C for 5 s with data collection, 35 cycles.
The primers for the telomere PCR were tel1b (5′-CGGTTT(GTTTGG)5GTT-3′), used at a final concentration of 100 nM, and tel2b (5′-GGCTTG(CCTTAC)5CCT-3′), used at a final concentration of 900 nM. The primers for the single-copy gene (human β-globin) PCR were hbg1 (5′-GCTTCTGACACAACTGTGTTCACTAGC-3′), used at a final concentration of 300 nM, and hbg2 (5′-CACCAACTTCATCCACGTTCACC-3′), used at a final concentration of 700 nM. The final reaction mix contained 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 200 μM each dNTP, 1% DMSO, 0.4 × Syber Green I, 22 ng Escherichia coli DNA per reaction, 0.4 U of Platinum Taq DNA polymerase (Life Technologies, Carlsbad, CA, USA) per 11 μl reaction and 7 ng of genomic DNA. Tubes containing 26, 8.75, 2.9, 0.97, 0.324 and 0.108 ng of a reference DNA (from HeLa cancer cells) were included in each PCR run so that the quantity of targeted templates in each research sample could be determined relative to the reference DNA sample by the standard curve method. The same reference DNA was used for all PCR runs.
To control for interassay variability, eight control DNA samples were included in each run. In each batch, the T/S ratio of each control DNA was divided by the average T/S for the same DNA from 10 runs to get a normalizing factor. This was carried out for all eight samples and the average normalizing factor for all eight samples was used to correct the participant DNA samples to get the final T/S ratio. The T/S ratio for each sample was measured two times. When the duplicate T/S value and the initial value varied by more than 7%, the sample was run a third time and the two closest values were reported. Using this method, the average CV for this study is 2.1%.
Stress task and cortisol collection
Daughters refrained from eating or drinking beginning 1 h before arriving at the laboratory for the stress task. The session began with a 30 min rest period, during which girls were allowed to listen to music or read magazines. Next, participants were given instructions about the upcoming stressor. The first part of the stressor was a 3-min serial subtraction task. Girls were given 3 min to count backward aloud from 400 to 0 in 7-step increments. When an error was made, they were interrupted by the experimenter and asked to start again at 400. Girls who moved quickly through the task were stopped and were told to start over at 4000 and count backwards in 17-step increments. Following the serial subtraction task, girls completed the 12-min Ewart Social Competence Interview,43 a semistructured interview designed to induce emotional stress in adolescents by having them talk about stressful life situations. Following the stressors, participants watched a neutral video about the Denali National Park (Denali Park, AK, USA).
Participants used Sarstedt Salivettes (Sarstedt, Numbrecht, Germany) to provide four saliva samples throughout the stress task protocol: immediately before the stressor onset, and at 15, 30 and 45 min after stressor onset. Cortisol collection times were selected based on meta-analytic findings that peak cortisol response to stress occurs 21–40 min after stressor onset and cortisol recovery to baseline occurs 41–60 min after stressor onset.44 On average, the first sample was collected at 14:37 hours (see Table 1 for Session Start Times); the low- and high-risk groups did not differ in their collection times, t(95)=1.768, P=0.080. Saliva samples were stored in a freezer chest until they could be transferred to a −20 °C freezer located at the General Clinical Research Center at Stanford University, where they were maintained until radioimmunoassay. Cortisol levels were assayed by luminescence immunoassay reagents using a commercial kit from Immuno-Biological Laboratories (Hamburg, Germany), and the assay sensitivity was set at 0.015 mg dl−1. Samples were assayed together in large batches to control for interassay error, and control samples were included to evaluate variability.
Results
Participant characteristics
Demographic and clinical characteristics of participants in the RSK and CTL groups are presented in Table 1. The RSK and CTL groups did not differ in age, ethnicity or Tanner stage (all Ps >0.05); participants in the RSK group had slightly but significantly higher scores on the CDI than did participants in the CTL group, t(95)=3.615, P<0.001). Importantly, however, CDI scores of both groups were far below the recommended clinical cutoff of 8 for the likely presence of depression.37
We examined the association between telomere length and demographic and clinical characteristics, both across the RSK and CTL groups and separately within each group. These correlations are presented in Table 2. Telomere length was not significantly correlated with age, Tanner stage or CDI scores, either across the RSK and CTL groups or within either group, all Ps >0.05.
Telomere length and cortisol responses to stress
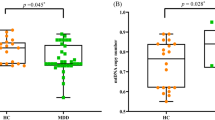
Mean telomere length of the RSK and CTL groups is presented in Figure 1. As we predicted, girls in the RSK group (M=1.524 T/S, s.d.=0.265) had significantly shorter telomeres than did girls in the CTL group (M=1.754T/S, s.d.=0.361), t(95)=3.582, P=0.001. This result did not change significantly when age, Tanner stage and CDI were included as covariates.

Telomere length (T/S) in the low-risk (CTL) and high-risk (RSK) groups. Girls in the RSK group had significantly shorter telomeres than did girls in the CTL group, t(95) = 3.582, P=0.001.
Next, we examined whether cortisol reactivity to stress differed as a function of risk group, telomere length and their interaction. Because the salivary cortisol data were positively skewed, we winsorized values 2 s.d. above the mean to the 2 s.d. value. Given the nested design of the study (i.e., time nested within people), we used hierarchical linear modeling45 to analyze the cortisol data. We used a piecewise linear growth model to fit the quadratic nature of the data and to test specific predictions concerning whether the slope of cortisol reactivity to stress (times 1–2) and the slope of cortisol recovery from stress (times 2–4; level 1) were influenced by person-level characteristics (level 2). The results of this model are presented in Table 3. Baseline cortisol levels did not differ significantly as a function of telomere length, risk group or their interaction. As predicted, however, individuals with shorter telomeres exhibited significantly greater cortisol reactivity to stress. There was no main effect of risk group on cortisol reactivity, and the nature of the association between telomeres and cortisol reactivity was similar across groups. In contrast, cortisol recovery from stress was not affected by telomere length, risk group or their interaction. These cortisol findings did not differ significantly when controlling for participants’ age, Tanner stage, CDI scores or the time of day at which the stress task was administered. In Figure 2, we depict the relation between telomere length and cortisol reactivity to stress for participants in the upper (>1.846 T/S; long) and lower (<1.391 T/S; short) quartiles of telomere length. Cortisol reactivity to stress was significantly greater for participants in the short versus long telomere group, t(44)=2.052, P=0.046.

Cortisol response to stress for participants in the upper (long) and lower (short) quartiles of telomere length. Cortisol reactivity is assessed via the slope from before to after the stress task; cortisol recovery is assessed via the slope from immediately after to 45 min after the stress task. Children with shorter telomeres exhibited significantly greater cortisol reactivity to stress than did children with longer telomeres, t(44) = 2.052, P=0.046.
Discussion
The present study was designed to address the formulation that shortened telomere length is a vulnerability marker for major depression that is present before illness onset. We addressed this hypothesis by examining whether shorter telomere length is evident in individuals who are at increased risk for the development of depression but have not yet experienced a depressive episode. This study is the first to examine telomere length, as well as the association between telomere length and cortisol reactivity to stress, in never-depressed children at risk for MDD. By investigating telomere length in healthy individuals at risk for the development of depression, we can examine whether accelerated biologic aging begins before the onset of disorder. This is also the first study to test the association between telomere length and cortisol reactivity to stress in a high-risk sample, enabling us to gain a better understanding of the association between two central markers of stress in the context of risk for psychopathology.
The results of this study indicate that healthy children at familial risk for depression have shorter telomeres than do their non-risk peers. Thus, telomere shortening appears to be an antecedent to, and potentially a risk factor for, the onset of depression. Telomere shortening in these young high-risk girls has important health implications. Telomere shortening is not only a marker of stress, but is also a mechanism of biological aging.46,47 Insufficient telomere maintenance can accelerate biological aging and increase individuals’ risk for experiencing age-related chronic diseases.48,49 Therefore, it is critical to identify individuals, such as the children of depressed mothers in the present study, who might be vulnerable to experience telomere shortening. Also, it is important to follow these girls longitudinally to examine whether telomere length influences the onset of MDD and whether developing depression, in turn, contributes to further shortening of telomeres.
It is important to note that both genetic and environmental mechanisms could have contributed to the shorter telomere length we documented in our high-risk participants; because the daughters in this study were all living at home with their biologic mothers, we cannot distinguish between these two types of mechanisms. A recent meta-analysis estimated a 70% heritability of telomere length, with figures from individual studies ranging from 34 to 82%.50 Although this analysis suggests that telomere length is strongly influenced by genetic factors, it is clear that a substantial amount of variability in telomere length is also explained by environmental factors. Indeed, there is growing evidence that shorter telomere length is associated with exposure to stress, including childhood adversity, stressful life events and chronic stressors.12,51, 52, 53 In the present study, we operationalized participants’ risk for psychopathology on the basis of their mothers’ history of recurrent depressive episodes during the daughters’ lifetime. Maternal depression has been associated with a stressful early environment for children (see Goodman and Gotlib22 and Lovejoy et al.54 for reviews); the chronic exposure of these children to this stress as a function of living with mothers who have experienced recurrent episodes of depression could represent a mechanism of accelerated biologic aging, operationalized as having shorter telomere length. It will be important in future research to attempt to elucidate the relative contributions of genetic versus environmental factors to telomere length in high-risk children.
Importantly, we found that girls with shorter telomeres exhibited greater cortisol reactivity to stress. The fact that we found this association in both the high- and low-risk participants suggests a universal relation between telomere length and HPA-axis regulation. This possibility is consistent with findings of an association between shorter telomere length and hypercortisolemia in healthy populations (for a review see Price et al.13) and across psychiatric and control groups.15 Interestingly, we found that telomere length was associated with cortisol reactivity to stress, but not with baseline levels of cortisol. This finding extends results of studies with other populations indicating that telomere length is associated more strongly with biologic responses to stress than with basal levels of cortisol (e.g., refs 30,55,56). Other investigators have also reported stronger relations between cortisol and telomere length when measuring dynamic than static aspects of cortisol secretion (e.g., waking-associated increases in cortisol or cortisol responses provoked by psychological stress31, 32, 33). Although this pattern of results may be because of the greater stability of cortisol measures for dynamic (i.e., within individuals) than for cross-sectional (i.e., across individuals) assessment, it is also possible that telomere shortening is related more consistently to cortisol reactivity to stress than to resting cortisol levels. It will be important in future research to conduct a more systematic examination of the relation of telomere length with basal levels of cortisol by sampling at various points in the diurnal cycle under the usual living conditions for participants, rather than in a laboratory setting.
Similarly, although an increasing number of studies have documented a relation between telomere length and cortisol secretion, the nature of this association is not well understood. For example, high levels of cortisol secretion may contribute to accelerated telomere shortening (e.g., Picard et al.57). In this context, as we noted earlier, Choi et al.34 presented in vitro data indicating that cortisol diminishes the activity of telomerase, the major enzyme responsible for preserving telomere length. It is also possible, however, that a third variable is responsible for both shorter telomere length and greater cortisol reactivity to stress. Oxidative stress, for instance, has been implicated as an important mechanism of telomere shortening (Epel et al.12 and Szebeni et al.,58 but also see Bull et al.59). Clearly, additional longitudinal research is needed to gain a more comprehensive understanding of the temporal and causal relations between these central markers of chronic stress.
We should note three limitations of the current study. First, the participants in our study were 10 to 14 years of age. We selected this relatively narrow age range, in part, to increase the likelihood that girls in the high-risk group had not yet experienced a depressive episode. As a consequence, however, we cannot examine whether our findings extend to a broader age range of individuals at risk for depression. This is an important direction for future research. Second, girls in the high-risk group had slightly, but significantly, higher CDI scores than did girls in the low-risk group. We should note, however, that there was no association between this narrow range of CDI scores and telomere length, and that our findings did not change significantly when we included CDI scores as a covariate in the analyses. Finally, we used Tanner staging as our measure of pubertal status, and found no relation between pubertal status and telomere length. Tanner staging is a self-report measure, and although researchers have demonstrated that it correlates highly with physicians’ physical examinations of pubertal development (e.g., Shirtcliff et al.60), it will be important in future research to conduct more extensive physical assessments of pubertal status.
In this study, we demonstrated that (1) daughters of mother with recurrent depression have shorter telomeres than do their low-risk counterparts and that (2) shorter telomere length is associated with greater cortisol reactivity to stress across the high- and low-risk groups. These findings suggest that healthy girls who are at risk of developing MDD are characterized by accelerated biologic aging before the onset of depression, which may predispose them to develop age-related medical illnesses. Indeed, MDD is associated with an increased risk of experiencing serious comorbid medical illnesses (see Freedland and Carney61 for a recent review of this literature). Interestingly, the specific medical illnesses that are observed most frequently in individuals with psychiatric conditions are those that are most commonly seen with advanced age (e.g., cardiovascular disease, stroke, dementia, diabetes, osteoporosis62), raising the possibility that psychiatric illnesses are associated with accelerated aging at the cellular or organismic level. Finally, our findings also contribute to biologic models of health and aging by documenting what appears to be a universal association between telomere length and cortisol reactivity to stress in children. Future work should examine the role of telomere length in risk for MDD and the subsequent effect of a depressive episode on telomere shortening.
References
Wolkowitz OM, Epel ES, Reus VI, Mellon SH . Depression gets old fast: do stress and depression accelerate cell aging?. Depress Anxiety 2010; 27: 327–338.
Garcia-Rizo C, Fernandez-Egea E, Miller BJ, Oliveira C, Justicia A, Griffith JK, et al. Abnormal glucose tolerance, white blood cell count, and telomere length in newly diagnosed, antidepressant-naive patients with depression. Brain. Behav Immun 2013; 28: 49–53.
Hartmann N, Boehner M, Groenen F, Kalb R . Telomere length of patients with major depression is shortened but independent from therapy and severity of the disease. Depress Anxiety 2010; 27: 1111–1116.
Hoen PW, de Jonge P, Na BY, Farzaneh-Far R, Epel E, Lin J, et al. Depression and leukocyte telomere length in patients with coronary heart disease: data from the Heart and Soul Study. Psychosom Med 2011; 73: 541–547.
Lung FW, Chen NC, Shu BC . Genetic pathway of major depressive disorder in shortening telomeric length. Psychiatr Genet 2007; 17: 195–199.
Simon NM, Smoller JW, McNamara KL, Maser RS, Zalta AK, Pollack MH, et al. Telomere shortening and mood disorders: preliminary support for a chronic stress model of accelerated aging. Biol Psychiatry 2006; 60: 432–435.
Wikgren M, Maripuu M, Karlsson T, Nordfjäll K, Bergdahl J, Hultdin J, et al. Short telomeres in depression and the general population are associated with a hypocortisolemic state. Biol Psychiatry 2012; 71: 294–300.
Blackburn EH . Switching and signaling at the telomere. Cell 2001; 106: 661–673.
De Lange T . Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 2005; 19: 2100–2110.
Mather KA, Jorm AF, Parslow RA, Christensen H . Is telomere length a biomarker of aging? A review. J Gerontol Ser A 2011; 66: 202–213.
Aubert G, Lansdorp PM . Telomeres and aging. Physiol Rev 2008; 88: 557–579.
Epel ES, Blackburn EH, Lin J, Dhabhar FS, Adler NE, Morrow JD, et al. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci USA 2004; 101: 17312–17315.
Price LH, Kao HT, Burgers DE, Carpenter LL, Tyrka AR . Telomeres and early-life stress: an overview. Biol Psychiatry 2013; 73: 15–23.
Wolkowitz OM, Mellon SH, Epel ES, Lin J, Dhabhar FS, Su Y, et al. Leukocyte telomere length in major depression: correlations with chronicity, inflammation and oxidative stress—preliminary findings. PLoS One 2011; 6: e17837.
Verhoeven JE, Révész D, Epel ES, Lin J, Wolkowitz OM, Penninx BW . Major depressive disorder and accelerated cellular aging: results from a large psychiatric cohort study. Mol Psychiatry 2013; 19: 895–901.
Monroe SM, Slavich G, Georgiades K . The social environment and depression: the roles of life stress. In: Gotlib, IH, Hammen CL. Handbook of Depression, 3rd edn. New York, NY, USA: Guilford Press, 2014.
Morris MC, Ciesla JA, Garber J . A prospective study of stress autonomy versus stress sensitization in adolescents at varied risk for depression. J Abnorm Psychol 2010; 119: 341.
Chopra KK, Ravindran A, Kennedy SH, Mackenzie B, Matthews S, Anisman H, et al. Sex differences in hormonal responses to a social stressor in chronic major depression. Psychoneuroendocrinology 2009; 34: 1235–1241.
Weinstein AA, Deuster PA, Francis JL, Bonsall RW, Tracy RP, Kop WJ . Neurohormonal and inflammatory hyper-responsiveness to acute mental stress in depression. Biol Psychol 2010; 84: 228–234.
Burke HM, Davis MC, Otte C, Mohr DC . Depression and cortisol responses to psychological stress: a meta-analysis. Psychoneuroendocrinology 2005; 30: 846–856.
Jarcho MR, Slavich GM, Tylova-Stein H, Wolkowitz OM, Burke HM . Dysregulated diurnal cortisol pattern is associated with glucocorticoid resistance in women with major depressive disorder. Biol Psychol 2013; 93: 150–158.
Goodman SH, Gotlib IH . Risk for psychopathology in the children of depressed mothers: a developmental model for understanding mechanisms of transmission. Psychol Rev 1999; 106: 458–490.
Holsboer F . The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 2000; 23: 477–501.
Gotlib IH, Colich NL . Children of depressed parents. In: Gotlib IH, Hammen CL. Handbook of Depression, 3rd edn. New York, NY, USA: Guilford Press, 2014. 240–258.
Azar R, Paquette D, Zoccolillo M, Baltzer F, Tremblay RE . The association of major depression, conduct disorder, and maternal overcontrol with a failure to show a cortisol buffered response in 4-month-old infants of teenage mothers. Biol Psychiatry 2007; 62: 573–579.
Mackrell SV, Sheikh HI, Kotelnikova Y, Kryski KR, Jordan PL, Singh SM, et al. Child temperament and parental depression predict cortisol reactivity to stress in middle childhood. J Abnorm Psychol 2014; 123: 106–116.
Lupien SJ, Parent S, Evans AC, Tremblay RE, Zelazo PD, Corbo V et al. Larger amygdala but no change in hippocampal volume in 10-year-old children exposed to maternal depressive symptomatology since birth. Proc Natl Acad Sci USA 2011; 108: 14324–14329.
Adam EK, Doane LD, Zinbarg RE, Mineka S, Craske MG, Griffith JW . Prospective prediction of major depressive disorder from cortisol awakening responses in adolescence. Psychoneuroendocrinology 2010; 35: 921–931.
Halligan SL, Herbert J, Goodyer I, Murray L . Disturbances in morning cortisol secretion in association with maternal postnatal depression predict subsequent depressive symptomatology in adolescents. Biol Psychiatry 2007; 62: 40–46.
Epel ES, Lin J, Wilhelm FH, Wolkowitz OM, Cawthon R, Adler NE, et al. Cell aging in relation to stress arousal and cardiovascular disease risk factors. Psychoneuroendocrinology 2006; 31: 277–287.
Kroenke CH, Epel E, Adler N, Bush NR, Obradović J, Lin J, et al. Autonomic and adrenocortical reactivity and buccal cell telomere length in kindergarten children. Psychosom Med 2011; 73: 533–540.
Révész D, Verhoeven JE, Milaneschi Y, de Geus EJ, Wolkowitz OM, Penninx BW . Dysregulated physiological stress systems and accelerated cellular aging. Neurobiol Aging 2013; 35: 1422–1430.
Tomiyama AJ, O’Donovan A, Lin J, Puterman E, Lazaro A, Chan J, et al. Does cellular aging relate to patterns of allostasis?: An examination of basal and stress reactive HPA axis activity and telomere length. Physiol Behav 2012; 106: 40–45.
Choi J, Fauce SR, Effros RB . Reduced telomerase activity in human T lymphocytes exposed to cortisol. Brain Behav Immun 2008; 22: 600–605.
Kaufman J, Birmaher B, Brent DA, Ryan ND, Rao U . K-Sads-PI. J Am Acad Child Adolesc Psychiatry 2000; 39: 1208.
First MB, Spitzer RL, Gibbon M, William JB . Structure Clinical Interview for DSM-IV-TR Axis I Disorders-Non-patient Edition (SCID-I/NP, 11/2002 revision). Biometric Research Department, New York, State Psychiatric Institute: New York, NY, USA, 2002.
Kovacs M The Children’s Depression Inventory (CDI). Multi-Health Systems, Toronto, Canada, 1992.
Tanner JM, Whitehouse RH . Clinical longitudinal standards for height, weight, height velocity, weight velocity, and stages of puberty. Arch Dis Childhood 1976; 51: 170–179.
Mitchell C, Hobcraft J, McLanahan SS, Siegel SR, Berg A, Brooks-Gunn J, Garfinkel I, Notterman D . Social disadvantage, genetic sensitivity, and children’s telomere length. Proc Natl Acad Sci USA 2014; 111: 5944–5949.
Daniali L, Benetos A, Susser E, Kark JD, Labat C, Kimura M, et al. Telomeres shorten at equivalent rates in somatic tissues of adults. Nat Commun 2013; 4: 1597.
Cawthon RM . Telomere measurement by quantitative PCR. Nucleic Acids Res 2002; 30: e47–e47.
Lin J, Epel E, Cheon J, Kroenke C, Sinclair E, Bigos M, et al. Analyses and comparisons of telomerase activity and telomere length in human T and B cells: insights for epidemiology of telomere maintenance. J Immunol Methods 2010; 352: 71–80.
Ewart CK, Jorgensen RS, Suchday S, Chen E, Matthews KA . Measuring stress resilience and coping in vulnerable youth: the Social Competence Interview. Psychol Assess 2002; 14: 339.
Dickerson SS, Kemeny ME . Acute stressors and cortisol responses: a theoretical integration and synthesis of laboratory research. Psychol Bull 2004; 130: 355.
Bryk AS, Raudenbush SW . Hierarchical Linear Models: Applications and Data Analysis. Sage Publications: Newbury Park, CA, USA, 1992.
Blackburn EH . Telomere states and cell fates. Nature 2000; 408: 53–56.
Blackburn EH, Greider CW, Szostak JW . Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med 2006; 12: 1133–1138.
Fitzpatrick AL, Kronmal RA, Gardner JP, Psaty BM, Jenny NS, Tracy RP, et al. Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am J Epidemiol 2007; 165: 14–21.
Njajou OT, Hsueh WC, Blackburn EH, Newman AB, Wu SH, Li R, et al. Association between telomere length, specific causes of death, and years of healthy life in health, aging, and body composition, a population-based cohort study. J Gerontol Ser A 2009; 64: 860–864.
Broer L, Codd V, Nyholt DR, Deelen J, Mangino M, Willemsen G, et al. Meta-analysis of telomere length in 19 713 subjects reveals high heritability, stronger maternal inheritance and a paternal age effect. Eur J Hum Genet 2013; 21 1163–1168.
Kiecolt-Glaser JK, Gouin JP, Weng NP, Malarkey WB, Beversdorf DQ, Glaser R . Childhood adversity heightens the impact of later-life caregiving stress on telomere length and inflammation. Psychosom Med 2011; 73: 16–22.
Lin J, Epel E, Blackburn E . Telomeres and lifestyle factors: roles in cellular aging. Mutat Res/Fund Mol Mech Mutagen 2012; 730: 85–89.
O’Donovan A, Epel E, Lin J, Wolkowitz O, Cohen B, Maguen S, et al. Childhood trauma associated with short leukocyte telomere length in posttraumatic stress disorder. Biol Psychiatry 2011; 70: 465–471.
Lovejoy MC, Graczyk PA, O’Hare E, Neuman G . Maternal depression and parenting behavior: a meta-analytic review. Clin Psychol Rev 2000; 20: 561–592.
Aschbacher K, O’Donovan A, Wolkowitz OM, Dhabhar FS, Su Y, Epel E . Good stress, bad stress and oxidative stress: insights from anticipatory cortisol reactivity. Psychoneuroendocrinology 2013; 38: 1698–1708.
O’Donovan A, Tomiyama AJ, Lin J, Puterman E, Adler NE, Kemeny M, et al. Stress appraisals and cellular aging: a key role for anticipatory threat in the relationship between psychological stress and telomere length. Brain Behav Immun 2012; 26: 573–579.
Picard M, Juster RP, McEwen BS . Mitochondrial allostatic load puts the 'gluc' back in glucocorticoids. Nat Rev Endocrinol 2014; 10: 303–310.
Szebeni A, Szebeni K, DiPeri T, Chandley MJ, Crawford JD, Stockmeier CA, et al. Shortened telomere length in white matter oligodendrocytes in major depression: potential role of oxidative stress. Int J Neuropsychopharmacol 2014; 1–11.
Bull CF, Christensen H, Fenech MF . 24. Cortisol increases DNA damage in lymphocytes in vitro, but is not associated with telomere shortening. Brain Behav Immun 2013; 32: e7.
Shirtcliff EA, Dahl RE, Pollak SD . Pubertal development: correspondence between hormonal and physical development. Child Dev 2009; 80: 327–337; PMID19466995.
Freedland K, Carney R . Depression and medical illness. In: Gotlib IH, Hammen CL (eds.), Handbook of Depression, 3rd edn, pp 122–141. Guilford Press: New York, NY, USA, 2014.
Evans DL, Charney DS, Lewis L, Golden RN, Gorman JM, Krishnan KR, et al. Mood disorders in the medically ill: scientific review and recommendations. Biol Psychiatry 2005; 58: 175–189.
Acknowledgements
This research was supported by Grant MH74849 from the National Institute of Mental Health to IHG. We thank Hannah Burley, Yamanda Wright, Maria Lemus and Brooke Gilbert for their help in recruiting the participants in this study, and Elizabeth H Blackburn for her help in analyzing the telomere data. Jue Lin is a cofounder and Director of Research of Telomere Diagnostics (formerly Telome Health). All other authors report no competing or financial interests.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
PowerPoint slides
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Gotlib, I., LeMoult, J., Colich, N. et al. Telomere length and cortisol reactivity in children of depressed mothers. Mol Psychiatry 20, 615–620 (2015). https://doi.org/10.1038/mp.2014.119
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2014.119
This article is cited by
-
A randomized controlled trial of a preventive intervention for the children of parents with depression: mid-term effects, mediators and moderators
BMC Psychiatry (2023)
-
Strong associations of telomere length and mitochondrial copy number with suicidality and abuse history in adolescent depressed individuals
Molecular Psychiatry (2023)
-
Leukocyte telomere dynamics across gestation in uncomplicated pregnancies and associations with stress
BMC Pregnancy and Childbirth (2022)
-
Stress reactivity elicits a tissue-specific reduction in telomere length in aging zebrafish (Danio rerio)
Scientific Reports (2021)
-
Telomere reprogramming during fetal life in low socioeconomic mothers
Egyptian Journal of Medical Human Genetics (2019)
