
Abstract
Analyzing undiluted whole human blood is a challenge due to its complex composition of hematopoietic cellular populations, nucleic acids, metabolites, and proteins. We present a novel multi-functional microfluidic acoustic streaming platform that enables sorting, enrichment and in situ identification of cellular subsets from whole blood. This single device platform, based on lateral cavity acoustic transducers (LCAT), enables (1) the sorting of undiluted donor whole blood into its cellular subsets (platelets, RBCs, and WBCs), (2) the enrichment and retrieval of breast cancer cells (MCF-7) spiked in donor whole blood at rare cell relevant concentrations (10 mL−1), and (3) on-chip immunofluorescent labeling for the detection of specific target cellular populations by their known marker expression patterns. Our approach thus demonstrates a compact system that integrates upstream sample processing with downstream separation/enrichment, to carry out multi-parametric cell analysis for blood-based diagnosis and liquid biopsy blood sampling.
Similar content being viewed by others
Introduction
Efficient separation of cellular populations from whole blood is an essential preparatory step for many biological and clinical assays1. Conventional macro-scale cell sorting techniques include density gradient based centrifugation, immunolabeling-based separation such as fluorescence activated or, magnetic-activated cell sorting (FACS, MACS, respectively) and CellSearch. However, these techniques are limited by separation sensitivity, large blood sample volume requirement, multi-step interventions prone to artifacts, processing time, and cost. Lab-on-a-chip and microfluidic systems developed for microscale separation techniques have been gaining importance over the past decade. Many types of unique cell sorting and enrichment devices have been devised but they are not usually able to work with unprocessed whole blood2–5. These technologies typically require pre-processed samples and cannot handle the physical properties and complex populations inherent in whole blood6–10. They are limited to either diluted 11,12 or lysed blood13,14, and in some cases, density based centrifugation to reduce blood complexity and cell-cell interaction15–21.
The microfluidic cell sorting technologies that can handle whole blood, in general fall into two categories: label free sorting based on physical characteristics of the target cells or biomarker labeling. For example, Davis et al.22 utilized deterministic lateral displacement (DLD) to separate whole blood components. Nagrath et al.23 used microposts and Stott et al.24 utilized herringbone structures to capture EpCAM+ CTCs from whole blood. In another study, Karabacack et al.25 integrated DLD with inertial focusing and magnetophoresis for negative isolation of CTCs from whole blood. Although they successfully demonstrated target cell sorting, they were unable to enrich in a small volume (<50 μL).
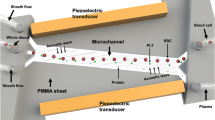
Acoustic microstreaming has been demonstrated to focus, sort and enrich target cells/particles within the microchannel26–32. Studies have shown negligible adverse effect of ultrasonic actuation on cellular phenotypes, viability and functional capacity9,21,27,28,33,34. Acoustic microstreaming is a phenomenon of localized streaming patterns occurring near an oscillating liquid interface29. In lateral cavity acoustic transducers (LCATs), liquid–gas interfaces are formed by trapped bubbles in patterned dead-end side channels that form the acoustic microstreaming sources. Oscillation of these bubble surfaces is actuated by an acoustic energy source, leading to a second-order characteristic streaming viscous flow velocity, Us, which is maximum at the outer edge of the microstreaming vortices. With the slanted LCAT angles, this streaming flow also induces a bulk flow in the microchannel that ‘squeezes’ through an open streamline gap (termed dgap) that is between the outer edge critical streamline of the vortex and the oscillating liquid-air interface. Particles trapped in the vortices form size-dependent orbits, where larger particles are pushed towards vortex center (Figures 1a and c). Particles with radii smaller than the dgap are released from the LCAT and are pumped along with the bulk flow. Therefore, LCATs are capable of bulk pumping of the sample while simultaneously separating particles/cells by size based on the bubble surface oscillation amplitude. The oscillation thus controls the trapping and releasing of particles, correlated roughly to the ratio between bulk and streaming velocities (Refs. 28, 30).

Device schematics. (a) Schematic illustrating the size-based separation, enrichment and in situ labeling in the lateral cavity acoustic transducer (LCAT) device. (b) Image of the polydimethylsiloxane (PDMS) based LCAT device. (c) Bright-field image showing the trapping of 25 μm particles in the closed circular streamlines in comparison to the 10 μm particles in open streamlines.
As depicted in Figure 1a, LCATs integrate the following sample processing steps (1) separate whole blood into its cellular constituents, (2) enrich rare cells based on size, and (3) deliver fluorescent biomolecules (monoclonal antibodies) to selectively label target cells. These functions allow one to identify target cells based on two important characteristics–size and surface biomarkers in a single device. Here we are utilizing enrichment ratio (ER, materials and methods) as a metric to analyze the device performance14,35,36. It is defined as the enhancement of target cell to background cell ratio from the device input to the output sample36. ER in the order of ~100× to 1000× is clinically significant for subsequent gene profiling by RT-PCR as discussed by Tong et al.14,37 In our device, we enhanced the ER to 170× for particle mixture (15 and 10 μm at initial ratio 1:1 00 000). We achieved 213× enrichment of MCF-7 cells with respect to WBCs when spiked at 10 mL−1 in whole blood while ensuring the capturing of all target cells and thus avoiding false negatives.
Materials and methods
Microfluidic device design
The LCAT chip is composed of 200 LCATs spaced 200 μm apart at an angle of 15° relative to the main channel. The main channel width and the side channel width were fixed to 500 and 100 μm, respectively. The height of the device was remained constant to 100 μm.
Microfluidic device fabrication
Microfluidic devices were fabricated using soft lithographic techniques. Silicon wafer was first cleaned with 2% HF to render it hydrophobic and dehydrated at 120 °C for 15 min. Negative photoresist, SU-8 2050 was spin coated per manufacturer’s (Microchem) protocol for 100 μm height. Following spin coating, it was soft-baked, exposed, post baked and developed. After hard baking at 200 °C, it was kept in silane overnight. Poly (dimethylsiloxane), PDMS (Sylgard 184, Dow Corning) base and curing agent at 11.5:1 ratio was mixed and poured on the mold. Following degassing in a desiccator, it was cured at 65 °C overnight. Hardened PDMS was cut and peeled carefully. Inlet hole was punched with a 1.5 mm biopsy punch and the outlet was cut. After cleaning the device, it was bonded on a thin cover slip (Fisher Scientific) using standard plasma procedure (Harrick). Following oxygen plasma, the bonded device was kept on a hot plate set at 65 °C overnight to make the device hydrophobic.
MCF-7 cell culture
MCF-7 breast adenocarcinoma (HTB-185) cell line was procured from ATCC (Manassas, VA, USA) and cultured per ATCC instruction in DMEM (Mediatech, Manassas, VA, USA) supplemented with fetal bovine serum (10%), penicillin (100U mL−1), streptomycin (100 g mL−1) (Mediatech, Manassas, VA, USA), and Novolin R insulin (44U L−1) (Novo Nordisk, Plainsboro, NJ, USA).
SKBR-3 cell culture
SKBR-3 breast adenocarcinoma (HTB-30) cell line was procured from ATCC (Manassas, VA, USA) and cultured as per ATCC instructions in RPMI (Mediatech, Manassas, VA, USA) supplemented with fetal bovine serum (10%), penicillin (100U mL−1), and streptomycin (100 g mL−1; Mediatech).
For biomarker staining on MCF-7 cells, anti-human EpCAM CD326 antibody conjugated with PE-Dazzle 594 (Biolegend) was used. After obtaining the cell pellet in an Eppendorf tube, they were blocked by adding 60 μL staining buffer (1% BSA, 0.1% NaN3 in 1X PBS, pH 7.4) to re-suspend cell pellets with 20 μL of FcR block (Miltenyi Biotech) for 10 min. Appropriate volumes of antibody stain (3 μg mL−1) was added and incubated for 30 min on ice. Following incubation, the cells were washed with staining buffer and collected by centrifugation at 1000 r.p.m. for 5 min. These pre-stained cells were observed under fluorescence and spiked in whole blood at appropriate concentrations.
For nuclear staining of WBCs, we added DAPI (4',6-diamidino-2-phenylindole) stain (Fisher Scientific) at the concentration of 1mg mL−1 and incubated the sample for 10 min. Following that, we imaged the stained cells using DAPI filter of the fluorescent microscope.
Blood sample collection and processing
Normal donor blood was obtained from Institution for Clinical Translation and Science, UCI under Institutional Review Board (IRB) approval. Whole blood with anti-coagulant was mixed with pre-stained MCF-7 cells to represent target cancer cells/rare cells. All normal donor blood was processed within 24 h after withdrawal. The blood cells after separation were stained using Wright-Giemsa Stain (Electron Microscopy Services). After making the smear of whole blood and its separated components (RBCs and WBCs), the cells were fixed and stained using Wright-Giemsa stain for 5 min. The glass slide is then washed in DI water carefully and dried for observation under the microscope.
Microfluidic device experimental setup
The blood sample and 1× PBS buffer were supplied to the microfluidic devices by 1 mL syringes connected to Harvard Apparatus syringe pump and NE-500 (New Era), respectively. The tubing of inner diameter 0.51 mm (Cole Parmer) was used to connect the microfluidic device with the syringe. This tubing was inserted in the inlet hole punch by a 1.5 mm biopsy punch and the outlet was cut to ensure the release of 20 μL trapped sample. The ultrasound gel (ultrasonic) was smeared on the piezoelectric transducer (Steminc piezo) to ensure the efficient transport of acoustic wave from PZT to the PDMS device. The PZT was connected to the function generator (Agilent 33220A) and the voltage amplifier (Krohn-Height 7500) to supply the square wave. To monitor the functionality of the microfluidic device, we connected a high-speed camera (Phantom, vision research) to a L150 Nikon Eclipse upright microscope. Fluorescence detection was performed using a 100 W, 488 nm laser for excitation and a yellow green laser of 561 nm for detection by a canon DSLR mounted on an Olympus IX 51 inverted microscope.
Results
Isolation and separation of cells from whole healthy blood
The LCAT device (Figure 1b) was fabricated by soft lithography techniques (materials and methods). To initiate the testing, the device was first primed with 1x PBS buffer to form air-liquid interfaces, and then placed on a piezoelectric transducer (PZT) with ultrasound gel. Undiluted normal donor whole blood (10 μL) was then introduced into the inlet of the LCAT microchannel. The mechanical vibration from the PZT (controlled by a function generator) is coupled through the gel into the LCAT device to actuate the air–liquid interfaces.
At 2Vpp, the PZT vibration induced the pumping of plasma and small platelets in the blood, but RBCs and WBCs were trapped in the vortices formed by the microstreaming flow (Us). This resulted in the collection of 2 μL of fluid enriched for plasma and platelets from the outlet. As the PZT voltage is increased to 2.5 Vpp, the increase in Ub/Us results in RBCs being released from the microstreaming vortices and collected from the outlet. To increase the purity of separated sample, excess blood was pipetted out of the inlet and replaced with 30 μL of PBS buffer. The device remained driven at 2.5 Vpp, until all RBCs were flushed out by the buffer and WBCs remain trapped as shown in Supplementary Video 1. After collecting all the RBCs from the outlet, the voltage was further increased to 4.5 Vpp to release remaining trapped WBCs. The separated samples were kept in individual vials for inspection. The plasma and platelet sample was viewed in bright field at 40× on a countess slide while the whole blood, and RBC samples were stained with Wright-Giemsa stain to distinguish them within (Figures 2a and b). WBC samples were spotted on a fluorescent microscope using DAPI nuclear stain (Figure 2b, right) followed by the morphological analysis as shown in Supplementary Figure S1. By utilizing imageJ software for enumeration, the plasma sample consists of 96.3% platelets, the RBC sample consists of 90.8% RBCs, and the WBC sample consists of 66.4% WBCs (Figure 2c).

Sample preparation of whole blood. (a) Smear of the whole blood stained with Wright-Giemsa stain (left), 10 mL vial of whole unprocessed blood. (b) Bright-field image of vial containing plasma containing platelets (left), Bright field image of the smear of the RBC sample after sorting from device (middle), Bright field and fluorescent image (DAPI) of WBC sample after device operation (right). (c) Bar graph showing the percent of cells in each sample after sorting in device (n=3).
Integration of size-based separation with biomarker-based immunolabeling
In some cases, rare cells such as circulating tumor cells or cancer-associated fibroblasts also exist in our circulatory system. Separation and enrichment of these rare cells are of prime interest for both prognosis and treatment monitoring. Post-separation analysis such as identification and enumeration can be performed by immunofluorescence and is unaffected by the presence of non-target cells in the output sample. Currently, despite considerable automation along with blood handling and separation, detection by immuno-staining of target cells is performed manually. The standard staining procedure takes at least one hour, and requires expensive centrifuges to collect cells maintained in suspension. To maximize the utility of lab-on-chip devices in point-of-care setting, we have integrated size-based separation with biomarker expression based immunolabeling. This enables our device to integrate complete hematological separation of cellular subsets and on-chip immunofluorescent labeling to confirm the identity of captured cells of interest based on both size and biomarker, reducing turnaround time, minimizing labor-intensive sample preparation, and limiting cost.
For proof of concept, MCF-7 breast cancer cells (2.1×105 mL−1) were prepared into a single cell suspension. The LCAT device excitation voltage was set to capture the larger MCF-7 cells, followed by on-chip staining with anti-human EpCAM mAb conjugated to PE/Dazzle 594 (Biolegend). 30 μL of MCF-7 cell suspension in PBS was pumped at 2.5 Vpp from the LCAT device inlet for 5 min. After the cells were trapped in the microstreaming vortices, 20 μL of Fc block (Miltenyi Biotec) was added in the inlet after removing the extra cell sample. Fc block was flown for 5min followed by pumping 4 μL of CD326 (0.8 μg anti-EpCAM) antibody for 2 min (2 μL min−1). To prevent the inlet from emptying and air entering the channel, 30 μL of staining buffer (1% BSA, 0.1% NaN3 in 1× PBS, pH 7.4) was added to the inlet for 3 min. The buffer also served to wash out any unbound antibodies in the channel. After the cells were immunolabeled in the device, the fluorescent cells were imaged on-chip using a fluorescent upright microscope (Figure 3a). Subsequently, stained MCF-7 cells were released at 4.5–5 Vpp to analyze the fluorescent intensity and the morphology. We compared the detected expression level of the MCF-7 cells stained using a standard benchtop procedure (0.8 μg, incubation time of 50–60 min) to the 15 min in situ chip-based staining procedure and a correlation of 91% demonstrates the similar expression level detected via on-chip staining regardless of the shortened staining time (Figure 3b).

In Situ labeling of MCF-7 cells from blood. (a) Fluorescent image of MCF-7 cells stained on device. Inset: bright-field image of the boxed region. (b) Plot of intensity correlation between staining protocol on device with benchtop staining (n=3). (c) Fluorescent image of MCF-7 cells after sorting, enriching, and on-chip labeling. (d) Bright-field image of the released sample containing WBCs (smaller) and MCF-7 cells.
After optimizing the immunostaining protocol, we sought to produce a more realistic case by spiking MCF-7 cells into normal donor whole blood samples (50 000 mL−1). The spiked blood samples were run through the device, with the intent to separate the cancer cells from the blood. Platelets and RBCs were first extracted, followed by pumping of 30 μL of RBC lysing buffer (Gibco) for 5 min to ensure complete removal of RBCs from the microstreaming vortices. As a result, a mixture of remaining WBCs and MCF-7 cells remained trapped at the microstreaming vortices. At this point, we immunostained the MCF-7 cells with anti-EpCAM antibody to discriminate them from the remaining WBCs on the device using the protocol described above and released the mixture at 4.5–5 Vpp. The released sample of 20μL was placed on a countess slide, and imaged using a fluorescent filter (Figure 3c), as well as bright field (Figure 3d). 10× ratio increment (ER 9.71×) for fluorescently labeled MCF-7 cells with respect to WBCs confirms the identification and enumeration of target cancer cells specifically based on both size and marker expression, which is particularly important for clinical blood samples.
Device optimization for analysis at rare concentration
At this point, we demonstrated a quick and portable technique for separating blood components and detecting target cancer cells by immunolabeling on a single device. While the size of circulating tumor cells ranges from 15–25 μm, the rate of occurrence is 1–10 cells per mL of whole blood, thus making them extremely difficult to track38. Since rare cell detection requires a large sample volume to process and to identify, enumerate and analyze, we used an external syringe pump. The set-up in Figure 4a shows two syringe pumps connected with a three-way valve for controlling the flow of undiluted blood sample and the wash buffer. The LCAT device was primed with an aqueous solution of 10% glycerol in lipid to maintain the stability of air-liquid interfaces and increase the time of operation39. Due to an external bulk flow, it was imperative to optimize the device with respect to flow rate and the voltage applied to PZT.

(a) Operational set-up with external syringe pump (b) Device optimization with respect to flow rate and voltage. (c) Bright field image showing pure vortices before (left) and after voltage switching (right). Plot of (d) trapping efficiency, and (e) enrichment ratio for 15 μm particles at different concentrations in the presence of million per ml non-target 10 μm particles (n=3).
As a model system, we evaluated the device sensitivity and efficiency by utilizing polystyrene microparticles of varying sizes that resembled cell diameters and concentrations of rare cells and WBCs in whole blood. Fifteen micrometer particles were spiked at 10 000 particles per mL concentration in a solution containing one million particles per mL of 10 μm particles. We obtained the highest ER of 8.5×at 25 μL min−1 and 2.75 Vpp. At constant flow rate, Ub by the syringe pump, enrichment ratio reduces at larger voltage values (reduced Ub/Us) due to non-specific trapping of 10 μm particles. However, at low voltage values (larger Ub/Us), due to partial release of target 15 μm particles, a reduced ER was observed (Figure 4b).
At 8.5×ER, there was still non-specific trapping in the microstreaming vortices. To increase the purity and ER, we devised a voltage switching procedure by applying a short pulse at low voltage while keeping bulk flow at the same rate (Ub) (Figure 1a). When Us decreases by reducing the PZT voltage to 2 Vpp, the increase in Ub/Us ratio allows 10 μm particles to be released. To prevent 15 μm particles from releasing, this voltage is maintained for only 30 s. After that, the flow from the syringe pump was switched off and the voltage was increased to 3.5 Vpp for 1.5 min to remove 10 μm particles by the LCATs own ability of generating bulk flow. We removed 78.2% of non-target 10 μm particles (Figure 4c), achieving 30× ER from the same starting material as above. To test at practical occurrence rate, we reduced the spiking concentration of 15 μm particles to 10 mL−1 and achieved 170× ER with ~100% trapping efficiency (Figures 4d and e).
Device validation with spiked particles and cultured cancer cells from unprocessed blood
After validating the device with microparticle mixtures, the ability to isolate particles from normal donor whole blood was tested. We spiked 25 μm and 15 μm particles at concentrations ranging from 10 000 mL−1 to 10 mL−1. Flow for the spiked sample was 25 μL min−1 and particles were trapped at 2.75Vpp. We imaged the device using an upright microscope equipped with a high-speed phantom camera, and observed at the beginning, the channel filled with RBCs as shown in Figure 5a. After 14 min of flow, we switched the valve to introduce the wash buffer (1×PBS) at the same flow rate for 20 min, with voltage switching to maximize the release of non-target particles and achieve highly pure vortices (Figures 5b and c).

Enrichment of 25/15 μm diameter particles in whole blood. (a) Bright-field image of whole spiked blood in the LCAT device. (b) Bright-field image during the washing step in the device. (c) Bright-field image of pure vortices with 25 μm particles trapped after the device operation. (d) Fluorescent image of initial spiked blood (with 25 μm particles) at 10 000 per mL concentration. (e) Fluorescent image of enriched 25 μm particles after releasing from device. (f) Plot of enrichment ratio with varying spiking concentration of 25 and 15 μm diameter particles; (n=3) for each spiking concentration.
One of the major requisites for the downstream processing of target cells, such as genomic analysis, is the enrichment of target cells in a small volume. Here after operating the device for 34min, the PZT was switched off and the trapped cells were released in a 20 μL volume of buffer. We analyzed the original and trapped sample on a countess slide using fluorescent imaging as shown in Figures 5d and e. At the higher concentration of particles, 10 000 mL−1 with WBCs at the original concentration of one million cells per mL, the ER of the particles with respect to WBCs was 70×. We repeated the same procedure at low concentrations and achieved 479× ER for 15 μm particles and 531× for 25 μm particles at 10 mL−1 spiking concentration (Figure 5f).
As a final proof-of-concept experiment, the separation and enrichment of MCF-7 breast cancer cells from whole blood was demonstrated. MCF-7 cancer cells in suspension were immunofluorescently stained with anti-EpCAM antibody and then spiked into normal donor whole blood at concentrations ranging from 1,000 mL−1 to 10 mL−1. The trapping and enrichment of MCF-7 cells was performed using the same protocol described in the sections above. After processing 400 μL of spiked blood sample (separately spiked with 25 μm, 15 μm and MCF-7 cells) for 34min, we could capture and image 3–4 particles (Figures 6a and b) and 4 cells (Figure 6c) when they were spiked at initial concentration of 10 mL−1 in whole blood, thereby giving ~100% trapping efficiency (Figure 6e). After counting the fluorescent MCF-7 cells in the final sample, we obtained 213× enrichment at 10 mL−1 spiking concentration (Figure 6f). In addition, we cultured the enriched MCF-7 and SKBR-3 cells for 3 days in conditioned media to ensure the preservation of phenotypic and genotypic characteristics after ultrasound exposure and obtained successful proliferation after acoustic exposure as shown in bright field images (Figure 6d). We immunostained MCF-7 and SKBR-3 cells in culture with DAPI antibody and obtained multiple nuclei as shown in Figure 6d (right). We further stained MCF-7 cells with anti-EpCAM antibody and observed intact cell membranes as shown in Supplementary Figure S2.

Enrichment of 25 μm, 15 μm and MCF-7 cells in whole blood. Fluorescent image of (a) 25 μm, (b) 15 μm and (c) MCF-7 cells released after analyzing 400 μL of whole blood sample in device at 10 mL−1 spiking level. (d) Bright field and fluorescent image (DAPI) showing the MCF-7 (top) and SKBR-3 cells (bottom) proliferating on the culture plate. (e) Plot of the trapping efficiency for 25 μm, 15 μm diameter particles and MCF-7 cells at 100 mL−1 and 10 mL−1 concentration in whole blood (n=3). (f) Plot of enrichment ratio with varying concentration of MCF-7 cells in whole unprocessed blood sample (n=3).
Discussion
Rare cells in the circulatory system carry significance for improving cancer prognosis and monitoring treatment response1. The ultimate goal of liquid biopsy based detection of rare cells requires a multi-modal system that can process raw samples by separating, sorting and enriching for specific cell populations, and finally identifying the targeted cells via known biological markers40. Typically, each of these processes require separate instruments and the transfer of samples across different platforms, resulting in lowered sensitivity, specificity, and added costs. As described in this paper, our LCAT rare cell isolation platform can process whole blood without dilution, separate out the cellular subcomponents, isolate and enrich the targeted size-based population, and perform in situ immunolabeling. There is no other liquid biopsy system that can achieve this level of integration and multi-modal functionality. For example, the first category of rare cell detection platforms relied on surface biomarkers (for example, EpCAM) and the binding to antibody-coated surfaces to filter and concentrate rare cells such as CTCs. However, it is now well known that cancer cells can undergo epithelial to mesenchymal transition (EMT) in circulation, causing undesired false negatives41. Another promising category of microfluidic systems is “label-free”, and they are able to sort blood cells based on their sizes using known techniques such as deterministic lateral displacement (DLD)22,42, inertial microfluidics43, and acoustophoresis20,21,44. However, recent developments in oncology have shown heterogeneity and great size overlap between rare cells such as CTCs and healthy WBCs19,45. Being able to separate blood cell sub-populations with fine resolution to define a size threshold that ensures ~100% capture of targeted rare cells, and then identify the targeted cells within this concentrated population via gentle immunolabeling in situ is a powerful combination of techniques needed for liquid biopsy. An integrated system that is multimodal, label-free, biomarker compatible, low cost and can be fully automated has enormous potential to make clinical impact. In this paper, our device has demonstrated the enrichment of MCF-7 cells from whole blood at concentrations as low as 10 mL−1, at ~100% capture efficiency, and verified by EpCAM biomarker staining in situ. In contrast, although hydrodynamic vortices based devices demonstrated cancer cell enrichment up to four orders of magnitude, it required significant blood dilution to avoid cell-cell interactions and the capture efficiency was only ~20%11. Another enormous advantage of this LCAT platform is in the speed of operation for cell enrichment and identification. Considering just the downstream analysis and enumeration of target cells after enrichment, benchtop immunostaining and fixation could take several hours. However, to develop an inexpensive and automated point-of-care device, we labelled the captured target cells in situ within 15 min and achieved 91% correlation in the fluorescence intensity by optimizing the LCAT pumped bulk flow.
While our system has great potential for liquid biopsy, it may be limited by throughput due to the low flow rate and the instability of LCATs over time. Inertial microfluidics based devices are high-throughput (mL min−1) but require significant dilution. Our strategy to improve throughput is to parallelize LCAT devices and increase the total blood sample volume it can handle. We are also optimizing the length of the sorting microchannels to maximize microfluidic channel length while minimizing hydrodynamic resistance. Another possibility is to flow a second pass of the enriched 20 μL sample in the device to reduce the volume required in the first place since it is likely the enrichment ratio will be greatly improved.
Overall, this work demonstrates a highly promising approach for integrated isolation, enrichment, release and identification of targeted blood cells, and ultimately could lead to the development of complete point-of-care blood diagnosis. The technology, based on acoustic microstreaming, processes whole blood and does not require centrifugation or benchside processing of separated blood components. Thus, an offshoot of this technology is to create a complete blood sample preparation front end device able to separate each of the blood cellular component (platelets, RBC and WBC), as each of these components contain critical information for the diagnosis of physiologic and pathologic conditions such as infectious diseases and inflammatory responses.
Conclusions
An acoustic microstreaming based LCAT device that can process whole blood and all its cellular constituents by size-based sorting, enrichment and subsequent biomarker-based identification is reported. This is, to our knowledge, the first demonstration of a single microfluidic device that combines the advantages of both label-free detection and in situ immunolabeling to select for targeted specific cell types. It is also the first device that can separate and collect the major components in whole blood (platelet rich plasma sample, RBCs and WBCs). Rapid immunolabeling (15 min) was demonstrated as targeted cells were trapped in microstreaming vortices. With these novel integrated functions, we demonstrated ~100% trapping efficiency of spiked polystyrene beads and MCF-7 cells in whole blood and achieved ~200× enrichment ratio in the sorted sample of 20 μL for concentrations as low as 10 mL−1.
References
Yu ZTF, Aw Yong KM, Fu J . Microfluidic blood cell sorting: now and beyond. Small 2014; 10: 1687–1703.
Mehmet Toner, Daniel Irimia . Blood-on-a-chip. Annual Review of Biomedical Engineering 2005; 7: 77–103.
Cui Francis, Rhee Minsoung, Singh Anup . Microfluidic sample preparation for medical diagnostics. Annual Review of Biomedical Engineering 2015; 17: 267–286.
Bhagat AAS, Bow H, Hou HW et al. Microfluidics for cell separation. Medical & Biological Engineering & Computing 2010; 48: 999–1014.
Jin C, McFaul S, Duffy SP et al. Technologies for label-free separation of circulating tumor cells: From historical foundations to recent developments. Lab on a Chip 2014; 14: 32–44.
Che J, Yu V, Dhar M et al. Classification of large circulating tumor cells isolated with ultra-high throughput microfluidic Vortex technology. Oncotarget 2016; 7: 12748–12760.
Gupta V, Jafferji I, Garza M et al. Apostream, a new dielectrophoretic device for antobody independent isolation and recovery of viable cancer cells from blood. Biomicrofluidics 2012; 6: 24133.
Lin B, McFaul S, Jin C et al. Highly selective biomechanical separation if cancer cells from leukocytes using microfluidic ratchets and hydrodynamic concentrator. Biomicrofluidics 2013; 7: 34114.
Li P, Mao Z, Peng Z et al. Acoustic separation of circulating tumor cells. Proceedings of the National Academy of Sciences of the United States of America 2015; 112: 4970–4975.
Yamada M, Seko W, Yanai T et al. Slanted, asymmetric microfluidic lattices as size-selective sieves for continuous particle/cell sorting. Lab on a Chip 2017; 17: 304–314.
Sollier E, Go D, Che J et al. Size-selective collection of circulating tumor cells using vortex technology. Lab on a Chip 2014; 14: 63–77.
Nivedita N, Papautsky I. . Continuous separation of blood cells in spiral microfluidic devices. Biomicrofluidics 2013; 7: 054101.
Gao JG, Riahi R, Sin MLY et al. Electrokinetic focusing and separation of mammalian cells in conductive biological fluids. Analyst 2012; 137: 5215–5221.
Augustsson P, Magnusson C, Nordin M et al. Microfluidic, label-free enrichment of prostate cancer cells in blood based on acoustophoresis. Analytical Chemistry 2012; 84: 7954–7962.
McFaul S, Lin B, Ma H . Cell separation based on size and deformability using microfluidic funnel ratchets. Lab on a Chip 2012; 12: 2369–2376.
Petersson F, Åberg L, Swärd-Nilsson A-M et al. Free flow acoustophoresis: Microfluidic-based mode of particle and cell separation. Analytical Chemistry 2007; 79: 5117–5123.
Kotz KT, Xiao W, Miller-Graziano C et al. Clinical microfluidics for neutrophil genomics and proteomics. Nature Medicine 2010; 16: 1042–1047.
Issadore D, Shao H, Chung J et al. Self-assembled magnetic filter for highly efficient immunomagnetic separation. Lab on a Chip 2011; 11: 147–151.
Kim MS, Sim TS, Kim YJ et al. SSA-MOA: A novel CTC isolation platform using selective size amplification (SSA) and a multi-obstacle architecture (MOA) filter. Lab on a Chip 2012; 12: 2874–2880.
Antfolk M, Magnusson C, Augustsson P et al. Acoustofluidic, label-free separation and simultaneous concentration of rare tumor cells from white blood cells. Analytical Chemistry 2015; 87: 9322–9328.
Ding X, Peng Z, Lin S-CS et al. Cell separation using tilted-angle standing surface acoustic waves. Proceedings of the National Academy of Sciences of the United States of America 2014; 111: 12992–12997.
Davis JA, Inglis DW, Morton KJ et al. Deterministic hydrodynamics: Taking blood apart. Proceedings of the National Academy of Sciences of the United States of America 2006; 103: 14779–14784.
Nagrath S. . Isolation of rare circulating tumor cells in cancer patients by microchip technology. Nature 2007; 450: 1235–1239.
Stott S, Hsu C, Tsukrov D et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proceedings of the National Academy of Sciences of the United States of America 2010; 107: 18392–18397.
Karabacack N, Spuhler P, Fachin F et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nature Protocols 2014; 9: 694–710.
Huang P-H, Nama N, Mao Z et al. A reliable and programmable acoustofluidic pump powered by oscillating sharp-edge structures. Lab on a Chip 2014; 14: 4319–4323.
Patel MV, Nanayakkara IA, Simon MG et al. Cavity-induced microstreaming for simultaneus on-chip pumping and size based separation of cells and particles. Lab on a Chip 2014; 14: 3860–3872.
Tovar AR, Lee AP. . Lateral cavity acoustic transducer. Lab on a Chip 2009; 9: 41–43.
Patel MV, Tovar AR, Lee AP. . Lateral cavity acoustic transducer as an on-chip cell/particle microfluidic switch. Lab on a Chip 2011; 12: 139–145.
Wang C, Jalikop SV, Hilgenfeldt S. . Efficient manipulation of microparticles in bubble streaming flows. Biomicrofluidics 2012; 6: 12801–1280111.
Rogers P, Neild A. . Selective particle trapping using an oscillating microbubble. Lab on a Chip 2011; 11: 3710–3715.
Wang C, Jalikop SV, Hilgenfeldt S. . Size-sensitive sorting of microparticles through control of flow geometry. Applied Physics Letters 2011; 99: 34101.
Burguillos MA, Magnusson C, Nordin M et al. Microchannel acoustophoresis does not impact survival or function of microglia, leukocytes or tumor cells. PLoS ONE 2013; 8: e64233.
Tovar AR, Patel MV, Lee AP. . Lateral air cavities for microfluidic pumping with the use of acoustic energy. Microfluidics and Nanofluidics 2011; 10: 1269–1278.
Hur S, Mach A, Di Carlo D. . High-throughput size-based rare cell enrichment using microscale vortices. Biomicrofluidics 2011; 5: 22206.
Chen Y, Li P, Huang P-H et al. Rare cell isolation and analysis in microfluidics. Lab on a Chip 2014; 14: 626–645.
Tong X, Yang L, Lang JC et al. Application of immunomagnetic cell enrichment in combination with RT-PCR for the detection of rare circulating head and neck tumor cells in human peripheral blood. Cytometry Part B: Clinical Cytometry. 2007; 72B: 310–323.
Dhar M, Pao E, Renier C et al. Label-free enumeration, collection and downstream cytological and cytogenetic analysis of circulating tumor cells. Science Reports. 2016; 6: 35474.
Nivedita N, Garg N, Lee A et al. A high throughput microfluidic platform for size-selective enrichment of cell populations in tissue and blood samples. Analyst 2017; 142: 2558–2569.
Yu M, Stott S, Toner M et al. Circulating tumor cells: approaches to isolation and characterization. The Journal of Cell Biology 2011; 192: 373–382.
Koumoutsakos P, Pivkin I, Milde F. . The fluid mechanics of cancer and its therapy. Annual Review of Fluid Mechanics 2013; 45: 325–355.
Liu Z, Huang F, Du J et al. Rapid isolation of cancer cells using microfluidic deterministic lateral displacement structure. Biomicrofluidics 2013; 7: 11801.
Carlo DD . Inertial microfluidics. Lab on a Chip 2009; 9: 3038–3046.
Chen Y, Wu M, Ren L et al. High-throughput acoustic separation of platelets from whole blood. Lab on a Chip 2016; 16: 3466–3472.
Marrinucci D, Bethel K, Bruce RH et al. Case study of the morphologic variation of circulating tumor cells. Human Pathology 2007; 38: 514–519.
Acknowledgements
This work was supported by the NSF Center for Advanced Design and Manufacturing of Integrated Microfluidics (CADMIM) (Award Nos. IIP-1362165 and IIP-1362048, Schlumberger Faculty for the Future Award (Award No. SF-202940), and the National Cancer Institute of the National Institutes of Health under award no. P30CA062203. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Microsystems & Nanoengineering website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Garg, N., Westerhof, T., Liu, V. et al. Whole-blood sorting, enrichment and in situ immunolabeling of cellular subsets using acoustic microstreaming. Microsyst Nanoeng 4, 17085 (2018). https://doi.org/10.1038/micronano.2017.85
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/micronano.2017.85
This article is cited by
-
Acoustic tweezers for high-throughput single-cell analysis
Nature Protocols (2023)
-
Manipulation of single cells via a Stereo Acoustic Streaming Tunnel (SteAST)
Microsystems & Nanoengineering (2022)
-
Nanodroplet-mediated catheter-directed sonothrombolysis of retracted blood clots
Microsystems & Nanoengineering (2021)
-
Plasma Isolation in a Syringe by Conformal Integration of Inertial Microfluidics
Annals of Biomedical Engineering (2021)
-
On-chip simultaneous rotation of large-scale cells by acoustically oscillating bubble array
Biomedical Microdevices (2020)
