
Abstract
Genetic defects that affect intestinal epithelial barrier function can present with very early-onset inflammatory bowel disease (VEOIBD). Using whole-genome sequencing, a novel hemizygous defect in NOX1 encoding NAPDH oxidase 1 was identified in a patient with ulcerative colitis-like VEOIBD. Exome screening of 1,878 pediatric patients identified further seven male inflammatory bowel disease (IBD) patients with rare NOX1 mutations. Loss-of-function was validated in p.N122H and p.T497A, and to a lesser degree in p.Y470H, p.R287Q, p.I67M, p.Q293R as well as the previously described p.P330S, and the common NOX1 SNP p.D360N (rs34688635) variant. The missense mutation p.N122H abrogated reactive oxygen species (ROS) production in cell lines, ex vivo colonic explants, and patient-derived colonic organoid cultures. Within colonic crypts, NOX1 constitutively generates a high level of ROS in the crypt lumen. Analysis of 9,513 controls and 11,140 IBD patients of non-Jewish European ancestry did not reveal an association between p.D360N and IBD. Our data suggest that loss-of-function variants in NOX1 do not cause a Mendelian disorder of high penetrance but are a context-specific modifier. Our results implicate that variants in NOX1 change brush border ROS within colonic crypts at the interface between the epithelium and luminal microbes.
Similar content being viewed by others

Introduction
In the gastrointestinal (GI) tract, the epithelium is the primary barrier separating the high density of microbes in the intestinal lumen from the immune cells in the lamina propria. Defects in epithelial barrier function predispose to inflammatory bowel disease (IBD) in humans and cause intestinal inflammation in animal models.1, 2
Genetic risk variants for IBD in loci linked to genes relevant for epithelial integrity have been identified by genome-wide association studies (e.g., GNA12, HNF4A, MUC19, or XPB1).3, 4 In addition, there is an expanding group of monogenic defects associated with significant functional impact on intestinal epithelial barrier function.5, 6 Defects in intestinal epithelial guanylcyclase gene GUCY2C or the solute carrier family 9 member 3 (SLC9A3) encoding the sodium hydrogen exchanger NHE3 cause congenital diarrhea and Crohn’s disease-like intestinal inflammation in ∼30% of cases.7, 8 Likewise, defects in IKBKG causing impaired nuclear factor-κB signaling result in epithelial apoptosis and intestinal inflammation.9 In patients with TTC7A deficiency, defects in epithelial PI4K and Rho signaling cause disturbed epithelial polarization leading to multiple intestinal atresia or very early-onset intestinal inflammation.10, 11, 12
Recently, we reported a common NOX1 missense mutation associated with ulcerative colitis (UC) in males of Ashkenazi Jewish ancestry, in addition to a rare NOX1 mutation in a patient with early onset of intestinal inflammation.13 NOX1 is the catalytic subunit of superoxide-generating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex 1, and is comprised of NOX1, p22phox, NOXA1, NOXO1, and Rac1-GTP.14, 15, 16 NOX1 is a close structural homolog of NOX2, the catalytic subunit of the multimeric NADPH oxidase complex 2 (NOX2, p22phox, p67phox, p47phox, p40phox, and Rac2-GTP), the enzyme responsible for the microbicidal “respiratory burst” in phagocytes. However, in contrast to NOX2, NOX1 is located in the membrane of the intestinal epithelial cells17, 18, 19, 20, 21 and, may have additional functions in endothelial cells and in non-GI epithelium.22
Here we investigate several loss-of-function missense variants in NOX1 that were identified by whole-genome and whole-exome sequencing in patients with very early-onset IBD (VEOIBD). We identified eight males with six hemizygous non-synonymous NOX1 variants, with variable functional impact upon reactive oxygen species (ROS) production as compared to the previously described p.P330S and the common p.D360N (rs34688635) variants. We identified two variants with nearly complete loss of function and a further four variants with reduced activity. Primary patient-derived intestinal colonic organoids of the loss-of-function variant p.N122H were used to validate those findings.
Results
Index patient
We studied a male patient of Caucasian origin who presented with bloody stools and failure to thrive at the age of 2 years (Table 1). He was diagnosed with IBD at the age of 5 years. His disease progressed from proctitis to pancolitis at the age of 8 years without upper GI involvement (Figure 1a). Histological examination of colonic specimens showed crypt architectural distortion, cryptitis, and crypt abscess formation (Figure 1a). Histologic assessment of epithelial cell proliferation or goblet cell formation did not reveal marked differences compared to inflammatory controls (Supplementary Figure S1 online). The patient required oral corticosteroid therapy at diagnosis, followed by thiopurine and then methotrexate therapy. After years of poorly controlled inflammation, mucosal healing was achieved with anti-tumor necrosis factor therapy (adalimumab). Nine years after diagnosis, his disease was complicated by Epstein–Barr virus-associated hemophagocytic lymphohistiocytosis whilst on azathioprine therapy, requiring intensive care therapy. Mutations in XIAP and SAP were excluded at that time. There was no family history of IBD.

Clinical phenotype of the NOX1 p.N122H patient and NOX1 contribution to baseline reactive oxygen species (ROS) within colonic crypts. (a) Endoscopy and histology from NOX1 mutant patient. Colonoscopy shows loss of vascular pattern, ulceration, and luminal bleeding (left). Hematoxylin and eosin stain of colonic biopsies shows crypt architectural distortion and dense inflammatory cell infiltrate in the lamina propria (scale bar=50 μm, middle). Magnification of NOX1 mutant epithelial crypt demonstrates elongated nuclei (black arrow head) and crypt abscesses (white arrow head, right). (b) Family pedigree and Sanger sequencing of NOX1 gene around the mutation site in a wild-type control and family members. (c) NOX1 protein sequence alignment of different genera showing the evolutionary-conserved asparagine at position 122. (d) The NOX1 variant p.N122H affects all three isoforms of NOX1. Amino-acid sequence was aligned using ClustalW. (e) Analysis of NOX1, NOXA1, and NOXO1 expression by quantitative PCR in biopsies of the gastrointestinal tract. Single biopsies per segment were obtained from four to seven non-inflamed individuals. Bars represent mean. Statistical significance was determined by Mann–Whitney U-test. (f) Ex vivo colonic biopsies (within 90 min of collection) were incubated with nitroblue tetrazolium (NBT) in phosphate-buffered saline-glucose at room temperature. Biopsy specimens were analyzed using light microscopy immediately after 60 min NBT incubation (top left, biopsy overview showing dark crypts containing reduced formazan; bottom, ring-like deposition of formazan at epithelial margin of crypts; top right, single colonic epithelial crypt captured longitudinally at high magnification). (g) Microscopy of ROS-producing epithelial cells in colonic crypts of ex vivo biopsies detected by NBT dye (× 200 magnification). After 60 min of incubation a ring-like deposition of formazan at the epithelial margin of crypts was evident. In indicated conditions biopsies were co-incubated with either a NOX1 inhibitor (ML171) or a non-specific NAPDH oxidase inhibitor (diphenyleneiodonium; DPI), to verify the signal. (h, left) DPI or ML171 inhibit superoxide generation and L-012-enhanced chemiluminescence on colonic biopsy specimens ex vivo. Following the detection of superoxide at baseline, DPI, ML171, or vehicle were added (black arrow) and chemiluminescence was recorded over 60 min. For each biopsy light emission was normalized to baseline reading. Data represent mean of nine biopsies per condition obtained from five non-inflamed controls. (h, right) Results of kinetic assay calculated as area under the curve per patient. Statistical significance was determined by Mann–Whitney U-test.
Whole-genome sequencing of the patient led to the identification of a novel hemizygous variant in the NADPH oxidase 1 NOX1; NM_007052; c.A364C; p.N122H (Supplementary Figure S2). The mutation was validated with Sanger sequencing and inherited in an X-linked manner from the patient’s mother (Figure 1b). The mutation is not present in 1,678 Crohn’s disease (CD), 1,089 UC patients, or 1,862 non-IBD controls, or in any public exome database covering an excess of 120,000 human alleles (e.g., 1000 Genomes and Exome Aggregation Consortium (ExAC)), thus supporting the view that this is an extremely rare genetic variant (Supplementary Table 1). The amino acid N122 is located in the third transmembrane domain and is evolutionary highly conserved across species (Figure 1c). In silico analysis by several tools, including SIFT, PolyPhen-2, and CADD,23 predicted that the mutation causes a loss-of-function (Supplementary Table 2). Furthermore, the p.N122H variant affects all known isoforms of NOX1 (Figure 1d). Other hemizygous, homozygous, or compound heterozygous variants in the index patient were considered unlikely pathogenic (Supplementary Table 3). None of these variants have been described as causative for IBD or Epstein–Barr virus-induced hemophagocytosis.
NOX1 expression and epithelial-derived ROS
To understand the relative distribution of ROS-generating NADPH oxidases in the human GI tract and to compare non-inflamed homeostasis with inflammatory conditions, we determined the mRNA expression pattern of several NADPH oxidases and their subunits. Consistent with previous literature,17, 18, 19, 20 NOX1 is highly expressed in the colonic epithelium with a gradient from ileum to the distal colon (Figure 1e). The subunits NOXA1 and NOXO1 are constitutively expressed along the human GI tract (Figure 1e). Whereas phagocytic NOX2 can be detected even in the non-inflamed mucosa, we observed no expression of NOX4 and very low expression of NOX5 (Supplementary Figure S3a). In parallel with the NOX1 expression gradient, we found increased ROS generation in ex vivo biopsies obtained from lower compared to the upper GI tract suggesting that NOX1 is the primary source of superoxide in the non-inflamed colonic epithelium (Supplementary Figure S3b,c).
We visualized superoxide production of colonic crypts ex vivo using nitroblue tetrazolium chloride. Reaction of nitroblue tetrazolium chloride with superoxide results in formation of water-insoluble blue formazan precipitates, which can be observed by light microscopy (Figure 1f). Following 60 min incubation, we observed the deposition of formazan at the outer margin of colonic crypts indicating the constitutive release of superoxide from the colonic epithelium into the crypt lumen (Figure 1f). Co-incubation with ML171 (ref. 24) or diphenyleneiodonium (DPI), both NADPH oxidase inhibitors, suggested a reduction of superoxide production (Figure 1g). This finding was confirmed by the luminescence assay (Figure 1h).
Ex vivo NOX1 biopsy and primary organoids
To assess the functional consequences of p.N122H mutation on colonic epithelial ROS production, we first assessed superoxide release in ex vivo colonic biopsies from the index patient and compared those to biopsies from patients with IBD and non-inflamed controls. Although biopsies from the NOX1 p.N122H variant patient generated low level of superoxide, this was within the range seen in other patients with quiescent IBD and non-inflamed controls (Figure 2a). However, as phagocytes that express NOX2 are present in non-inflamed biopsies (Supplementary Figure S3a) and lamina propria infiltrating phagocytes likely contribute to ROS measurements in IBD patient biopsies, the assay specificity is limited and an epithelial NOX1 defect is potentially masked.

NOX1 p.N122H diminishes epithelial reactive oxygen species in primary organoids. (a) Superoxide generation of colonic epithelial biopsies measured over 60 min by L-012-enhanced chemiluminescence and total light emission quantified by area under the curve (AUC). Results are derived from 17 patients with colonic Crohn’s disease (active disease n=10), 11 patients with ulcerative colitis (active disease n=6), 16 non-inflamed controls, and the patient with NOX1 p.N122H mutation. One symbol represents the mean of 1–3 pan-colonic biopsy specimens (both right- and left-sided) per patient. Active vs. quiescent disease dichotomized according to composite assessment, including endoscopic appearance (mucosal ulceration, loss of vascular pattern, and bleeding), biomarkers (C-reactive protein (CRP)), and histology (density of inflammatory infiltrate, cryptitis, and crypt abscesses). Statistical significance was determined by Mann–Whitney U test. (b) Organoids from patient with NOX1 mutation p.N122H and inflammatory bowel disease (IBD) control patient were generated from colonic biopsies obtained during quiescent disease state. Representative images of endoscopy, histology (hematoxylin and eosin staining, scale bar=50 μm), and organoids are displayed. (c) Analysis of NADPH oxidases and certain subunits by quantitative PCR in NOX1 p.N122H patient and IBD control organoids. (d) Kinetic assay of phorbol 12-myristate 13-acetate (PMA)-activated L-012-enhanced chemiluminescence on NOX1 p.N122H mutant, IBD control, and non-inflamed control organoid cells. Relative light units were measured over 60 min. Results are representative of two independent experiments measured in replicates. Statistical significance was determined by Wilcoxon signed-rank test comparing the AUC.
NOX1 p.N122H reduces ROS production in primary organoids
To validate the functional impact of the genetic defect in epithelial cells, we generated primary epithelial organoids from colonic biopsy specimens of the NOX1 p.N122H patient, a male IBD control patient, and a healthy female control without IBD (Figure 2b). Both IBD patients were in clinical remission at the time of the endoscopy. Colonic organoids express NOX1 mRNA, along with its subunits NOXA1 and NOXO1, and this expression did not differ between the index p.N122H patient and the IBD control patient (Figure 2c). We found high DUOX2 mRNA expression but only minimal NOX2 expression in the colonic epithelial organoids reflecting the epithelial origin and the absence of phagocytes (Supplementary Figure S3a).
Colonic epithelial organoid cells carrying NOX1 p.N122H mutation produced significantly less constitutive superoxide than control organoid cells derived from the IBD patient or a patient without inflammatory disease (Figure 2d). Of note, whereas phorbol 12-myristate 13-acetate (PMA) stimulation increased superoxide generation by control organoids, PMA stimulation of NOX1 p.N122H organoids did not. Even after stimulation with PMA, superoxide production by NOX1 p.N122H organoids was significantly less than baseline ROS of IBD control organoids (Figure 2d).
Functional characterization of multiple NOX1 hemizygous variants in VEOIBD
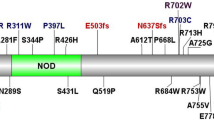
To find additional NOX1 variants in patients with VEOIBD we screened exomes of 1,878 pediatric IBD patients. We identified 5 additional non-synonymous variants in NOX1 in separate patients in whom no other known or plausible genetic monogenic diagnosis was established (Table 1). The missense variants were predicted to be damaging by SIFT, PolyPhen-2, or CADD scores, and were rare (minor allele frequency <1%) genetic variants (Supplementary Tables 1 and 2). All variants changed conserved amino acids (Supplementary Figure S4) and were found in regions with significant homology between NOX1 and NOX2 (Supplementary Figure S5).
To characterize the functional impact of those variants we transfected the multimeric NADPH oxidase complex 1 in the colonic epithelial cell line HCT116. Alongside the rare NOX1 variants p.N122H, p.I67M, p.R287Q, p.Q293R, p.Y470H, and p.T497A that we identified in this study, we tested the previously described p.P330S variant13 and the common polymorphism p.D360N (allele frequency European-non-Finnish, 2.4%). Three variants in patients P14, P15, and P16 affected potential splice sites, which we could not assess functionally (Supplementary Tables 1, 2, and 4). We used HCT116 cells as these cells express only low levels of NOX1 subunits (data not shown) and endogenous ROS generation cannot be detected (Figure 3a). Co-expression of wild-type NOX1 together with the subunits NOXA1 and NOXO1 resulted in superoxide generation in unstimulated and PMA-stimulated condition (Figure 3a,b). In contrast, co-expression with NOX1 variants variably diminished ROS generation. Whereas NOX1 variants p.N122H (index patient) and p.T497A completely abrogated both spontaneous and PMA-induced ROS production, the NOX1 variants p.I67M, p.R287Q, p.Q293R, p.P330S, p.D360N (common polymorphism), or p.Y470H were observed to either decrease spontaneous ROS and/or diminish PMA-stimulated ROS (Figure 3a,b). Expression levels of wild-type and variant NOX1 constructs were comparable (data not shown). This suggests that the rare genetic variants affected the ROS production to a variable degree between complete loss-of-function and moderate reduction comparable to polymorphisms.

Comparative analysis of NOX1 variants. (a, b) Generation of superoxide in colonic epithelial cell line HCT116 reconstituted with multimeric NADPH oxidase complex and expressing wild-type or variant NOX1. ROS was analyzed by L-012-enhanced chemiluminescence in the absence (a) or presence of phorbol 12-myristate 13-acetate (PMA) (b). Chemiluminescence was measured 48 h post transfection and recorded over 60 min. Results represent eight independent experiments with 3–4 replicates. Each dot represents one replicate. Experiments were normalized to mean relative light units (RLUs) without PMA. Line represents mean. Statistical significance was determined with analysis of variance and post-test correction performed with Dunnett (multiple comparisons to one single control).
Characteristics of patients with loss-of-function NOX1 variants
We analyzed the IBD phenotype in all male patients with confirmed loss-of-function NOX1 variants (Table 1 and Supplementary Table 4). Out of the 10 male patients with hemizygous NOX1 mutations, 4 presented with CD, 3 with UC, and 3 with IBD unclassified. In congruency with NOX1 expression, disease location was always colonic, with two patients also having perianal disease. Median age of onset was 7.9 years (range 1.8–10.5 years). Medical treatment included corticosteroids, aminosalicylates, azathioprine, methotrexate, or anti-tumor necrosis factor (infliximab or adalimumab in 4 of 8 patients) and 1 patient underwent ileocecal resection for stricture. No infections with intestinal pathogens were recorded. With exception of the index patient, no other severe hemophagocytic lymphohistiocytosis / Epstein–Barr virus-driven pathologies were recorded.
Population-based characteristics of genetic loss-of-function NOX1 variants
In addition to those rare variants in NOX1, common polymorphisms might similarly contribute to IBD susceptibility. Of note, the common NOX1 polymorphism p.D360N has recently been associated with UC in Ashkenazi Jewish males.13 To determine whether this is Ashkenazi Jewish-specific or an association across several populations, we performed a genetic analysis of individuals of non-Jewish European ancestry. Although we observed a trend in the US European ancestry cohort (Table 2; Supplementary Tables 5 and 6), neither analysis of allele counts in UK cohorts nor analysis of summed allele counts from a total of 9,513 healthy controls and 11,140 IBD patients revealed an association (Table 2). This suggests that the association of NOX1 p.D360N with UC in males is not present across different populations. Variation is also reflected by a variable distribution of p.D360N allele frequency across populations (Figure 4a).

NOX1 population frequencies and genetic variation. (a) Allele frequency of NOX1 p.D360N in different populations according to Exome Aggregation Consortium (ExAC) data and published literature.13 (b) Number of hemizygotes found for a given variant (missense, nonsense, and frameshift) in NOX2 and NOX1 is blotted according to amino-acid position (x-axis). Frequency is based on ExAC data. (c) Probability of loss of-function intolerance (pLI) of 727 human X-chromosomal genes based on ExAC data. X-linked primary immunodeficiency genes and NOX1 gene are highlighted.
To further understand the importance of NOX1 variation, we compared the frequency of hemizygous NOX1 missense and NOX1 loss-of-function variants (STOP codon, frameshift, and predicted splice site), with those in NOX2, the causative gene of most patients with chronic granulomatous disease. The frequency of genetic missense variation in NOX1 is higher as compared to the homologous NOX2 (Figure 4b). Similarly, the constraint metric based on the ExAC data set indicates that loss-of-function variants in NOX1 are tolerated as indicated by a probability of being loss-of-function intolerant25 of 0 (Figure 4c). In contrast NOX2 is extremely intolerant to any loss-of-function variation (probability of loss-of-function intolerant 0.998)25 as are many other genes causing X-linked primary immunodeficiencies (Figure 4c). This suggests that NOX1 underwent less evolutionary pressure as compared to NOX2 or other X-linked immunodeficiency genes.
In summary, our data suggest that NOX1 is the key NADPH oxidase for baseline ROS in human colonic crypt epithelium. Complete loss-of-function variants in NOX1 are rare and not associated with a distinct IBD sub-phenotype. Population statistics suggest that loss-of-function variants in NOX1 are rare but likely tolerated.
Discussion
In this study we investigated rare X-linked variants in NOX1 in eight male IBD patients. Functional analysis suggests a variant-dependent loss of NOX1-mediated superoxide generation. The clinical phenotype of affected patients was early onset of IBD with progressive and severe colonic disease, refractory to conventional therapy. The affected cohort demonstrates that NOX1-dependent ROS production is essential for superoxide production in colonic crypts.
Our characterization of six rare variants in NOX1 (p.I67M, p.N122H, p.R287Q, p.Q293R, p.Y470H, and p.T497A) expands the recent description of the rare NOX1 variant p.P330S and the common NOX1 polymorphism p.D360N in two male VEOIBD patients.13 Furthermore, the NOX1 variant p.R287Q, which is functionally characterized in this study, has also been described but not characterized in an independent cohort of children with VEOIBD, adding further weight to the significance of epithelial NOX1.26
Diverse mechanisms maintain intestinal epithelial integrity, which serves as a critical barrier to luminal bacteria, viruses, and fungi.1, 2 Amongst these, NOX1 is likely to have a relevant role.27, 28 The generation of superoxide by NOX1 at the colonic epithelial border may have an anti-adhesive or anti-invasive effect on bacteria, contributing to the barrier function of the inner mucus layer within colonic crypts. Indeed, our data confirm previous findings that the gene expression of NOX1 follows the density of bacterial colonization in the GI tract.17, 18, 19, 20 Reduced generation of superoxide due to genetic variants in NOX1 may affect crypt homeostasis allowing subsequent crypt colonization by luminal microbiota. NOX1-derived superoxide also influences intestinal epithelial differentiation and goblet cell function.29, 30, 31 Colonic mucus produced by goblet cells provides an important barrier between the intestinal microbiota and the colonic epithelium, maintaining sterility at the epithelial border.32
The importance of NADPH oxidases for barrier function is also exemplified by DUOX2, an NADPH oxidase family member, which is expressed throughout the GI tract. Notably, defects in DUOX2 have been recently described as a potential susceptibility factor in patients with VEOIBD.13, 33 In animal models, defects in the Drosophila duox2 homolog or in Zebrafish increased susceptibility against enteropathogens.34, 35 Whereas Duoxa−/− mice on thyroid hormone replacement showed normal growth and no evident phenotype,36 recent studies suggested that colonization of segmented filamentous bacteria in the ileum, bacterial translocation into mesenteric lymph nodes, and Salmonella typhimurium systemic dissemination, is dependent on DUOX activity.37 Although DUOX2 expression is inducible and is increased in patients with intestinal inflammation33, 38 thereby potentially contributing to epithelial stress and intestinal tissue damage, these data indicate that DUOX activity may a functional mechanism that restricts bacterial colonization at the mucosal interface.
Interestingly, NOX1 activity regulates DUOX2 expression in the intestinal epithelium during Citrobacter rodentium infection.39 Although loss of epithelial Cyba protected against C. rodentium and Listeria monocytogenes infection, this is likely an indirect effect due to the resulting overgrowth of H2O2-producing lactobacilli.39 This suggests an unexpected level of functional hierarchy and complex collaboration between NOX1 and DUOX2.39
Despite the complete loss of brush border ROS that has been observed in two NOX1 variants (p.N122H and p.T497A), we have not observed an increased susceptibility to intestinal infections such as Salmonella or Yersinia. This is in agreement with recent data suggesting that lack of Nox1 does not affect infection susceptibility in a mouse model of Salmonella typhimurium-induced typhlitis.40 Nonetheless, larger patient numbers with complete loss-of-function NOX1 variants are required to more definitively determine the impact of these mutations on susceptibility to infection. Interestingly, the patient with the p.N122H mutation developed severe hemophagocytic lymphohistiocytosis in the context of Epstein–Barr virus infection whilst on azathioprine therapy.41 Viral spreading has been shown to be increased in Nox1-deficient mice infected with influenza virus,42 implicating a role for NOX1 in viral control. Again, larger patient numbers are required to fully understand the penetrance of the NOX1 variant phenotype.
Given the relatively high load of non-synonymous mutations of NOX1 compared to NOX2 and the large number of DUOX2 mutations in humans, it is likely that NOX/DUOX enzymes have partially redundant functions. It is therefore likely that both NOX1 and DUOX2 have a role in susceptibility to intestinal inflammation, and loss-of-function variants alone cause not highly penetrant Mendelian-type disorders. In keeping with a second hit hypothesis, Nox1 deficiency in interleukin-10-deficient mice resulted in earlier and more severe intestinal pathology, but loss of Nox1 alone did not cause intestinal inflammation.30 Similarly, patients with congenital secretory diarrhea due to activating mutations in the guanylate cyclase C (GC-C)7, 43 or inactivating mutations in SLC9A3 encoding for the sodium/proton exchanger NHE3,8 a downstream target of GC-C, may also present with an IBD phenotype. In these patients the decreased epithelial uptake of sodium results in changes of mucosal ion content and pH, and Nhe3 deficiency in mice leads to an altered composition of the intestinal microbiota.44, 45 This dysbiosis drives intestinal inflammation in a T-cell-dependent model of intestinal inflammation.46 Our studies suggest that loss of ROS at the colonic crypt may have a similar effect on barrier function.
Altogether these data suggest a highly context specific role of epithelial NADPH oxidases in intestinal homeostasis. Whilst in some animal models, a lack of interleukin-10 and Nox1 increases epithelial stress and susceptibility to colitis,30 in other instances, lack of NADPH oxidase activity is protective. Indeed a protective role of Nox1 and Duox2 is suggested in a mouse model of ileocolitis caused by lack of protective glutathione peroxidase factors.47, 48, 49 In this model GPx1- and GPx2-knockout leads to oxidative stress caused by Nox1 and Duox2 with a dominant role of Nox1 for crypt epithelial apoptosis.48, 49 This oxidative stress is dependent on the presence of the intestinal microbiota as germ-free mice are protected and colitis develops within 1 week of bacterial colonization.50 Given the role of NOX1 as a modifier of intestinal microbiota, we can only speculate whether IBD patients with loss-of-function NOX1 variants would benefit from therapeutic microbiome-altering strategies (e.g., antibiotics, nutrition, probiotics, or fecal transplant). Further studies characterizing the human gut microbiota in the setting of NOX1 deficiency are required.
It is possible that the human population genetics reflect this context specific role of NOX1. Whereas in populations like Ashkenazi Jews, the hemizygous NOX1 p.D360N variant is associated with UC, in European descent populations it is not. Substantially stronger genetic variation of NOX1 compared to NOX2 suggests that the overall impact on disease susceptibility of these genes is significantly different despite the high degree of protein conservation and similarity. It is even possible that the degree of genetic variation in NOX1 is caused by environmental factors such as infections where reduced ROS is protective.
In conclusion, we report six novel variants in the NOX1 gene in patients with IBD, two of which are associated with complete loss of function of the gene product and with loss of ROS production. Our study illustrates the complexity of epithelial barrier function in the pathogenesis of IBD and the need better to understand the functional impact of ROS production at the colonic crypt epithelium as a cause of colonic inflammation.
Methods
Human subjects
The recruitment of participants was approved by relevant ethics committees or institutional review boards at the individual contributing institution. Written informed consent was obtained from all patients/legal guardians and healthy controls.
Patients with IBD (including the index patient P1) and controls without intestinal inflammation were prospectively recruited as a part of the Oxford IBD cohort study or the Oxford Gastrointestinal Illness Biobank (REC 09/H1204/30 and REC 11/YH/0020). Replication genotyping of the p.N122H variant was performed in 2,767 IBD patients and 1,862 non-IBD controls. These individuals were enrolled by the UK IBD Genetics Consortium (http://www.ibdresearch.co.uk/). Exomes of 1,878 pediatric-onset IBD patients without genetic diagnosis were screened for additional rare NOX1 variants. Samples were previously genotyped as part of the COLORS in IBD study (n=150; Oxford, UK), the NEOPICS consortium (n=545; Toronto, ON, Canada; www.neopics.org), or the RISK cohort51 (n=1,183; Atlanta, GA, USA). Genetic association testing of NOX1 p.D360N (rs34688635) was conducted on genotype data from IBD patients of non-Jewish European ancestry from the following cohorts: the European ancestry exome chip data set52 (n=10,523); the Oxford IBD cohort study (n=513); and the COLORS in IBD study (n=104). Sequence data from healthy individuals were obtained from the European ancestry exome chip data set52 (n=5,726) and the INTERVAL study (n=4,436; www.intervalstudy.org.uk). Population controls were also obtained from the UK10K project (www.uk10k.org).
Genotyping
Whole-genome sequencing and NOX1 variant p.N122H identification
The index mutation in NOX1 was identified by whole-genome sequencing as part of the WGS500 project.53 DNA was analyzed from peripheral blood mononuclear cells. In summary, whole-genome sequencing was performed on 3.5–7.5 ng DNA on either the Illumina HiSeq2000 (Cambridge, UK) or the HiSeq2500 run in standard mode using v2.5 or v3 sequencing chemistry. The genomic DNA was fragmented, end-paired, A-tailed, and adapter-ligated before size selection and amplification for a multiplexed library preparation as described in Taylor et al.53 The libraries were paired-end sequenced and sequenced to an average coverage between 27 and 40 quality reads per base. Resulting 100 base pair reads were mapped to the GRCh37d5/hg19 human reference sequence using Stampy and the single-nucleotide variants and short InDels were called with Platypus.54 Variant files were analyzed using Qiagen’s Ingenuity Variant Analysis software (www.qiagen.com/Ingenuity; Qiagen Redwood City, CA, USA). High-quality calls were analyzed. For candidate analysis, we investigated a gene list of known variants associated with monogenic forms of IBD.5 Filters for variant frequency (<1%; 1000 Genomes Project) and predicted deleterious filters (SIFT and PolyPhen) were applied to prioritize high quality calls for non-synonymous and likely pathogenic variants.
Replication genotyping of the p.N122H variant
Replication genotyping on the Sequenom plex was carried out at the Sanger Institute.
Whole-exome sequencing
Regeneron performed whole-exome sequencing on a cohort of 1,183 probands with pediatric-onset IBD (ages 0–18.5 years), including their affected and unaffected parents and siblings, where available (total samples=2,704). Sample preparation, whole-exome sequencing, and sequence data production were performed as previously described.55 The exomes of RISK cohort study participants (n=545) were captured using SureSelect Human All Exon kit v2 (Agilent Technologies, Oxford, UK), using 3 μg of genomic DNA. Subsequently, captured libraries were sequenced on Illumina HiSeq2000 by 100-base pair paired-end sequencing. Image processing and base calling was performed using standard Illumina software. Raw sequence reads were mapped relative to the human genome reference sequence (GRCh38/hg38) using the PEMapper software tool (Cutler DJ et al., submitted) to identify single-nucleotide variants.
Sanger sequencing
NOX1 variants were validated using Sanger sequencing according to the standard techniques.
Histology
Historic routine hematoxylin and eosin histology sections from the patient identified to harbor the NOX1 p.N122H mutation and IBD controls were analyzed. Staining of goblet cells or the proliferation marker Ki67 was performed on archived paraffin-embedded colonic epithelial biopsies. Specimens were stained using an Alcian blue and periodic acid-Schiff’s reagent staining kit (Clin-Tech Limited, Guildford, UK) or rabbit monoclonal antibody to Ki67 (clone SP66, AbCam, Cambridge, USA), respectively. Nuclear fast red and hematoxylin (Sigma-Aldrich, Gillingham, UK) were used to counterstain cell nuclei.
Protein sequence alignment
Multiple sequences were aligned using ClustalW2.56 Data were obtained from the National Center for Biotechnology Information. Sequence alignment is based on the following accession numbers: Danio rerio, NP_001095857.1; Xenopus tropicalis, XP_002939249.2; Gallus gallus, NP_001095300.1; Mus musculus, NP_757340.1; Homo sapiens, NOX1 isoform 1–3, NP_008983.2, NP_039249.1, and NP_001258744.1; Homo sapiens, NOX2, NP_000388.2.
Expression analysis
Colonic biopsies were stored in RNAlater (Qiagen) before analysis. RNA was extracted using RNAeasy kit (Qiagen), transcribed in cDNA with High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA), and expression data were obtained using TaqMan primers (Life Technologies, Paisley, UK) for NOX 1-5, NOXO1, NOXA1, DUOX1, and DUOX2. Results were normalized to GAPDH expression. Relative gene expression was calculated using the ΔCt method.
Colonic epithelial organoid cultures
Human colon crypts were isolated and organoids were cultured as previously described.57 Briefly, crypts were embedded in Cultrex Reduced Growth Factor Basement Membrane Extract, Type 2 RGF PathClear (Amsbio, Abingdon, UK) and seeded in 48-well plates. The Basement Membrane Extract was polymerized for 10 min at 37 °C, and 250 μl per well of organoid culture media was added. Organoid culture media consisted of advanced Dulbecco’s modified eagle medium/F12 supplemented with penicillin/streptomycin (10 mmol l−1), HEPES (1 M), Glutamax (10 mmol l−1), and the following growth factors: Wnt conditioned media (50%) B27 (1 ×), epidermal growth factor (50 ng ml−1), Noggin (10%), R spondin (20%), gastrin (10 nM), nicotinamide (10 mM), A83-01 (500 nM), and SB202190 (10 μM). Y-27632 (10 μmol l−1; Sigma) was included in the medium for the first 5 days in culture. Once organoid growth was established, the culture media was refreshed every 3 days.
Transfection of HCT116 cells
The colonic epithelial cell line HCT116 was maintained in McCoy’s 5A Modified (Gibco, Paisley, UK) supplemented with 10% fetal bovine serum. Cell systems were based on reconstitution of the multimeric NOX1 complex containing NOX1, NOXO1, and NOXA1 in HCT116 cells. PcDNA3.1-hNox1, pcDNA3.1-hNoxa1, and pcDNA3.1-hNoxo1 were kind gifts from Botond Banfi & Karl-Heinz Krause (Addgene, Cambridge, MA, plasmid # 58344, # 58531, and # 58530).19, 58 Mutations were introduced using site-directed mutagenesis and were verified by sequencing (Eurofins Genomics, Ebersberg, Germany). HCT116 cells were transfected with Lipofectamine 2000 (Invitrogen, Paisley, UK) following the manufacturer’s instructions. Expression of wild-type and NOX1 variants was confirmed by quantitative PCR. ROS production was analyzed by L-012-enhanced chemiluminescence 48 h post transfection.
ROS assays
ROS generation by colonic epithelial biopsies, organoid cells, and transiently transfected HCT116 cells was measured using L-012-enhanced chemiluminescence (Wako Laboratories, Neuss, Germany). After washing with phosphate-buffered saline, cells or biopsies were pre-incubated in L-012 (100 μM) for 10 min, followed by stimulation with PMA (5 μg ml−1, Sigma) or addition of diphenyleneiodonium (DPI, 10 μM, Sigma) and 2-acetylphenothiazine (ML171, 10 μM, Tocris, Bristol, UK) in indicated conditions. Emission of light was recorded at baseline and thereafter as a kinetic assay every 1–10 min in opaque white 96-well plates (FLUOstar OPTIMA, BMG Labtech, Aylesbury, UK).
For standardization, all intestinal epithelial biopsies were collected using Olympus (Southend-on-Sea, UK). Endojaw needle forceps with a cup-opening size of 7.2 mm and analyzed within 90 min of collection with minimal handling. Organoids were trypsinized into single-cell suspension using TrypLETM express (Gibco), counted, and equal cell numbers dispensed into white 96-well plates.
For microscopy of ROS-producing epithelial cells, ex vivo colonic biopsies were incubated with 100 μg ml−1 nitroblue tetrazolium chloride (Sigma) in phosphate-buffered saline-glucose (1 g l−1) with or without NADPH oxidase inhibitors. After 60 min incubation, formation of formazan was analyzed by light microscopy (Carl Zeiss Axioskop2, Cambridge, UK, × 200 magnification) on glass slides without coverslips. Images were photographed using Q-Imaging software (Surrey, BC, Canada). Microscopy images were cropped and analyzed using ImageJ software.59
Disease activity amongst IBD patients was characterized as active or quiescent on the basis of clinical symptoms (diarrhea, rectal bleeding, and abdominal pain), endoscopic features (mucosal vascular pattern, bleeding, or ulceration), histological features (cryptitis and crypt abscesses, and lamina propria polymorphomuclear cell infiltrate), and serum biomarkers (C-reactive protein).
Data and statistical analysis
All the data analyses were performed using GraphPad Prism version 5.02 (GraphPad Software, San Diego, CA) or R (The Foundation for Statistical Computing, R version 3.2.2, Vienna, Austria). Unpaired data were compared using Mann–Whitney U-test and paired data were compared with Wilcoxon signed-rank test. Significance of differences between multiple groups was performed with analysis of variance and post-test correction with Dunnet. Genetic association testing in cases and controls was conducted with the ALLELIC test.60 Pooled analyses were performed on the summed allele counts of different cohorts. P-values of <0.05 after correction for multiple testing were considered significant.
References
Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9, 799–809 (2009).
Pastorelli, L., De Salvo, C., Mercado, J.R., Vecchi, M. & Pizarro, T.T. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front. Immunol. (2013) 4, 280.
Khor, B., Gardet, A. & Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature (2011) 474, 307–317.
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Uhlig, H.H. Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut 62, 1795–1805 (2013).
Uhlig, H.H. et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 147, 990–1007 e1003 (2014).
Fiskerstrand, T. et al. Familial diarrhea syndrome caused by an activating GUCY2C mutation. N. Engl. J. Med. 366, 1586–1595 (2012).
Janecke, A.R. et al. Reduced sodium/proton exchanger NHE3 activity causes congenital sodium diarrhea. Hum. Mol. Genet. 24, 6614–6623 (2015).
Cheng, L.E. et al. Persistent systemic inflammation and atypical enterocolitis in patients with NEMO syndrome. Clin. Immunol. 132, 124–131 (2009).
Chen, R. et al. Whole-exome sequencing identifies tetratricopeptide repeat domain 7A (TTC7A) mutations for combined immunodeficiency with intestinal atresias. J. Allergy Clin. Immunol. 132, 656–664 e617 (2013).
Samuels, M.E. et al. Exome sequencing identifies mutations in the gene TTC7A in French-Canadian cases with hereditary multiple intestinal atresia. J. Med. Genet. 50, 324–329 (2013).
Avitzur, Y. et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology 146, 1028–1039 (2014).
Hayes, P. et al. Defects in NADPH oxidase genes and in very early onset inflammatory bowel disease. Cell. Mol. Gastroenterol. Hepatol. 1, 489–502 (2015).
Bedard, K. & Krause, K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313 (2007).
O’Neill, S., Brault, J., Stasia, M.J. & Knaus, U.G. Genetic disorders coupled to ROS deficiency. Redox Biol. 6, 135–156 (2015).
Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4, 181–189 (2004).
Suh, Y.A. et al. Cell transformation by the superoxide-generating oxidase Mox1. Nature 401, 79–82 (1999).
Szanto, I. et al. Expression of NOX1, a superoxide-generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J. Pathol. 207, 164–176 (2005).
Banfi, B., Clark, R.A., Steger, K. & Krause, K.H. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J. Biol. Chem. 278, 3510–3513 (2003).
Glebov, O.K. et al. Distinguishing right from left colon by the pattern of gene expression. Cancer Epidemiol. Biomarkers Prev. 12, 755–762 (2003).
Jones, R.M. & Neish, A.S. Redox signaling mediated by the gut microbiota. Free Radic. Biol. Med. 105, 41–47 (2016).
Konior, A., Schramm, A., Czesnikiewicz-Guzik, M. & Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 20, 2794–2814 (2014).
Kircher, M., Witten, D.M., Jain, P., O’Roak, B.J., Cooper, G.M. & Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Gianni, D. et al. A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem. Biol. 5, 981–993 (2010).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Kelsen, J.R. et al. Exome sequencing analysis reveals variants in primary immunodeficiency genes in patients with very early onset inflammatory bowel disease. Gastroenterology 149, 1415–1424 (2015).
Jones, R.M. et al. Symbiotic lactobacilli stimulate gut epithelial proliferation via Nox-mediated generation of reactive oxygen species. EMBO J. 32, 3017–3028 (2013).
Kawahara, T. et al. Role of nicotinamide adenine dinucleotide phosphate oxidase 1 in oxidative burst response to Toll-like receptor 5 signaling in large intestinal epithelial cells. J. Immunol. 172, 3051–3058 (2004).
Patel, K.K. et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J. 32, 3130–3144 (2013).
Treton, X. et al. Combined NADPH oxidase 1 and interleukin 10 deficiency induces chronic endoplasmic reticulum stress and causes ulcerative colitis-like disease in mice. PLoS ONE 9, e101669 (2014).
Coant, N. et al. NADPH oxidase 1 modulates WNT and NOTCH1 signaling to control the fate of proliferative progenitor cells in the colon. Mol. Cell. Biol. 30, 2636–2650 (2010).
Johansson, M.E. et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 63, 281–291 (2014).
Levine, A.P. et al. Genetic complexity of Crohn’s disease in two large Ashkenazi Jewish families. Gastroenterology 151, 698–709 (2016).
Ha, E.M., Oh, C.T., Bae, Y.S. & Lee, W.J. A direct role for dual oxidase in Drosophila gut immunity. Science 310, 847–850 (2005).
Flores, M.V., Crawford, K.C., Pullin, L.M., Hall, C.J., Crosier, K.E. & Crosier, P.S. Dual oxidase in the intestinal epithelium of zebrafish larvae has anti-bacterial properties. Biochem. Biophys. Res. Commun. 400, 164–168 (2010).
Grasberger, H. et al. Mice deficient in dual oxidase maturation factors are severely hypothyroid. Mol. Endocrinol. 26, 481–492 (2012).
Grasberger, H. et al. Increased expression of DUOX2 is an epithelial response to mucosal dysbiosis required for immune homeostasis in mouse intestine. Gastroenterology 149, 1849–1859 (2015).
Haberman, Y. et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Invest. 124, 3617–3633 (2014).
Pircalabioru, G. et al. Defensive mutualism rescues NADPH oxidase inactivation in gut infection. Cell Host Microbe 19, 651–663 (2016).
Chu, F.F., Esworthy, R.S., Doroshow, J.H. & Shen, B. NADPH oxidase-1 deficiency offers little protection in Salmonella typhimurium-induced typhlitis in mice. World J. Gastroenterol. 22, 10158–10165 (2016).
Biank, V.F. et al. Association of Crohn’s disease, thiopurines, and primary epstein-barr virus infection with hemophagocytic lymphohistiocytosis. J. Pediatr. 159, 808–812 (2011).
Selemidis, S. et al. Nox1 oxidase suppresses influenza a virus-induced lung inflammation and oxidative stress. PLoS ONE 8, e60792 (2013).
Muller, T. et al. Congenital secretory diarrhoea caused by activating germline mutations in GUCY2C. Gut 65, 1306–1313 (2015).
Larmonier, C.B. et al. Reduced colonic microbial diversity is associated with colitis in NHE3-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G667–G677 (2013).
Kumar, A. et al. Lactobacillus acidophilus counteracts inhibition of NHE3 and DRA expression and alleviates diarrheal phenotype in mice infected with Citrobacter rodentium. Am. J. Physiol. Gastrointest. Liver Physiol. 311, G817–G826 (2016).
Laubitz, D. et al. Reduced epithelial Na+/H+ exchange drives gut microbial dysbiosis and promotes inflammatory response in T cell-mediated murine colitis. PLoS ONE 11, e0152044 (2016).
Esworthy, R.S., Aranda, R., Martin, M.G., Doroshow, J.H., Binder, S.W. & Chu, F.F. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G848–G855 (2001).
Chu, F.F. et al. Deficiency in Duox2 activity alleviates ileitis in GPx1- and GPx2-knockout mice without affecting apoptosis incidence in the crypt epithelium. Redox Biol. 11, 144–156 (2016).
Esworthy, R.S., Kim, B.W., Chow, J., Shen, B., Doroshow, J.H. & Chu, F.F. Nox1 causes ileocolitis in mice deficient in glutathione peroxidase-1 and -2. Free Radic. Biol. Med. 68, 315–325 (2014).
Esworthy, R.S., Binder, S.W., Doroshow, J.H. & Chu, F.F. Microflora trigger colitis in mice deficient in selenium-dependent glutathione peroxidase and induce Gpx2 gene expression. Biol. Chem. 384, 597–607 (2003).
Cutler, D.J. et al. Dissecting allele architecture of early onset IBD using high-density genotyping. PLoS ONE 10, e0128074 (2015).
Li, D. et al. A pleiotropic missense variant in SLC39A8 is associated with Crohn’s disease and human gut microbiome composition. Gastroenterology 151, 724–732 (2016).
Taylor, J.C. et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat. Genet. 47, 717–726 (2015).
Rimmer, A. et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 46, 912–918 (2014).
Li, Q. et al. Variants in TRIM22 that affect NOD2 signaling are associated with very-early-onset inflammatory bowel disease. Gastroenterology 150, 1196–1207 (2016).
Larkin, M.A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Sato, T. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009).
Banfi, B. et al. A mammalian H+ channel generated through alternative splicing of the NADPH oxidase homolog NOH-1. Science 287, 138–142 (2000).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Clarke, G.M., Anderson, C.A., Pettersson, F.H., Cardon, L.R., Morris, A.P. & Zondervan, K.T. Basic statistical analysis in genetic case-control studies. Nat. Protoc. 6, 121–133 (2011).
Moore, C. et al. The INTERVAL trial to determine whether intervals between blood donations can be safely and acceptably decreased to optimise blood supply: study protocol for a randomised controlled trial. Trials 15, 363 (2014).
Levine, A. et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: the Paris classification. Inflamm. Bowel Dis. 17, 1314–1321 (2011).
Acknowledgements
We thank all our patients and their families for participation in this study. We thank all contributors of the Oxford IBD cohort study in particular Carolina Arancibia and Sophia Bennett for help with Sanger sequencing.
Participants in the INTERVAL randomized controlled trial were recruited with the active collaboration of NHS Blood and Transplant England (www.nhsbt.nhs.uk), which has supported field work and other elements of the trial. DNA extraction and genotyping was funded by the National Institute of Health Research (NIHR), the NIHR BioResource (http://bioresource.nihr.ac.uk/), and the NIHR Cambridge Biomedical Research Centre (www.cambridge-brc.org.uk). The academic coordinating centre for INTERVAL was supported by core funding from the following: NIHR Blood and Transplant Research Unit in Donor Health and Genomics; UK Medical Research Council (G0800270); British Heart Foundation (SP/09/002); and NIHR Research Cambridge Biomedical Research Centre. A complete list of the investigators and contributors to the INTERVAL trial is provided in reference.61
R.V.B. and H.H.U. were supported by the Medical Research Fund Oxford. Part of this work has formed the MScR thesis of R.V.B. T.S. is supported by the Deutsche Forschungsgemeinschaft (SCHW1730/1-1). H.H.U. and F.P. are supported by the Crohn’s & Colitis Foundation of America (CCFA); A.M.M., H.H.U., D.P.B.M., and F.P. by The Leona M. and Harry B. Helmsley Charitable Trust; and H.H.U. by Crohn’s and Colitis UK—for Crohns—;H.H.U. and D.C.W. by ESPGHAN. S.P. was supported by a Boehringer Ingelheim Foundation grant. F.P. is supported by the Wellcome Trust. U.G.K. is supported by Science Foundation Ireland. The WGS500 Project was funded in part by Illumina. The Oxford IBD cohort is supported by Oxford BRC. L.M. and V.S.W.L. are supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK, the UK Medical Research Council, and the Wellcome Trust. S.K. and L.D. are supported by R01 DK098231. R.K.R. is supported by a NHS Research Scotland Senior Fellowship. D.C.W. is supported by Medical Research Council and Medical Research Foundation for PICTS; Crohn’s and Colitis UK. J.R.K. is supported by the National Institutes of Health (K23DK100461-01A1). D.P.B.M. is supported by P01 DK046763 and DK062413. This work was funded by a Wellcome Trust Core Award (090532/Z/09/Z) and a Medical Research Council Hub grant (G0900747 91070) to Peter Donnelly, the NIHR Biomedical Research Centre Oxford, the UK Department of Health’s NIHR Biomedical Research Centres funding scheme, and Illumina. D.J.M. was supported by the General Sir John Monash Foundation, Australia.
Web Resources
The URLs for data presented herein are as follows:
Plasmids, https://www.addgene.org/
1000 Genomes, http://browser.1000genomes.org
dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/
ExAC Browser, http://exac.broadinstitute.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
HGMD, http://www.hgmd.cf.ac.uk/ac/index.php
RefSeq, http://www.ncbi.nlm.nih.gov/RefSeq
SIFT, http://sift.jcvi.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
PROVEAN, http://provean.jcvi.org/index.php
Combined Annotation Dependent Depletion (CADD), http://cadd.gs.washington.edu/
Author contributions
T.S., R.V.B., S.P., M.C., L.M., J.J., V.S.W.L., and H.H.U. designed and performed experiments, evaluated and interpreted data. K.M., K.F., P.B.S., A.R., S.P.L.T., R.K.R., D.C.W., J.R.K., L.A.D., S.K., A.M.M., R.H.D., D.P.B.M., C.G.M., J.S., W.H.O., D.J.R., J.D., and J.C. contributed patients, collected clinical data and samples, or screened cohorts. J.-B. C., M.P., C.G.M., R.C., E.G.S., C.A.A., R.H.D., D.P.B.M., and J.C. conducted bioinformatics and statistical analyses. U.G.K., F.P., V.S.W.L., A.M.M., E.G.S., C.A.A., R.H.D., D.P.B.M., D.J.M., and J.C. gave conceptual advice. H.H.U. drafted the manuscript and directed the study. All authors reviewed and contributed to the writing of the manuscript.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
H.H.U. declares industrial project collaboration with Eli Lilly and UCB Pharma. Travel support was received from GSK foundation, Actelion, and MSD. T.S. received speaker’s fees from MSD and travel support from Nestlé Nutrition. R.V.B. has received conference travel support from Janssen. R.V.B. has received speaker’s fees from Janssen, Takeda, and Shire, and travel support from Takeda. S.P.L.T. has received Grants/Research Support from AbbVie, IOIBD, Lilly, UCB, Vifor, and Norman Collisson Foundation; Consulting Fees and or travel support from AbbVie, Amgen, Asahi, Biogen, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Chemocentryx, Cosmo, Ferring, Giuliani SpA, GlaxoSmithKline, Janssen, Lilly, MSD, Neovacs, NovoNordisk, Norman Collison Foundation, Novartis, NPS Pharmaceuticals, Pfizer, Proximagen, Receptos, Shire, Sigmoid Pharma, Takeda, Topivert, UCB, VHsquared, and Vifor; and Speaker fees from AbbVie, Amgen, Biogen, Ferring, and Takeda. R.K.R. has received speaker’s fees, travel support, and participated in medical board meetings with MSD Immunology, Abbvie, Nestle, and Takeda. D.C.W. received investigator-initiated research funding from MSD, speaker’s fees and travel support from AbbVie, and consultancy fee from Pfizer.
Additional information
WGS500 Consortium: Names and Affiliations of Authors Steering Committee P Donnelly (Chair)5, J Bell32, D Bentley33, G McVean5, P Ratcliffe5, J Taylor5,34, AOM Wilkie34,35 Operations Committee P Donnelly5 (Chair), J Broxholme5, D Buck5, J-B Cazier37, R Cornall5, L Gregory5, J Knight5, G Lunter5, G McVean5, J Taylor5,34, I Tomlinson5,34, AOM Wilkie34,35 Sequencing and Experimental Follow-up D Buck5 (Lead), C Allan5, M Attar5, A Green5, L Gregory5, S Humphray33, Z Kingsbury33, S Lamble5, L Lonie5, A Pagnamenta5, P Piazza45, G Polanco46,47, A Trebes5 Data Analysis G McVean5 (Lead), P Donnelly5, J-B Cazier37, J Broxholme5, R Copley38, S Fiddy39, R Grocock33, E Hatton5, C Holmes5, L Hughes5, P Humburg5, A Kanapin40, S Lise41, G Lunter5, H Martin42, L Murray33, D McCarthy43, A Rimmer44, N Sahgal5, B Wright5, C Yau37 32Office of the Regius Professor of Medicine, Richard Doll Building, Roosevelt Drive, Oxford OX3 7LF, UK; 33Illumina Cambridge, Chesterford Research Park, Little Chesterford CB10 1XL, UK; 34NIHR Oxford Biomedical Research Centre, Oxford, UK.; 35Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Headington, Oxford OX3 9DS, UK; 36Imperial College London, South Kensington Campus, London SW7 2AZ, UK; 37Centre for Computational Biology, Haworth Building, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK; 38UPMC Paris 6, CNRS UMR 7009, Villefranche-sur-Mer; 39Oxford Nanopore Technology, Edmund Cartwright House, 4 Robert Robinson Avenue, Oxford Science Park, Oxford OX4 4GA, UK; 40CRUK Oxford Centre, Department of Oncology, Old Road Campus Research Building, Roosevelt Drive, Oxford OX3 7DQ, UK; 41Centre for Evolution and Cancer, The Institute of Cancer Research, Brookes Lawley Building, 15 Cotswold Road, Sutton SM2 5NG, UK; 42Sanger Institute, Wellcome Genome Campus, Hinxton CB10 1SA, UK; 43EMBL-EBI, Wellcome Genome Campus, Hinxton CB10 1SD, UK; 44Genomics plc, King Charles House, Park End Street, Oxford OX1 1JD, UK; 45Imperial College London, Commonwealth Building, Hammersmith Campus, Du Cane Road, London W12 0NN, UK; 46Genome Center and Department of Biochemistry and Molecular Medicine, School of Medicine, University of California, Davis, CA, USA; 47Grupo de Citogenética, Filogenia y Evolución de Poblaciones, Facultades de Ciencias y Facultad de Ciencias de la Salud, Universidad del Tolima, Ibagué, Colombia
Carolina Arancibia, Adam Bailey, Ellie Barnes, Beth Bird-Lieberman, Oliver Brain, Barbara Braden, Jane Collier, James East, Alessandra Geremia, Lucy Howarth, Satish Keshav, Paul Klenerman, Simon Leedham, Rebecca Palmer, Fiona Powrie, Astor Rodrigues, Alison Simmons, Peter B Sullivan, Simon P L Travis, Holm H Uhlig
Holm H. Uhlig, Astor Rodrigues, Peter B. Sullivan, Children s Hospital, John Radcliffe Hospital, Oxford, UK and Department of Paediatrics, University of Oxford, UK Rob Heuschkel, Matthias Zilbauer, Addenbrooke's Hospital, Cambridge, UK Marcus KH Auth, Alder Hey Children's Hospital, Liverpool, UK Neil Shah, Jochen Kammermeier, Great Ormond Street Hospital, London, UK Nick Croft, Farah Barakat, The Royal London Children's Hospital, Barts Health NHS Trust. London UK and Barts and the London School of Medicine, Queen Mary University of London, London UK Richard K. Russell, Royal Hospital for Children, Glasgow, UK David C. Wilson, Paul Henderson, Royal Hospital for Sick Children, Edinburgh, UK and Child Life and Health, University of Edinburgh, Edinburgh, UK Christian P. Braegger, University Children's Hospital Zurich, Zurich, Switzerland Carsten Posovszky, University Children's Hospital Ulm, Ulm, Germany Krzysztof Fyderek, Andrzej Wędrychowicz, University Children's Hospital Krakow, Krakow, Poland Marlen Zurek, St. Georg Hospital, Leipzig, Germany Caterina Strisciuglio, Department of Translational Medical Sciences, Section of Pediatrics, University of Naples, Italy Mamoun Elawad, Bernice Lo, Sidra Medical and Research Center, Doha, Qatar Miles Parkes, Inflammatory Bowel Disease Research Group, Addenbrooke's Hospital, Cambridge, UK Jack Satsangi, Gastrointestinal Unit, Western General Hospital University of Edinburgh, Edinburgh, UK Carl A. Anderson, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, UK
L Jostins5, NA Kennedy48, CA Lamb49, T Ahmad48, C Edwards50, A Hart51, C Hawkey52, JC Mansfield53, C Mowat54, WG Newman55, J Satsangi56, A Simmons1, M Tremelling57, HH Uhlig1,11, DC Wilson20,21, JC Lee7, NJ Prescott8, CW Lees56, CG Mathew8, M Parkes7, JC Barrett16, CA Anderson16 48Precision Medicine Exeter, University of Exeter, Exeter, UK; 49Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, UK; 50Department of Gastroenterology, Torbay Hospital, Torbay, UK; 51Department of Medicine, St Mark’s Hospital, Harrow, UK; 52Nottingham Digestive Diseases Centre, Queens Medical Centre, Nottingham, UK; 53Institute of Human Genetics, Newcastle University, Newcastle upon Tyne, UK; 54Department of Medicine, Ninewells Hospital and Medical School, Dundee, UK; 55Genetic Medicine, Manchester Academic Health Science Centre, Manchester, UK; 56Gastrointestinal Unit, Western General Hospital University of Edinburgh, Edinburgh, UK; 57Gastroenterology & General Medicine, Norfolk and Norwich University Hospital, Norwich, UK
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Schwerd, T., Bryant, R., Pandey, S. et al. NOX1 loss-of-function genetic variants in patients with inflammatory bowel disease. Mucosal Immunol 11, 562–574 (2018). https://doi.org/10.1038/mi.2017.74
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2017.74
