
Abstract
Mast cells and basophils are innate immune cells with overlapping functions that contribute to anti-helminth immunity. Mast cell function during helminth infection was previously studied using mast cell-deficient Kit-mutant mice that display additional mast cell-unrelated immune deficiencies. Here, we use mice that lack basophils or mucosal and connective tissue mast cells in a Kit-independent manner to re-evaluate the impact of each cell type during helminth infection. Neither mast cells nor basophils participated in the immune response to tissue-migrating Strongyloides ratti third-stage larvae, but both cell types contributed to the early expulsion of parasitic adults from the intestine. The termination of S. ratti infection required the presence of mucosal mast cells: Cpa3Cre mice, which lack mucosal and connective tissue mast cells, remained infected for more than 150 days. Mcpt5Cre R-DTA mice, which lack connective tissue mast cells only, and basophil-deficient Mcpt8Cre mice terminated the infection after 1 month with wild-type kinetics despite their initial increase in intestinal parasite burden. Because Cpa3Cre mice showed intact Th2 polarization and efficiently developed protective immunity after vaccination, we hypothesize that mucosal mast cells are non-redundant terminal effector cells in the intestinal epithelium that execute anti-helminth immunity but do not orchestrate it.
Similar content being viewed by others


INTRODUCTION
Helminth parasites infect approximately one-third of the world’s population.1 Strongyloides ratti is a rodent-specific parasite with a rapid life cycle that includes tissue-migrating and intestinal parasitic stages.2 Thus, infective third-stage larvae (L3i) actively penetrate the skin of their mammalian host and migrate within 2 days to the mouth, predominantly via the head but also via the lung.3, 4, 5 The L3 are swallowed and arrive in the small intestine 3 days after infection. They moult via fourth-stage larvae to become parasitic adults on day 5 post infection (p.i.). The adults live embedded in the mucosa closely attached to intestinal epithelial cells6 and reproduce by parthenogenesis. The eggs and already hatched first-stage larvae (L1) are excreted with the feces. Immunocompetent mice efficiently terminate the infection: the intestinal numbers of parasitic adults decline rapidly after day 6 p.i.3 Although there are few detectable adults after day 10 p.i., mice remain infected at a low level and continue to release eggs and L1, as shown by a sensitive quantitative-PCR (qPCR) method, for approximately 30 days, until the infection is completely terminated.5 A completed first S. ratti infection elicits a typical Th2 response and renders mice semi-resistant to subsequent infections.3, 5
Therefore, S. ratti-infected mice can be used to analyze the mechanisms of efficient anti-helminth immunity in tissues and the intestine. Although the effector mechanisms that eradicate the tissue-migrating L3 have been studied extensively by using human pathogenic S. stercoralis L3,7 the mechanisms of intestinal immunity are less well understood.8 Mast cells are thought to mediate the expulsion of parasitic adults from the intestine because injections with the mast cell-activating cytokine interleukin (IL)-3 promoted parasite expulsion and mastocytosis during infection with S. ratti.9, 10 Moreover, IL-3-deficient mice infected with the closely related S. venezuelensis showed delayed expulsion of the parasites. Furthermore, when WBB6F1-KitW/W-v (W/Wv) mice, which carry a mutation in the stem cell factor receptor Kit that results in mast cell deficiency, were infected with S. venezuelensis or S. ratti, they had a higher intestinal parasite burden during infection.11, 12 However, because W/Wv mice have several immunological and non-immunological defects, including reduced basophil numbers and lack of interstitial pacemaker cells of Cajal,13 their phenotypes may not solely reflect the mast cell deficiency.14 Re-evaluation of mast cell function using novel Kit-independent models for mast cell deficiency failed to reproduce the involvement of mast cells in different immune pathologies such as autoimmune arthritis and experimental autoimmune encephalomyelitis,15 wound healing,16 postoperative ileus,17 or diet-induced obesity and insulin resistance18 that was proposed using Kit-mutant mice. Thus, the role of mast cells in Strongyloides elimination remains unclear. Moreover, a differentiation between the impact of the two mast cell subsets, the constitutive connective tissue mast cells that are predominantly located in the skin and submucosa and the mucosal mast cells that are located within mucosal epithelium19 was not possible using Kit-mutant mice. Finally, because basophils and mast cells have overlapping functions,20 are both induced and activated by IL-3,21 and contribute to immunity during gastrointestinal helminth infection,22 it remains unclear to what extent each cell type contributes to anti-Strongyloides immunity.
Here, we used the mast cell-deficient Cpa3Cre mice along with two other murine models of mast cell or basophil deficiency15, 23, 24 to elucidate the overlapping and distinct functions of these cells during S. ratti infection in the tissue and intestine. Neither mast cells nor basophils contributed to control of migrating larvae during the first 2–3 days of infection, but both cell types promoted the early expulsion of S. ratti from the intestine. Moreover, the presence of mucosal mast cells was found to be essential for the timely termination of S. ratti infection: mice that lacked all mast cells had prolonged infection, whereas deficiency of connective tissue mast cells alone or basophils alone did not affect the timely termination of infection at about day 30 p.i.
RESULTS
Mucosal mast cells are essential for termination of S. ratti infection
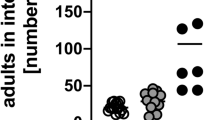
Heterozygous expression of Cre recombinase under the control of the Cpa3 promoter deleted all mucosal and connective tissue mast cells in Cpa3Cre mice by genotoxic mechanisms.15 To analyze the importance of mast cells in the immunological control of S. ratti, we infected Cpa3Cre mice and their mast cell-intact Cpa3wt littermates on either the C57BL/615 or the BALB/c25 background by subcutaneously injecting 2,000 L3i. Mast cell-deficient and -intact mice did not differ in terms of tissue-migrating L3 numbers in the head and lungs during the first 4 days of infection, regardless of the genetic background (Figure 1a,b). The L3 numbers in the head generally peaked on day 2 p.i., as described before,3 and L3 started to appear in the small intestine 1 day later. The mast cell-deficient and -intact mice did not differ in intestinal L3 numbers until day 3 p.i. However, on day 4 p.i., the mast cell-deficient mice tended to have higher larval burden in the intestine, although this difference did not achieve statistical significance. On day 6 p.i., that is the time point of maximal S. ratti burden, the mast cell-deficient mice had significantly more parasitic adults in the intestine than their mast cell-competent littermates, regardless of genetic background (Figure 1c,d). Thus, although mast cells were dispensable for controlling tissue-migrating L3, they contributed to the intestinal immunity against S. ratti.

Mast cell deficiency increases S. ratti adult burden in the intestine. Cpa3Cre (black symbols) and Cpa3wt (open symbols) mice were infected with 2,000 S. ratti L3i by s.c. injection. (a, b) Numbers of larvae in the head, lung, and intestine on days 1–4 p.i. in mice on the C57BL/6 (a) and BALB/c (b) genetic backgrounds. (c, d) Numbers of parasitic adults in the intestine on day 6 p.i. (c) C57BL/6 background. (d) BALB/c background. The data are the means of two (a, b) and three (c, d) independent experiments (a, b: n=3 (day 1) or n=11 (days 2–4) mice per group; c, d: n≥16 mice per group). ***P<0.001: the groups differed significantly at the indicated time points, as determined by using Mann–Whitney U test (a–c) or Student’s t-test (d). L3i, infective third-stage larvae; p.i., post infection; s.c., subcutaneous; S. ratti, Strongyloides ratti.
BALB/c mice are less susceptible to S. ratti infection than C57BL/6 mice.26 Indeed, BALB/c Cpa3wt mice had fewer parasitic adults than C57BL/6 Cpa3wt mice after infection with the same number of L3i (Figure 1c,d: open symbols). Notably, however, the two strains did not differ in terms of the L3 numbers in the head and lungs at any time point (Figure 1a,b). Thus, the strain-specific differences in susceptibility were established in the intestine.
To compare the kinetics of infection in the presence and absence of mast cells, we quantified S. ratti DNA in the feces as correlate for fecal egg and L1 release5 (Figure 2). Wild-type mice displayed maximal S. ratti DNA output during days 6–8 and terminated the infection after 1 month as described before.5 By contrast, the mast cell-deficient mice released higher S. ratti DNA levels in the first weeks of infection, correlating with the higher numbers of parasitic adults in their intestine. The quantity of S. ratti DNA declined after approximately 60 days in mast cell-deficient C57BL/6 Cpa3Cre mice (Figure 2a) and after 14 days in BALB/c Cpa3Cre mice (Figure 2b), thus highlighting the different susceptibility of these strains. However, both mast cell-deficient strains continued to pass S. ratti DNA in the feces for more than 150 days (Figure 2a,b: insets). The continuous release of S. ratti DNA in the mast cell-deficient mice was accompanied by the presence of vital and fertile S. ratti adults and L1 in their intestine on day 90 p.i. (Supplementary Figure S1 and Movie online).

Mast cell deficiency prolongs S. ratti infection. Cpa3Cre (black bars) and Cpa3wt (open bars) mice on the C57BL/6 (a) and BALB/c (b) background were s.c. infected with 2,000 S. ratti L3i and the release of S. ratti 28S RNA-coding DNA in their feces at the indicated time points until termination of infection was analyzed by qPCR. The data are the means of two independent experiments (n=6–12 mice per group and time point). *P<0.05, **P<0.01, and ***P<0.001: the groups differed significantly at the indicated time point, as determined by Student’s t-test or Mann–Whitney U test. Once wild-type mice terminated the infection and all values of this group became zero, e.g., at day 48 (a) or day 42 (b) p.i., statistical analysis with Student’s t-test or Mann–Whitney U test were no longer performed. L3i, infective third-stage larvae; ns, not significant; p.i., post infection; qPCR, quantitative-PCR; s.c., subcutaneous; S. ratti, Strongyloides ratti.
To determine whether mucosal and connective tissue mast cells both contribute to controlling intestinal parasite burden, we used mice that expressed Cre recombinase under the control of the mast cell protease (Mcpt) 5 promoter and were crossed with R-DTA mice (Mcpt5Cre R-DTA) leading to diphtheria toxin expression and subsequent deletion of connective tissue mast cells.23 In line with the results obtained using complete mast cell-deficient Cpa3Cre mice, the numbers of tissue-migrating L3 and intestinal L3 at day 2 p.i. were alike in wild-type and Mcpt5Cre R-DTA mice lacking connective tissue mast cells only (Figure 3a). Numbers of parasitic adults in the intestine at day 6 p.i. (Figure 3b) and fecal output of S. ratti DNA during the first 2 weeks (Figure 3c) increased in connective tissue mast cell-deficient Mcpt5Cre R-DTA mice compared with their wild-type littermates. Thus, the absence of connective tissue mast cells compromised initial intestinal immunity to S. ratti even in the presence of mucosal mast cells. Despite this, however, the Mcpt5Cre R-DTA mice cleared the infection by day 30 p.i. with similar kinetics as their wild-type littermates, which indicates that connective tissue mast cells were not needed for terminating S. ratti infection. Thereby, stable deletion of connective tissue mast cells in the small intestine of Mcpt5Cre R-DTA mice also at this late time point of infection was controlled by histology and RT-PCR (Supplementary Figure S2).

Role of connective tissue mast cells during S. ratti infection. Mcpt5Cre R-DTA (Mcpt5Cre: black triangles/bars) and Mcpt5wt R-DTA (Mcpt5wt: open triangles/bars) mice on C57BL/6 background were s.c. infected with 2,000 S. ratti L3i. (a) Number of L3 in head, lung, and intestine on day 2 p.i. (b) Number of parasitic adults in the intestine on day 6 p.i. (c) Release of S. ratti 28S RNA-coding DNA in feces at the indicated time points until termination of infection, as measured by qPCR. The data are the means of two (a) and three (b, c) independent experiments (a: n≥9; b: n≥14; c: n≥10; mice per group). *P<0.05 and **P<0.01: the groups differed significantly, as determined by Mann–Whitney U or Student’s t-test. L3i, infective third-stage larvae; ns, not significant; p.i., post infection; qPCR, quantitative-PCR; s.c., subcutaneous; S. ratti, Strongyloides ratti.
In addition to their mast cell deficiency, Cpa3Cre mice have a 60% reduction in the numbers of splenic basophils.15 To assess whether the higher adult parasite burden in the Cpa3Cre mice was caused by the absence of mast cells and/or the reduction in basophil numbers, basophil-deficient Mcpt8Cre mice were infected with S. ratti. In these mice, heterozygous BAC transgenic expression of Cre recombinase under control of the regulatory elements of the basophil-specific protease Mcpt8 results in constitutive and efficient basophil deletion.24 The basophil-deficient mice and their wild-type littermates had similar numbers of tissue-migrating L3 on day 2 p.i. (Figure 4a). Numbers of parasitic adults in the intestine at day 6 p.i. increased in basophil-deficient Mcpt8Cre mice on BALB/c and C57BL/6 genetic background (Figure 4b). Increased intestinal parasite burden was correlated to increased fecal release of S. ratti DNA (Figure 4c,d). Despite this, the infection was terminated with similar kinetics in basophil-deficient and -intact mice before day 30 p.i. (Figure 4c,d). Thus, both connective tissue mast cells and basophils contributed to the early intestinal immunity against S. ratti and specifically presence of mucosal mast cells was needed for timely termination of infection. The impact of mucosal mast cells on early intestinal immunity remains to be elucidated, as no mice lacking selectively mucosal mast cells in the presence of connective tissue mast cells are presently available.

Role of basophils during S. ratti infection. Mcpt8Cre (black symbols/bars) and Mcpt8wt (open symbols/bars) mice were s.c. infected with 2,000 S.ratti L3i (a) Number of larvae in the head and lung on day 2 p.i. in Mcpt8Cre and Mcpt8wt C57BL/6 mice (n≥14 mice per group). (b) Number of parasitic adults in the intestine on day 6 p.i. (n≥12 mice per group). (c, d) Release of S. ratti 28S RNA-coding DNA in the feces at the indicated time points until termination of infection, as measured by qPCR. (c) C57BL/6 background. (d) BALB/c background. (n≥6 mice per group and time point). The means of two (a, c, d) and three (b) independent experiments are shown. *P<0.05 and **P<0.01: the groups differed significantly, as determined by Student’s t-test or Mann–Whitney U test. L3i, infective third-stage larvae; ns, not significant; p.i., post infection; qPCR, quantitative-PCR; s.c., subcutaneous; S. ratti, Strongyloides ratti.
Next, we intended to evaluate the impact of adaptive immunity on early intestinal immunity to S. ratti as well as during infection termination. Previous studies showed that T-cell-deficient nude mice remained infected with S. ratti for more than 6 weeks27 and nude rats carried parasitic S. ratti adults in the intestine for more than 1 year.28 S. ratti-infected RAG1−/− mice, which lack all cells expressing adaptive immune receptors (i.e., B cells, T cells, and NKT cells), had similar numbers of parasitic adults in the intestine on day 6 p.i. as the wild-type C57BL/6 control mice (Figure 5a). Nevertheless, the RAG1−/− mice remained infected for more than 300 days and displayed increased fecal release of S. ratti DNA at later time points when immune competent mice already cleared the infection (Figure 5b).

Role of adaptive immunity during S. ratti infection. RAG1−/− (black symbols/bars) and C57BL/6 (Wt: open symbols/bars) were s.c. infected with 2,000 S. ratti L3i (a) Number of adult parasites in the intestine on day 6 p.i. (n≥10 mice per group). (b) Release of S. ratti DNA in the feces, as measured by qPCR (n≥4 mice per group and time point). The data show the mean of two independent experiments (a) or a representative of two experiments (b). **P<0.01: the groups differed significantly, as determined by Student’s t-test (a) or Mann–Whitney U test (b). Once wild-type mice terminated the infection and all values of this group became zero, e.g., day 27 p.i. (b), statistical analyzes with Student’s t-test or Mann–Whitney U test were no longer performed. L3i, infective third-stage larvae; ns, not significant; p.i., post infection; qPCR, quantitative-PCR; s.c., subcutaneous; S. ratti, Strongyloides ratti.
In summary, these experiments show that intestinal parasite numbers were initially controlled by innate immune effector mechanisms also in the absence of a functional adaptive immune system. During this early phase, mast cells and basophils contributed to the parasite control on day 6 p.i. Final eradication of S. ratti from the intestine, however, required neither connective tissue mast cells nor basophils, but strictly relied on the presence of a functional adaptive immune system. Additional absence of mucosal mast cells in Cpa3Cre mice, however, abrogated timely termination of infection even in the presence of adaptive immunity.
Intact generation of S. ratti-specific immune response in mast cell-deficient mice
The incapability of complete mast cell-deficient Cpa3Cre mice to terminate S. ratti infection suggests that mucosal mast cells may be key terminal effector cells that execute the immune-driven eradication of S. ratti. Alternatively, mucosal mast cells may promote S. ratti eradication indirectly by helping to initiate the adaptive immune response to S. ratti as observed in another gastrointestinal nematode infection recently.29 To distinguish between these alternatives, we compared the cellular composition and local immune responses of complete mast cell-deficient (Cpa3Cre) and wild-type mice during infection (Figure 6). Histological analysis of the intestinal cellular composition in day 6 S. ratti-infected mice revealed no differences with regard to numbers of T cells, B cells, macrophages, eosinophils, neutrophils, or goblet cells in Cpa3Cre and Cpa3wt mice, while mast cells were absent in Cpa3Cre mice, as expected (Figure 6a and Supplementary Figure S3). Also, the local immune response reflected by infection-induced upregulation of the Th2-associated cytokine mRNA for IL-13, IL-4, IL-5, IL-3, IL-9, and IL-10 in intestinal tissues did not differ significantly in Cpa3Cre and Cpa3wt mice (Figure 6b). When mesenteric lymph node cells from mast cell-deficient Cpa3Cre and mast cell-competent Cpa3wt mice on either BALB/c or C57BL/6 genetic background were stimulated with anti-CD3 monoclonal antibody, they produced mostly comparable amounts of the respective Th2-associated cytokines (Figure 6c and Supplementary Figure S4a). The mast cell-deficient mice had comparable S. ratti-specific IgM concentrations and even higher serum concentrations of S. ratti-specific IgG1 and polyclonal IgE than the control mice (Figure 6d and Supplementary Figure S4b). This increased IgG1 and IgE titers in S. ratti-infected Cpa3Cre mice may reflect a deregulation of the Th2 immune response initiated in the absence of mast cells. It is also possible that the increased Ig response was due to increased and prolonged intestinal parasite burden in mast cell-deficient mice, as vaccination with irradiated L3i that do not establish a patent infection in the intestine and thus lead to comparable antigen loads resulted in comparable humoral responses in mast cell-deficient and -intact mice (Supplementary Figure S5a). Furthermore, connective tissue mast cell-deficient Mcpt5Cre R-DTA mice that terminated infection with wild-type kinetics displayed comparable Ig responses as their wild-type littermates at later time points of infection, when parasite burden was also comparable (Supplementary Figure S5b).

Cellular composition and immune response in S. ratti-infected Cpa3Cre mice. Cpa3Cre (black dots/bars) and Cpa3wt (open dots/bars) on BALB/c genetic background were infected with 2,000 S. ratti L3i. (a) Quantification of the cellular composition in the intestine of infected Cpa3Cre and Cpa3wt mice at day 6 p.i. by histopathologic analysis (n=7–13 mice per group); MΦ: macrophages. (b) Relative expression of cytokine mRNA in the small intestine of day 6 infected mice to naive mice was measured by qPCR and calculated using the comparative Ct method. Results are expressed as 2−(ΔΔCt) (n=7–9). (c) Cytokine production by α-CD3 activated MLNs derived from day 6 infected mice was quantified by ELISA (n=6–9 mice per group). (d) S. ratti-specific IgM and IgG1 titers and IgE concentration in sera taken from day 28 infected mice was measured by ELISA (n=5–8). Means of two independent experiments are shown in each graph. *P<0.05, ***P<0.001, ****P<0.0001: the groups differed significantly, as determined by Student’s t-test or Mann–Whitney U test. L3i, infective third-stage larvae; MLNs, mesenteric lymph nodes; p.i., post infection; qPCR, quantitative-PCR. S. ratti, Strongyloides ratti.
Although classic Th2 polarization during S. ratti infection was not abrogated by complete mast cell deficiency, the increased parasite burden in mast cell-deficient mice correlated with increased IgG and IgE responses, while cytokine production was not increased. Normalized to parasite numbers, this finding could be interpreted as a reduced and thus impaired cytokine response in mast cell-deficient mice. Therefore, we intended to further test the quality of the immune response that was initiated in the absence of mast cells. Immunocompetent mice, which terminate S. ratti infection within 30 days, are semi-resistant to second infections; they have only 10–20% of the initial fecal egg and L1 output, and the adults in the intestine are almost undetectable.3, 5 Because the mast cell-deficient Cpa3Cre mice could not terminate the infection before day 150 p.i. (Figure 2), we achieved protective immunity by vaccinating these mice with irradiated L3i. This vaccination regimen reduced the number of tissue migrating L3 present in the head 2 days after challenge infection (Figure 7a,b) and parasitic adults in the intestine 6 days after challenge infection (Figure 7c–e) with 2,000 live S. ratti L3i in comparison with non-vaccinated mice (first infection to challenge). The number of migrating L3 in heads of vaccinated Cpa3Cre and Cpa3wt mice did not differ during challenge infection, indicating that mast cell-deficient C57BL/6 Cpa3Cre mice (Figure 7a) and BALB/c Cpa3Cre mice (Figure 7b) benefited just as well from vaccination as their mast cell-intact littermates. Subsequent parasite burden in the intestine on day 6 after challenge infection was also significantly reduced in vaccinated Cpa3Cre and Cpa3wt mice compared with non-vaccinated mice (Figure 7c,d). However, vaccinated mast cell-deficient Cpa3Cre mice still displayed significantly elevated intestinal parasite burden compared with their vaccinated, mast cell-intact Cpa3wt littermates that carried only 0–2 detectable adults on day 6 after challenge infection. By contrast, vaccinated connective tissue mast cell-deficient Mcpt5Cre R-DTA mice showed the same almost complete protection against challenge infection on day 6 in the intestine (Figure 7e), although intestinal parasite burden during first infection was elevated in the absence of connective tissue mast cells.

Vaccine-induced protection against S. ratti challenge infection in mast cell-deficient mice. (a–d) Cpa3Cre (black symbols) and Cpa3wt (open symbols) mice were vaccinated with 2,000 irradiated S. ratti L3i or left naive. After 4 weeks, the naive and vaccinated mice were challenged by infection with 2,000 vital L3i. (a, b) Number of L3 in the head on day 2 p.i. (a) C57BL/6 background. (b) BALB/c background. (c, d) Number of parasitic adults in the intestine on day 6 p.i. (c) C57BL/6 background. (d) BALB/c background (a–d: n≥3). (e) Mcpt5Cre and Mcpt5wt mice were vaccinated and challenged as described above, the numbers of parasitic adults in the intestine on day 6 p.i. are shown (n≥4). Each symbol represents one mouse, the line indicates the mean, shown are pooled results from two independent experiments (a–d) or one representative of two experiments (e). *P<0.05 and **P<0.01: the groups differed significantly, as determined by Student’s t-test or Mann–Whitney U test. L3i, infective third-stage larvae; ns, not significant; p.i., post infection; S. ratti, Strongyloides ratti.
In summary, adaptive immunity to tissue-migrating larvae and intestinal parasitic adults was readily established in the absence of all mast cells or only connective tissue mast cells. Thereby, the quality of protection against tissue migrating larvae in absence and presence of mast cells was alike. The quality of protection against intestinal adults was reduced in mast cell-deficient mice compared with mast cell-intact mice and mice lacking selectively connective tissue mast cells.
Fc-receptor and IL-9 receptor-mediated signaling contribute to controlling S. ratti infection in the intestine but are not essential
Our experiments above showed that mucosal mast cells and adaptive immunity have central roles in terminating S. ratti infection. We then sought to define the factors of adaptive immunity that triggered the mucosal mast cells to eradicate S. ratti.
Mast cells are activated by cross-linking IgE that binds to their high affinity Fcɛ receptor.30, 31 Recent studies showed that both Strongyloides-specific IgG and IgE and their respective Fc receptors FcɛRI and FcγRIII contributed to expulsion of S. venezuelensis from the intestine.32 Moreover, in mice lacking the FcɛRIγ chain (FcRγ−/−) that is common to all activating Fc receptors and some C-type lectin receptors, the numbers of S. venezuelensis adults in the intestine and the fecal egg release on day 13 p.i. were increased.33 However, the latter study did not determine the fecal egg output up until the clearance of infection. To address this question, we compared parasite burden of FcRγ−/−34 mice and 5KO mice,35 which lack five Fc receptors due to deletion of the α chains of FcɛRI, FcɛRII, FcγRI, FcγRIIB, and FcγRIII to C57BL/6 wild-type mice (Figure 8). Intestinal parasite burden on day 6 p.i. was unchanged by Fc receptor deficiency either in FcRγ−/− mice or in 5KO mice (Figure 8a,c). Although fecal release of S. ratti DNA increased in FcRγ−/− at later time points of infection, the infection was terminated by day 30 in both Fc receptor-deficient mouse strains (Figure 8b,d). Thus, mucosal mast cells can be activated to terminate S. ratti infection with wild-type kinetics in the absence of activating IgE or IgG receptors.

Role of Fc receptor- and IL-9 receptor-mediated signaling in timely termination of S. ratti infection. (a, b) FcRγ−/−, (c, d) 5KO, and (e, f) IL9RKO mice (black symbols/bars) and control C57BL/6 mice (wt: open symbols/bars) were s.c. infected with 2,000 S. ratti L3i. (a, c, e) Number of parasitic adults in the intestine on day 6 p.i. are shown. (b, d, f) Release of S. ratti DNA in the feces, as measured by qPCR. Pooled results of two independent experiments (a, b, e; n≥7 mice per group; c, d, f; n≥6 mice per group) are shown. *P<0.05, **P<0.01, ***P<0.001: the groups differ significantly, as determined by Student’s t-test or Mann–Whitney U test. L3i, third-stage larvae; p.i., post infection; qPCR, quantitative-PCR; s.c., subcutaneous; S. ratti, Strongyloides ratti.
We have shown recently that IL-9-driven mast cell activation contributes to protecting the host during the first weeks of S. ratti infection.36 However, we did not investigate the impact of IL-9-induced mast cell activation on the termination of infection. To this end, we analyzed the course of S. ratti infection in IL-9 receptor-deficient mice.37 In line with our previous findings, IL-9 receptor-deficient BALB/c mice displayed increased parasite burden on day 6 p.i. (Figure 8e). Increased intestinal parasite numbers did not result into increased fecal release of S. ratti DNA during the peak of infection (Figure 8f) as observed for the mast cell-deficient or basophil-deficient mouse strains (Figures 1, 2, 3 and 4). Nevertheless, the absence of the IL-9 receptor increased the fecal release of S. ratti DNA at later time points starting on day 14 p.i. and prolonged infection by 2 weeks (Figure 8f). Still infection termination occurred earlier in IL-9 receptor-deficient mice than in the complete mast cell-deficient mice (Figure 2) suggesting that IL-9-mediated activation of mast cells contributed to the timely termination of S. ratti infection, but was not essential.
DISCUSSION
Analyzing the impact of connective tissue mast cells, mucosal mast cells and basophils to the protective immune response during S. ratti infection, we demonstrate different requirements for (i) control of tissue migrating L3, (ii) initial control of adults in the small intestine during the first week of infection, and (iii) final eradication of adults from the intestine.
-
i)
Mast cells and basophils were completely dispensable for control of migrating L3 in head and lung during first infection. By contrast, elevated numbers of tissue-migrating L3 were recorded in the head of S. ratti-infected W/Wv mice.12 As we did not observe a significant change in the number of tissue-migrating L3 in three mast cell-deficient nor in two basophil-deficient mouse strains (BALB/c Cpa3Cre, C57BL/6 Cpa3Cre, Mcpt5Cre R-DTA, and Mcpt8Cre mice), we argue that the different outcome observed in S. ratti-infected W/Wv mice was not due to mast cell deficiency or reduction in basophil numbers but rather caused by the multiple other immunological and non-immunological defects observed in Kit-mutant mice.13 This is further supported by several studies showing that S. stercoralis L3 are opsonized by complement and subsequently eradicated predominantly by eosinophils and neutrophils during first infection in the tissue.7
-
ii)
Absence of adaptive immunity in RAG1−/− mice did not influence early intestinal parasite burden, whereas total mast cell deficiency (BALB/c Cpa3Cre and C57BL/6 Cpa3Cre mice), connective tissue mast cell deficiency (Mcpt5cre R-DTA mice), and basophil deficiency (Mcpt8Cre mice) led to higher numbers of adult parasites in the intestine at day 6 p.i. This suggests that innate effector cells such as mast cells and basophils have an important role in controlling this stage of the infection. It is principally conceivable that absence of connective tissue mast cells and/or basophils and not absence of mucosal mast cells in Cpa3Cre mice caused the increased parasite burden on day 6 p.i. Because no mouse strain specifically deficient for mucosal mast cells in the presence of connective tissue mast cells and basophils is available to date, it is not possible to formally test the specific contribution of mucosal mast cells to the control of intestinal parasite burden during the first week of infection.
Elevated parasite burden and increased larval output during the first 2 weeks of infection were also observed in W/Wv mice during S. venezuelensis11 and S. ratti12 infection. Mice that lacked gastrointestinal and peritoneal mast cells while still containing dermal mast cells due to deletion of the phosphatidylinositol-3 kinase (PI3K) subunit p85α38 displayed elevated S. venezuelensis fecal egg release, further supporting the importance of mast cells in intestinal immunity to Strongyloides.
-
i)
Despite the initially overlapping functions of basophils and mast cells, final termination of S. ratti infection in the presence of adaptive immunity was selectively dependent on the presence of mucosal mast cells. We draw this conclusion because infection was terminated with wild-type kinetics by mice lacking selectively connective tissue mast cells or basophils but was prolonged for more than 150 days in the absence of all mast cells in Cpa3Cre mice. Given the lack of mice with a specific deficiency in mucosal mast cells, we cannot differentiate whether mucosal mast cells mediate the timely termination of S. ratti infection alone or by acting in synergy with basophils and/or connective tissue mast cells.
We compared mast cell- and basophil-deficient mice on BALB/c and C57BL/6 background to exclude strain-specific variations in the importance of a certain cell type for host defense. Although it is long known that BALB/c mice are less susceptible to S. venezuelensis39 and S. ratti26 infection, we show that this strain-specific difference is achieved shortly after the parasites arrive in the intestine. BALB/c mice had similar numbers of larvae in the head but lower numbers of intestinal parasites from day 3 p.i. until termination of infection than C57BL/6 mice. Because the early intestinal immunity was independent of adaptive immunity but dependent on mast cells and basophils, we hypothesize that strain-specific differences in the hematopoiesis of mast cells19 and nematode-induced modulation of mast cell activation36, 40 during the first days of infection contributed to the disparate susceptibilities of these strains.
IgE-activated basophils directly contribute to Th2 polarization by production of IL-4 and IL-13 during infection of immune mice with the gastrointestinal parasites Heligmosomoides polygyrus and Nippostrongylus brasiliensis.22 Also, mast cells have been implicated in the initiation of protective anti-helminth immune responses. Kit-mutant mice showed reduced alarmin (IL-33, IL-25, and TSLP) production resulting in defective Th2 polarization and increased parasite burden during H. polygyrus infection.29 As prevention of mast cell degranulation by cromolyn replicated the phenotype, it was suggested that degranulating mast cells initiated the release of tissue-derived alarmins in the first place. A similar role for alarmin-triggered induction of ILC2 and promotion of Th2-driven immunity was shown during S. venezuelensis infection, although here parasite-derived chitin was shown to trigger IL-33 release and the role of mast cells was not investigated.41 In the present study, our data did not support the notion that mast cells are essential to initiate appropriate immune responses during nematode infection. First, neither intestinal eosinophilia, IL-13, IL-5, and IL-4 responses to infection nor titers of the Th2-associated Ig isotypes IgE and IgG1 were reduced in the Cpa3Cre mice on both the BALB/c or C57BL/6 genetic backgrounds in comparison with their wild-type littermates. Second, vaccination against S. ratti in Cpa3Cre mice and Cpa3wt mice on the BALB/c and C57BL/6 backgrounds efficiently established protective immunity to tissue-migrating L3. This indicates that the initiation of the memory type 2 immune response, which mediates L3 eradication in immune mice, was intact in the absence of mast cells. Notably, however, vaccinated mucosal mast cell-deficient Cpa3Cre mice still bore more S. ratti adults in their intestine than vaccinated wild-type mice, while vaccinated connective tissue mast cell-deficient Mcpt5Cre DTA mice showed complete protection in the intestine as well. This suggests that mucosal mast cells functioned as important terminal effector cells as their absence resulted in elevated intestinal parasite burden in vaccinated mice even in the presence of an otherwise intact Th2 immune response. Our results are consistent with those of a recent study showing that the establishment of a Th2 response in Leishmania major-infected BALB/c mice as well as the strain-specific Th2/Th1 polarization in L. major-infected BALB/c vs. C57BL/6 mice was independent of mast cells.25
During Strongyloides infection, mast cells are activated by IL-3,9, 10, 42 IL-9,36 and Strongyloides antigen-specific IgG and IgE.32 When we tested two independent models of Fc receptor deficiency, we found that, at later time points, the fecal release of S. ratti eggs and L1 increased in the deficient mice relative to the control mice. This is consistent with a study showing that S. venezuelensis-infected FcRγ−/− mice had an increased fecal egg release and increased parasite burden at later time points.33 However, in both the mice lacking the FcɛRI-γ chain and the mice lacking 2 Fcɛ and 3 Fcγ receptors, the infection was terminated with wild-type kinetics by day 30 p.i. Thus, S. ratti-specific IgE and IgG may facilitate mast cell activation and parasite elimination via Fc-receptor engagement as demonstrated for S. venezuelensis-infected mice.32 Fc receptor-mediated activation of mast cells, however, did not have an indispensable role in the timely termination of S. ratti infection. Similarly, IL-3 was not central in infection termination because a previous study showed that IL-3−/− mice terminated S. venezuelensis infection with an 8-day delay compared with wild-type mice.42 When we infected IL-9 receptor-deficient mice with S. ratti, we observed increased parasite burden at day 6 p.i. and a delay in the resolution of infection by 2 weeks. Although the infection was only cleared after day 42 p.i., this was still earlier than when the complete mast cell-deficient mice terminated the infection, namely 140–160 days p.i. Redundant functions for IL-9 and IL-3 in activating mast cells have been shown before,43 and our combined results suggest that both cytokines in concert with Fc receptor-mediated signals contributed to activation of mucosal mast cells and partially compensated for each other’s absence.
The fact that specifically mucosal mast cells and not basophils or connective tissue mast cells are needed to terminate infection is striking. One explanation for this relates to the exclusive localization of mucosal mast cells between the intestinal epithelial cells.19 Strongyloides adults dwell embedded between the intestinal epithelial cell layer but never penetrate the basal lamina.6 This tight attachment to the intestinal cells is apparently central for the survival of the parasite because interference with this attachment by implanting the worms pre-incubated with proteoglycans promotes S. venezuelensis expulsion.44 Thus, it is conceivable that only intraepithelial localized mucosal mast cells can successfully attack embedded worms at later time points of infection. Support for this hypothesis is provided by the phenotype of mice with a conditional bone marrow-specific Notch2 knockout.45 Upon S. venezuelensis infection of these Notch 2−/− mice, mast cells were predominantly induced in the lamina propria and not in the intestinal epithelium as in wild-type mice. This altered mast cell localization in the intestine was accompanied by increased fecal egg release at later time points of the infection. However, the exact mechanism by which mast cells eradicate S. ratti from the intestine and which mast cell-derived effector molecules participate in this phenomenon remain to be elucidated.
METHODS
Ethics and mice. Animal experiments were conducted in agreement with the German animal protection law and experimental protocols were approved by Federal Health Authorities of the State of Hamburg (permission-number 55/13). Cpa3Cre 15, Mcpt8Cre 24 (C57BL/6 and BALB/c background), and Mcpt5Cre R-DTA23 (C57BL/6 background) mice have been described previously. The Mcpt5Cre R-DTA, Cpa3Cre, and Mcpt8Cre mice were bred heterozygously. The RAG1−/− mice and BALB/c IL-9R−/− mice,37 a kind gift from Dr Jean-Christophe Renauld, were bred homozygously. The FcRγ−/−34 and 5KO35 mice were provided by Friederike Jönsson. Wistar rats and C57BL/6 mice were obtained from Charles River (Sulzfeld, Germany). All mice were bred in house and kept in individually ventilated cages under specific pathogen-free conditions. For all experiments, male and female mice were used at 7–10 weeks of age, but experimental groups were matched for gender and age with maximally 7 days variance.
Parasite. The S. ratti cycle was maintained in Wistar rats and infections were performed by subcutaneous infection of 2,000 L3i in the hind footpad of mice.5 Mice were vaccinated with 2,000 irradiated L3i (160 Gy) 4 weeks before challenge infection with 2,000 vital L3i. Parasite burden in the intestine and quantification of the S. ratti 28S RNA-coding DNA in the feces of infected mice was performed as described.46
Characterization of the immune response. Mice were killed on day 6 p.i. Leukocyte populations in the small intestine were quantified by histopathology as described in Supplementary Methods. cDNA was prepared from 1-cm intestinal sections of naive and day 6 infected mice. Relative expression of cytokine mRNA was measured by qPCR as described in Supplementary Methods using the comparative Ct method. Cytokine production by ex vivo stimulated mesenteric lymph node cells derived from day 6 infected mice and Ig concentration in sera from day 28 infected mice was measured by ELISA as described.5
Statistical analysis. All data were assessed for normality. Groups were compared by using Student’s t-test (parametric) or Mann–Whitney U test (non parametric) test using GraphPad Prism software (San Diego, CA). P values≤0.05 were considered to indicate statistical significance.
References
Hotez, P.J., Brindley, P.J., Bethony, J.M., King, C.H., Pearce, E.J. & Jacobson, J. Helminth infections: the great neglected tropical diseases. J. Clin. Invest. 118, 1311–1321 (2008).
Viney, M.E. & Lok, J.B. The biology of Strongyloides spp. WormBook: The Online Review of C elegans Biology 1–17 (2015).
Dawkins, H.J. & Grove, D.I. Kinetics of primary and secondary infections with Strongyloides ratti in mice. Int. J. Parasitol. 11, 89–96 (1981).
Dawkins, H.J., Thomason, H.J. & Grove, D.I. The occurrence of Strongyloides ratti in the tissues of mice after percutaneous infection. J. Helminthol. 56, 45–50 (1982).
Eschbach, M.L., Klemm, U., Kolbaum, J., Blankenhaus, B., Brattig, N. & Breloer, M. Strongyloides ratti infection induces transient nematode-specific Th2 response and reciprocal suppression of IFN-gamma production in mice. Parasite Immunol. 32, 370–383 (2010).
Dawkins, H.J., Robertson, T.A., Papadimitriou, J.M. & Grove, D.I. Light and electron microscopical studies of the location of Strongyloides ratti in the mouse intestine. Z. Parasitenkd. 69, 357–370 (1983).
Bonne-Annee, S., Hess, J.A. & Abraham, D. Innate and adaptive immunity to the nematode Strongyloides stercoralis in a mouse model. Immunol. Res. 51, 205–214 (2011).
Breloer, M. & Abraham, D. Strongyloides infection in rodents: immune response and immune regulation. Parasitology 1–21 (e-pub ahead of print 24 February 2016).
Abe, T. & Nawa, Y. Worm expulsion and mucosal mast cell response induced by repetitive IL-3 administration in Strongyloides ratti-infected nude mice. Immunology 63, 181–185 (1988).
Abe, T., Sugaya, H., Ishida, K., Khan, W.I., Tasdemir, I. & Yoshimura, K. Intestinal protection against Strongyloides ratti and mastocytosis induced by administration of interleukin-3 in mice. Immunology 80, 116–121 (1993).
Khan, A.I., Horii, Y., Tiuria, R., Sato, Y. & Nawa, Y. Mucosal mast cells and the expulsive mechanisms of mice against Strongyloides venezuelensis. Int. J. Parasitol. 23, 551–555 (1993).
Nawa, Y., Kiyota, M., Korenaga, M. & Kotani, M. Defective protective capacity of W/Wv mice against Strongyloides ratti infection and its reconstitution with bone marrow cells. Parasite Immunol. 7, 429–438 (1985).
Reber, L.L., Marichal, T. & Galli, S.J. New models for analyzing mast cell functions in vivo. Trends Immunol. 33, 613–625 (2012).
Rodewald, H.R. & Feyerabend, T.B. Widespread immunological functions of mast cells: fact or fiction? Immunity 37, 13–24 (2012).
Feyerabend, T.B. et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity 35, 832–844 (2011).
Antsiferova, M. et al. Mast cells are dispensable for normal and activin-promoted wound healing and skin carcinogenesis. J. Immunol. 191, 6147–6155 (2013).
Gomez-Pinilla, P.J. et al. Mast cells play no role in the pathogenesis of postoperative ileus induced by intestinal manipulation. PloS One 9, e85304 (2014).
Gutierrez, D.A., Muralidhar, S., Feyerabend, T.B., Herzig, S. & Rodewald, H.R. Hematopoietic Kit deficiency, rather than lack of mast cells, protects mice from obesity and insulin resistance. Cell Metab. 21, 678–691 (2015).
Gurish, M.F. & Austen, K.F. Developmental origin and functional specialization of mast cell subsets. Immunity 37, 25–33 (2012).
Voehringer, D. Protective and pathological roles of mast cells and basophils. Nat. Rev. Immunol. 13, 362–375 (2013).
Lantz, C.S., Min, B., Tsai, M., Chatterjea, D., Dranoff, G. & Galli, S.J. IL-3 is required for increases in blood basophils in nematode infection in mice and can enhance IgE-dependent IL-4 production by basophils in vitro. Lab. Invest. 88, 1134–1142 (2008).
Schwartz, C., Turqueti-Neves, A., Hartmann, S., Yu, P., Nimmerjahn, F. & Voehringer, D. Basophil-mediated protection against gastrointestinal helminths requires IgE-induced cytokine secretion. Proc. Natl Acad. Sci. USA 111, E5169–E5177 (2014).
Dudeck, A. et al. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity 34, 973–984 (2011).
Ohnmacht, C., Schwartz, C., Panzer, M., Schiedewitz, I., Naumann, R. & Voehringer, D. Basophils orchestrate chronic allergic dermatitis and protective immunity against helminths. Immunity 33, 364–374 (2010).
Paul, C. et al. Mast cells have no impact on cutaneous leishmaniasis severity and related Th2 differentiation in resistant and susceptible mice. Eur. J. Immunol. 46, 114–121 (2015).
Dawkins, H.J., Grove, D.I., Dunsmore, J.D. & Mitchell, G.F. Strongyloides ratti: susceptibility to infection and resistance to reinfection in inbred strains of mice as assessed by excretion of larvae. Int. J. Parasitol. 10, 125–129 (1980).
Dawkins, H.J., Mitchell, G.F. & Grove, D.I. Strongyloides ratti infections in congenitally hypothymic (nude) mice. Aust. J. Exp. Biol. Med. Sci. 60 (Pt 2), 181–186 (1982).
Gardner, M.P., Gems, D. & Viney, M.E. Extraordinary plasticity in aging in Strongyloides ratti implies a gene-regulatory mechanism of lifespan evolution. Aging Cell 5, 315–323 (2006).
Hepworth, M.R. et al. Mast cells orchestrate type 2 immunity to helminths through regulation of tissue-derived cytokines. Proc. Natl Acad. Sci. USA 109, 6644–6649 (2012).
Jonsson, F. & Daeron, M. Mast cells and company. Front. Immunol. 3, 16 (2012).
Wernersson, S. & Pejler, G. Mast cell secretory granules: armed for battle. Nat. Rev. Immunol. 14, 478–494 (2014).
Matsumoto, M. et al. IgG and IgE collaboratively accelerate expulsion of Strongyloides venezuelensis in a primary infection. Infect. Immun. 81, 2518–2527 (2013).
Onah, D.N., Uchiyama, F., Nagakui, Y., Ono, M., Takai, T. & Nawa, Y. Mucosal defense against gastrointestinal nematodes: responses of mucosal mast cells and mouse mast cell protease 1 during primary strongyloides venezuelensis infection in FcRgamma-knockout mice. Infect. Immun. 68, 4968–4971 (2000).
Takai, T., Li, M., Sylvestre, D., Clynes, R. & Ravetch, J.V. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell 76, 519–529 (1994).
Mancardi, D.A., Iannascoli, B., Hoos, S., England, P., Daeron, M. & Bruhns, P. FcgammaRIV is a mouse IgE receptor that resembles macrophage FcepsilonRI in humans and promotes IgE-induced lung inflammation. J. Clin. Invest. 118, 3738–3750 (2008).
Blankenhaus, B. et al. Foxp3(+) regulatory T cells delay expulsion of intestinal nematodes by suppression of IL-9-driven mast cell activation in BALB/c but not in C57BL/6 mice. PLoS Pathog. 10, e1003913 (2014).
Steenwinckel, V. et al. IL-13 mediates in vivo IL-9 activities on lung epithelial cells but not on hematopoietic cells. J. Immunol. 178, 3244–3251 (2007).
Fukao, T. et al. Selective loss of gastrointestinal mast cells and impaired immunity in PI3K-deficient mice. Nat. Immunol. 3, 295–304 (2002).
Sato, Y. & Toma, H. Strongyloides venezuelensis infections in mice. Int. J. Parasitol. 20, 57–62 (1990).
Breloer, M., Hartmann, W., Blankenhaus, B., Eschbach, M.L., Pfeffer, K. & Jacobs, T. Cutting Edge: the BTLA-HVEM regulatory pathway interferes with protective immunity to intestinal Helminth infection. J. Immunol. 194, 1413–1416 (2015).
Yasuda, K. et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc. Natl Acad. Sci. USA 109, 3451–3456 (2012).
Lantz, C.S. et al. Role for interleukin-3 in mast-cell and basophil development and in immunity to parasites. Nature 392, 90–93 (1998).
Sasaki, Y. et al. IL-18 with IL-2 protects against Strongyloides venezuelensis infection by activating mucosal mast cell-dependent type 2 innate immunity. J. Exp. Med. 202, 607–616 (2005).
Maruyama, H., Yabu, Y., Yoshida, A., Nawa, Y. & Ohta, N. A role of mast cell glycosaminoglycans for the immunological expulsion of intestinal nematode, Strongyloides venezuelensis. J. Immunol. 164, 3749–3754 (2000).
Sakata-Yanagimoto, M. et al. Notch2 signaling is required for proper mast cell distribution and mucosal immunity in the intestine. Blood 117, 128–134 (2011).
Nouir, N.B. et al. Vaccination with Strongyloides ratti heat shock protein 60 increases susceptibility to challenge infection by induction of Th1 response. Vaccine 30, 862–871 (2012).
Acknowledgements
M.B. is supported by DFG grant BRE 3754/2-1 and 3754/2-2; F.J. is an employee of the Centre National de La Recherche Scientifique (CNRS). A.R. is supported by the DFG grant RO 2133/7-1. H.R.R. is supported by ERC Advanced Grant 233074. T.B.F. and H.R.R. are supported by DFG: SFB in Transregio 156 project A07. We thank Dr Jean-Christophe Renauld, Ludwig Cancer Research Institute, for sharing the IL-9 receptor-deficient mice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Reitz, M., Brunn, ML., Rodewald, HR. et al. Mucosal mast cells are indispensable for the timely termination of Strongyloides ratti infection. Mucosal Immunol 10, 481–492 (2017). https://doi.org/10.1038/mi.2016.56
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2016.56
This article is cited by
-
Mast cells link immune sensing to antigen-avoidance behaviour
Nature (2023)
-
Elucidating different pattern of immunoregulation in BALB/c and C57BL/6 mice and their F1 progeny
Scientific Reports (2021)
-
Basophil-derived tumor necrosis factor can enhance survival in a sepsis model in mice
Nature Immunology (2019)
-
Interleukin-9 promotes early mast cell-mediated expulsion of Strongyloides ratti but is dispensable for generation of protective memory
Scientific Reports (2018)
