
Abstract
Bacterial pneumonia is a leading cause of morbidity and mortality worldwide. Host responses to contain infection and mitigate pathogen-mediated lung inflammation are critical for pneumonia resolution. Aspirin-triggered resolvin D1 (AT-RvD1; 7S,8R,17R-trihydroxy-4Z,9E,11E,13Z,15E,19Z-docosahexaenoic acid) is a lipid mediator (LM) that displays organ-protective actions in sterile lung inflammation, and regulates pathogen-initiated cellular responses. Here, in a self-resolving murine model of Escherichia coli pneumonia, LM metabololipidomics performed on lungs obtained at baseline, 24, and 72 h after infection uncovered temporal regulation of endogenous AT-RvD1 production. Early treatment with exogenous AT-RvD1 (1 h post infection) enhanced clearance of E. coli and Pseudomonas aeruginosa in vivo, and lung macrophage phagocytosis of fluorescent bacterial particles ex vivo. Characterization of macrophage subsets in the alveolar compartment during pneumonia identified efferocytosis by infiltrating macrophages (CD11bHi CD11cLow) and exudative macrophages (CD11bHi CD11cHi). AT-RvD1 increased efferocytosis by these cells ex vivo, and accelerated neutrophil clearance during pneumonia in vivo. These anti-bacterial and pro-resolving actions of AT-RvD1 were additive to antibiotic therapy. Taken together, these findings suggest that the pro-resolving actions of AT-RvD1 during pneumonia represent a novel host-directed therapeutic strategy to complement the current antibiotic-centered approach for combatting infections.
Similar content being viewed by others


Introduction
One critical function of the acute inflammatory host response to infection is delivery of leukocytes to sites of tissue injury to protect the host from microbial invasion and restore homeostasis.1 If unrestrained in both amplitude and duration, persistent lung and airway infiltration by neutrophils can result in collateral injury to healthy bystander tissue, leading to organ damage and loss of function.2 In health, inflammatory responses are self-limited with essential fatty-acid-derived specialized pro-resolving mediator (SPM) signaling pivotal to the arrest of inflammation and initiation of resolution.1 Aspirin-triggered resolvin D1 (AT-RvD1; 7S,8R,17R-trihydroxy-4Z,9E,11E,13Z,15E,19Z-docosahexaenoic acid (DHA)) was first identified as a product of aspirin-acetylated cyclooxygenase-2.3, 4 It is now clear that AT-RvD1 is a DHA-derived SPM that can also be produced endogenously in the absence of aspirin by a second pathway initiated by cytochrome P450 enzymes.5 AT-RvD1 potently decreases acute lung inflammation after mucosal injury;6 however, its role in pneumonia remains to be characterized.
Bacterial pneumonia is a leading cause of mortality and morbidity worldwide, accounting for over 110 million years of life lost in 2010.7 Gram-negative bacterial pneumonia is the most common life-threatening hospital-acquired infection.8 In the United States, pneumonia causes more disease and death than any other infection, and there has been little change in pneumonia-associated mortality for more than five decades.9 Currently, antibiotics are the cornerstone of therapy; however, the rise of antibiotic-resistant microorganisms emphasizes a need for a new approach that augments host defense mechanisms to microbial invasion.10 Stimulation of bacterial clearance and counter-regulation of inflammatory responses are defining features of SPMs.11 Given its potent actions in regulating sterile lung inflammation,6 and pathogen-initiated cellular responses in vitro,12 we hypothesized that the SPM AT-RvD1 would be produced during pneumonia to limit infection and ensuing inflammatory responses.
Alveolar macrophages (AM) are tissue-resident cells with key functions in pathogen recognition, initiation of host defense via protective inflammation, and in clearance of invading microbes.11 Additional macrophage subsets infiltrate the alveolar space during the course of pathogen-mediated acute lung inflammation13 to augment the resident macrophage protective actions.14 Of interest, macrophages carry the biosynthetic and molecular circuitry to produce and respond to SPM.1 Here, in a self-resolving murine model of Escherichia coli pneumonia, AT-RvD1 was produced in a temporally regulated and enhanced gram-negative bacterial clearance and resolution of pathogen-initiated lung inflammation.
Results
Endogenous AT-RvD1 is produced during self-resolving E. coli pneumonia
To determine endogenous SPM production during lung infection, we used a self-resolving model of E. coli pneumonia. After intra-bronchial (IB) instillation of E. coli (see Methods), the resultant CFU at 6 h (154.5 × 103±26.4 × 103 CFU, mean±s.d.) and 24 h (3.0 × 103±2.7 × 103, mean±s.d.) were consistent with effective bacterial clearance (Figure 1a). Neutrophil counts peaked at 24 h (450.3 × 103±161.1 × 103 cells/BALF, mean±s.d.) and returned to near baseline within 72 h (15.2 × 103±3.9 × 103 cells/BALF, mean±s.d.) (Figure 1a). At 48 h post infection, neutrophil counts were approximately half-maximal (210.8 × 103±84.4 × 103 cells/BALF, mean±s.d.) and macrophage counts peaked (353.5 × 103±50.0 × 103 cells/BALF, mean±s.d.). Within 72 h, macrophage numbers also returned to near baseline (124.4 × 103±40.3 × 103 cells/BALF, mean±s.d.) (Figure 1a). E. coli was not detected in murine peripheral blood (data not shown). Murine lungs were obtained 0, 24, and 72 h after E. coli infection and SPM were extracted from lung homogenates for lipid mediator (LM) metabololipidomic analysis (see Methods). In this murine model of bacterial pneumonia, LM metabololipidomic profiling of lung tissue revealed temporal regulation of SPM production with several mediators increasing after infection (Table 1). Each of the mediators was identified in accordance with established criteria, including liquid chromatography retention time and diagnostic ions in MS/MS. A PCA (see Methods and ref. 15) gave distinct clustering for LM profiles obtained from 24 and 72 h (Figure 1b). The loading plot demonstrated that SPM levels were selectively regulated and associated with the resolution phase (i.e., 72 h) (Figure 1c). Of note, AT-RvD1 showed tight clustering among samples, as indicated by little deviation along principal component 2 (Figure 1c). After E. coli infection, lung AT-RvD1 was determined by liquid chromatography–MS/MS criteria (Figure 1d,e) and progressively increased at 24 and 72 h (∼11.2-fold increase 72 vs. 0 h) (Figure 1f and Table 1).

Endogenous AT-RvD1 is produced during bacterial pneumonia. (a) Time course of murine lung CFU, and BAL neutrophils and macrophages, at indicated time points after infection (E. coli, 106 CFU, IB). Results expressed as mean±s.d., n>5 mice in two independent experiments, *P<0.05 vs. 0 and 72 h. (b–f) Lipids were extracted from murine lungs 0, 24, and 72 h after infection and LM-SPM profiles were obtained using liquid chromatography (LC)–MS/MS metabololipidomics as in Methods. (b) Two-dimensional score plot and (c) loading plot of a principal component analysis of murine LM-SPM signature profiles. The white ellipse in the score plot denotes 95% confidence regions. (d) Representative multiple reaction-monitoring (MRM) chromatogram (m/z=375>141) and (e) MS/MS spectrum, obtained from infected murine lung, used for the identification of AT-RvD1 (7S,8R,17R-trihydroxy-docosa-4Z,9E,11E,13Z,15E,19Z-hexaenoic acid). (f) AT-RvD1 levels in murine lungs during pneumonia (n=4 per time point, mean±s.d., *P<0.05 vs. 0 h). AT-RvD, aspirin-triggered resolvin D1; BAL, bronchoalveolar lavage; CFU, colony-forming unit; LM, lipid mediator; MS/MS, tandem mass spectroscopy; SPM, specialized pro-resolving mediator.
AT-RvD1 enhances gram-negative bacterial clearance after lung infection
Temporal regulation of endogenous AT-RvD1 during pneumonia suggested it might exert host protective mechanisms during lung infection, so we next investigated its actions on bacterial clearance in vivo. Treatment of animals with AT-RvD1 (100 ng IV,∼4 μg kg−1) 1 h after infection led to a marked decrease in lung E. coli compared with untreated animals (Figure 2a, Supplementary Figure S1a online) and significant reductions in lung bacterial burden at 6 h (25.6% decrease, Figure 2b) and 24 h (90.3% decrease, Figure 2c) after infection. The increased clearance of E. coli was associated with increased physical activity in AT-RvD1-treated animals as early as 6 h after infection (see Supplementary Movie 1 in the online supplement). AT-RvD1 had no direct antimicrobial activity on bacterial cultures (data not shown). Of interest, AT-RvD1 also selectively regulated BALF cytokine levels with significant decreases in IL-6 (Figure 2d) and TNFα (Figure 2e). Levels of IL-1α, MCP-1, and IL-10 were not significantly changed by AT-RvD1 (Supplementary Figure S1b). In addition to E. coli, AT-RvD1 significantly increased lung P. aeruginosa bacterial clearance (61.6% decrease in CFU 6 h after infection, Figure 2f).

AT-RvD1 enhances bacterial clearance. (a) Representative lung sections from control mice, and 24 h after infection and treatment with AT-RvD1 or vehicle stained with an anti-E. coli antibody. Arrowheads indicate E. coli, bar=50 μm, inset bar=10 μm. Lung E. coli CFU counts (b) 6 h and (c) 24 h after infection (106 CFU, IB) in mice treated with AT-RvD1 or vehicle. Levels of (d) IL-6 and (e) TNF-α 6 h after infection determined by bead-based immunoassay. (f) Lung P. aeruginosa CFU counts 6 h after infection (106 CFU, IB) in mice treated with AT-RvD1 or vehicle. In all experiments, AT-RvD1 (100 ng, IV) or vehicle (<0.1% EtOH in saline vol/vo, IV) were given 1 h after E. coli. Results are expressed as mean±s.d. n=4–7 per group. *P<0.05. AT-RvD, aspirin-triggered resolvin D1; CFU, colony-forming unit; IB, intrabronchial.
AT-RvD1 increases macrophage phagocytosis of E. coli particles in situ
To determine mechanisms for AT-RvD1’s antimicrobial actions, we examined lung macrophage-mediated killing. To this end, fresh ultra-thin sections of perfused lungs from naive mice were stained with a fluorochrome-labeled anti-CD11c antibody and treated with AT-RvD1 or vehicle. These lung slices from naive animals have preserved airway and alveolar architecture and tissue-resident cells, including macrophages. No granulocytes were present (see Methods). E. coli particles conjugated with a pH-sensitive reporter were incubated with the lung sections for 60 min. The reporter is designed to indicate engulfment of the E. coli particles into acidic phagolysosomes.16 A dilute concentration of particles was used to minimize background phagocytosis of E. coli particles during this time interval in vehicle-exposed control lung sections (Figure 3a,c). Incubation with AT-RvD1 (10 nM, 60 min) significantly increased phagocytosis by CD11c+ lung macrophages (Figure 3b,c).

AT-RvD1 increases bacterial phagocytosis by CD11c+ lung cells. Ultra-thin perfused lung sections were obtained from naive mice, stained with anti-CD11c, and treated with AT-RvD1 (10 nM, 45 min, 37 °C) or vehicle. Sections were then co-incubated with E. coli particles labeled with a pH-sensitive probe (100 μg ml−1, 60 min, RT) and imaged using a fluorescence microscope. Particle dose was chosen to give minimal phagocytosis in vehicle-treated sections. Representative images of (a) control and (b) AT-RvD1-treated lung sections. (c) Proportion of CD11c+ lung cells participating in phagocytosis of E. coli particles. *P<0.05. ND, not detected. (d) Lipocalin 2 levels in BAL fluid 6 h after E. coli infection determined by ELISA. Results are expressed as mean±s.d. n⩾8 sections per group in two independent experiments. *P<0.05 vs. vehicle-treated un-infected mice, #P<0.05 vs. vehicle-treated infected mice. AT-RvD, aspirin-triggered resolvin D1; ELISA, enzyme-linked immunosorbent assay; RT, room temperature.
AT-RvD1 increases levels of the antimicrobial peptide lipocalin 2
Since Aderem and colleagues17 found that the antimicrobial peptide lipocalin 2 plays important roles in host responses to E. coli, we next determined the impact of AT-RvD1 on BAL fluid lipocalin 2 levels. Six hours after E. coli infection, lipocalin 2 was significantly increased (336.4 ng ml−1±158.2, mean±s.d., P⩽0.05) relative to uninfected mice (38.27 ng ml−1±20.56, mean±s.d., Figure 3d). AT-RvD1 further increased BAL lipocalin 2 levels (553.0 ng ml−1±346.6, P⩽0.05, Figure 3d).
Macrophage subsets during self-resolving lung infection
Neutrophil clearance occurred concomitant with increased AT-RvD1 levels and lung macrophage numbers (Figure 1), suggesting regulation of macrophages by AT-RvD1 for resolution of pathogen-initiated lung inflammation. CD11b and CD11c surface expression by BALF macrophages after E. coli infection was determined using flow cytometry, leading to the identification of three lung macrophage subsets in the alveolar compartment (Figure 4a); inflammatory macrophages (iMacs, CD11cLow CD11b+), exudate macrophages (ExMacs, CD11cHi CD11b+), and rAM (CD11cHi CD11b−) (Figure 4b). The presence of iMacs and ExMacs was transient, peaking 48 h after infection (7.1 × 103±5.2 iMacs/BALF, 35.2±17.23 ExMacs/BALF at 48 h, mean±s.d.), while rAM numbers did not significantly change (Figure 4c,d). iMacs and ExMacs were also identified in self-resolving acid-induced sterile lung injury (Supplementary Figure S2a), and LPS-induced lung inflammation (Supplementary Figure S2b). IB LPS resulted in a higher maximal number of macrophages compared with E. coli infection and hydrochloric acid-induced lung injury (Supplementary Figure S2c). Specific intracellular Ly6G immunostaining of BALF macrophages obtained 48 h after E. coli infection (Figure 5a,b) and 72 h after IB LPS (Supplementary Figure S3a,b) showed a significant increase in Ly6G+ iMacs and ExMacs, but not in rAM.

Macrophage subsets during E. coli pneumonia. BAL was obtained from mice at determined time points after E. coli infection (106 CFU, IB) for macrophage subset enumeration by flow cytometry. (a) Representative flow cytometry dot plots from BAL gated on macrophages (SScHi CD45+ F4/80+). (b) Identification of macrophage subsets based on CD11b and CD11c surface expression as infiltrating (iMacs, CD11cLow CD11bHi), exudative (ExMacs, CD11cHi CD11bHi), and resident alveolar macrophages (rAM, CD11cHi CD11b−). Time course of BAL macrophage subset (c) fractions and (d) counts. Results are expressed as mean±s.d. n=10 mice per group from two independent experiments, *P<0.05. BAL, bronchoalveolar lavage; CFU, colony-forming unit; IB, intrabronchial.

Recruited macrophages participate in efferocytosis of neutrophils during resolution of pathogen-mediated inflammation. (a) Representative dot plots and (b) bar-graph of intracellular Ly6G staining gated on macrophage subsets in BAL obtained 48 h after infection. Results are expressed as mean±s.d. n=4 sections per group in two independent experiments. (c) Percent increase in efferocytosis of apoptotic neutrophils by macrophage subsets after treatment with AT-RvD1 compared with vehicle (10 nM, 30 min, 37 °C). Macrophage and apoptotic neutrophil preparations are detailed in Methods. Results are expressed as mean±s.d. n⩾10 per group. #P<0.05 vs. vehicle-treated (d) neutrophil count in BAL obtained from mice 48 h after high-dose infection (E. coli, 2 × 106 CFU, IB) and treatment with AT-RvD1 (100 ng, IB) 24 and 36 h after E. coli. Results are expressed as mean±s.d. n=10 mice per group from two independent experiments. In all figures, *P<0.05. AT-RvD, aspirin-triggered resolvin D1; BAL, bronchoalveolar lavage; CFU, colony-forming unit; IB, intrabronchial.
AT-RvD1 promotes efferocytosis by macrophages
To determine if AT-RvD1 regulates this pro-resolving macrophage action,11 macrophage subsets were sorted from BALF obtained 72 h after LPS, exposed to AT-RvD1 (10 nM, 30 min, 37 °C) then incubated with fluorescent apoptotic neutrophils for 1 h. Phagocytosis of apoptotic neutrophils was then determined by measuring intracellular macrophage fluorescence (see Methods). AT-RvD1 significantly increased efferocytosis by iMacs, and to a lesser extent by ExMacs (Figure 5c). We next examined the impact of AT-RvD1 on neutrophil clearance in vivo. Animals were treated with IB AT-RvD1 (100 ng) for increased alveolar delivery to macrophages. When given at 24 and 36 h, AT-RvD1 significantly decreased BAL neutrophils after E. coli (25.0%, Figure 5d). In separate experiments, IB AT-RvD1 significantly decreased BAL neutrophils after IB LPS (38.1%, Supplementary Figure S3c). Collectively, these findings demonstrate that AT-RvD1 promotes clearance of apoptotic neutrophils by macrophage subsets and accelerates alveolar neutrophil clearance for resolution of pathogen-mediated lung inflammation.
AT-RvD1 enhances bacterial clearance in combination with antibiotics
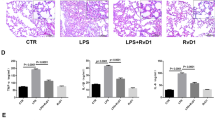
To determine if AT-RvD1 actions on host responses to E. coli infection provide additional benefit to antibiotics, mice were treated 1 h after E. coli with AT-RvD1 (100 ng, IV), ciprofloxacin at its IC50 dose for E. coli (0.2 mg kg−1, intraperitoneal) (Supplementary Figure S4a), or both AT-RvD1 and ciprofloxacin (see Methods). Lung CFUs were significantly decreased in mice treated with AT-RvD1 (24.9% reduction compared with untreated mice) (Figure 6a). In addition, mice that received both AT-RvD1 and ciprofloxacin had significantly less CFU than mice treated with antibiotics alone (Figure 6a). Of note, AT-RvD1 significantly decreased BAL fluid LPS (2.7±0.7 endotoxin units (EU) per ml BAL fluid, mean±s.d., P⩽0.05) compared with untreated animals (5.5±0.7 EU ml−1 BAL fluid, mean±s.d., Figure 6b). In sharp contrast, ciprofloxacin alone did not decrease BAL fluid LPS (5.6±0.7 EU ml−1 BAL fluid, mean±s.d.) despite the decrease in CFU (Figure 6a,b). In combination with ciprofloxacin, AT-RvD1 decreased the LPS levels (4.1±0.6 EU ml−1 BAL fluid, mean±s.d., P⩽0.05, Figure 6b). Moreover, AT-RvD1 treatment significantly decreased BALF neutrophils 48 h after infection (55.0% reduction in BALF neutrophil counts compared with untreated animals, Figure 6c), whereas antibiotic therapy had no significant impact on BALF neutrophils (Figure 6c). Combined AT-RvD1 and antibiotic treatment was additive and resulted in the lowest BALF neutrophil count (70% reduction in BALF neutrophil count compared with untreated animals, Figure 6c). At 48 h post infection, BALF neutrophil numbers were half-maximal and declining with macrophages rising to roughly equal numbers (Figure 1a). As an index to tissue resolution, the BALF macrophage-to-neutrophil ratio was 1.3 in vehicle control mice and increased to 1.7 with AT-RvD1 (P=0.06, Figure 6d). Ciprofloxacin alone did not significantly impact the macrophage-to-neutrophil ratio (1.2); however, the ratio was significantly increased to 2.6 by combination of ciprofloxacin and AT-RvD1 (P<0.05, Figure 6d). Histological examination of animals at this time point after infection revealed decreased cellularity and alveolar wall edema in animals treated with AT-RvD1 compared with vehicle-exposed animals and animals treated with ciprofloxacin alone (Figure 6e). Collectively, these results indicate that AT-RvD1 enhanced antibiotic-mediated bacterial clearance, and decreased pathogen-initiated lung inflammation.

AT-RvD1 enhances bacterial clearance in combination with antibiotics. (a) Lung CFU counts and (b) BAL fluid LPS concentrations (endotoxin units per ml BAL fluid) 6 h after E. coli infection, and (c) BALF neutrophils and (d) BALF macrophage-to-neutrophil ratio (a resolution index) 48 h after infection with E. coli (106 CFU, IB) in mice treated with AT-RvD1 (100 ng, IV, 1 h after infection) or vehicle (<0.1% EtOH in saline vol/vol), with or without ciprofloxacin (0.2 mg kg−1, IP, 1 h after infection). Results are expressed as mean±s.d. n=5–7 per group. *P<0.05. (e) Representative hematoxylin and eosin stains of murine lungs 48 h after infection. n=3 per group. AT-RvD, aspirin-triggered resolvin D1; BAL, bronchoalveolar lavage; CFU, colony-forming unit; IB, intrabronchial; IP, intraperitoneal.
Discussion
Bacterial clearance and counter-regulation of pathogen-mediated inflammation define resolution of bacterial pneumonia.9 While antibiotics are crucial for the former, they have little impact on clearance of inflamed tissue. Results presented herein demonstrate that endogenous AT-RvD1 levels increased during self-resolving E. coli pneumonia, suggesting a role for AT-RvD1 during lung catabasis. Exogenous AT-RvD1 treatment enhanced clearance of two gram-negative bacteria in vivo, in part via increased bacterial particle phagocytosis by resident macrophages and increased tissue levels of the antimicrobial peptide lipocalin 2. In addition, AT-RvD1 engaged macrophage subsets for phagocytosis of apoptotic neutrophils, increased alveolar neutrophil clearance, and decreased alveolar LPS levels. When combined with antibiotic therapy, AT-RvD1 further enhanced bacterial clearance, mitigated antibiotic-mediated LPS release, and promoted resolution of E. coli-initiated lung inflammation.
LM metabololipidomics profiling of the resolving exudates allows for sensitive detection of endogenous SPM production.1 In the present work, metabololipidomic profiling identified temporal and selective regulation of SPM during bacterial pneumonia. Levels of several SPM were increased during the resolution phase (from 24 to 72 h after infection, Figure 1a), likely a result of increased precursor availability in the resolving exudate18, 19 and biosynthetic enzyme activation.20 PCA identified several SPM that contributed to time-point separation, in particular AT-RvD1. Biosynthesis of AT-RvD1 in the absence of aspirin exposure is likely initiated via oxygenation at the carbon-17 position of DHA by cytochrome p450 enzymes,5 which are abundant in the lungs.21 Indeed, animals deficient in CYP1 monooxygenases demonstrate decreased levels of 17-hydroxy-DHA.22 Increased resistance to metabolic inactivation of AT-RvD1 relative to its 17 carbon position epimer RvD1 may account for its more consistent levels in the PCA loading plot and a pharmacological advantage for pneumonia.4
Resolution of lung mucosal inflammation is a defining characteristic of SPM.23 Eicosapentaenoic acid-derived resolvin E1 promotes the resolution of allergic airway inflammation,24 arachidonic acid-derived 15-epi-lipoxin A4 enhances resolution of acute lung injury,25 and DHA-derived maresin 1 protects lung function during acid-induced lung injury.26 AT-RvD1 decreases nuclear factor-kB signaling for organ protection after sterile acute lung inflammation,6 and attenuates pro-inflammatory signaling in human airway epithelial cells.27 These anti-inflammatory and pro-resolving actions are not immune-suppressive during bacterial infection.11 Indeed, select DHA-derived resolvins and their counter-regulatory circuits enhance bacterial clearance by leukocytes,28 and protect from polymicrobial murine sepsis.28, 29 In the lung, resolvin E1 treatment attenuates mucosal injury and protects animals from aspiration pneumonia.30 Results presented herein uncover direct AT-RvD1 actions on host-mediated anti-bacterial responses, and concomitant pro-resolving actions, emphasizing that SPMs are leads for multi-pronged therapy against bacterial pneumonia. Mechanisms for enhancing bacterial clearance include stimulating leukocyte phagocytosis of the offending agent.11 In allergic airway inflammation, AT-RvD1 increases macrophage phagocytosis of antigen-coated particles,31 and RvD1 increases neutrophil phagocytosis of bacteria during extra-pulmonary infection.32 Our results demonstrated an increase in macrophage phagocytosis of bacterial particles in live lung tissue. In addition to enhancing phagocyte function, AT-RvD1 increased levels of the antimicrobial peptide lipocalin 2 to provide an additional mechanism for mucosal host defense. Of note, metabololipidomic profiling of infected lungs demonstrated regulation of additional SPMs. Their contribution to host protection during pneumonia is under investigation.
In addition to their defensive role against bacterial invasion, macrophages are crucial effectors of resolution of inflammation.1 Examination of peritoneal macrophages during peritonitis identified a subset of macrophages with a M2 signature present only during resolution,33 and a subset of CD11bLo macrophages that emigrate to nearby lymphoid organs for termination of inflammation.34 Both peritoneal macrophage subtypes had decreased phagocytic ability. In the lung, macrophage phenotyping during resolving inflammation has identified two distinct subsets in addition to rAM.13, 14 Their activation markers are intermediate between M1 and M2, and both demonstrate increased CD11b expression. Findings presented here indicate that these macrophage subsets appear during resolution of pathogen-initiated lung inflammation and efferocytose apoptotic neutrophils. As such, iMacs and ExMacs are distinct from resolving macrophages identified during peritoneal inflammation. In our model of pathogen-initiated lung inflammation, their time course positions them for roles in the resolution of inflammation, as they infiltrate into the alveolar space during neutrophil clearance. Towards this end, ExMacs release IL1ra for organ protection during bacterial pneumonia.14 In addition, results presented here demonstrate efferocytosis of apoptotic neutrophils by iMacs and ExMacs. Furthermore, AT-RvD1 regulation of iMacs and ExMacs efferocytosis of apoptotic neutrophils uncovered a role for these macrophages as SPM cellular targets to promote pneumonia resolution. Of note, the analyses here were focused on BALF macrophages; however, myeloid-derived suppressor cell-like cells are in lung interstitium after bacterial pneumonia and also participate in tissue resolution by efferocytosis.35
Pneumonia continues to have a substantial impact on the global burden of disease with elevated mortality7 and increasing prevalence of antibiotic-resistant bacteria,36 in particular gram-negative bacteria in nosocomial infections.8 Therapies targeting host responses to infection offer the possibility of augmenting antibiotics to improve outcomes.9 Anti-inflammatory therapies, such as corticosteroids, have not proven successful,37 likely secondary to concomitant immune suppression. In sharp contrast to immunosuppressive agents, SPM engage endogenous resolution pathways that enhance bacterial host defense. Macrophages carry the dual effector functions of host defense and pro-resolution,11 thus represent ideal cell-based therapeutic targets. Results presented here show that antibiotic treatment alone had little effect on the resolution of lung inflammation, in part related to persistent increases in LPS likely induced by the bactericidal actions of ciprofloxacin.38 AT-RvD1 significantly promoted resolution through macrophage activation for phagocytosis, decreased BAL fluid LPS, and increased BAL fluid lipocalin 2 (an important antimicrobial peptide for E. coli host defense17). In conjunction with AT-RvD1’s protective actions on lung barrier integrity and mechanics,6 these new pro-resolving mechanisms in bacterial defense emphasize roles for SPM in promoting resolution of pathogen-initiated tissue inflammation,28, 32 and help further distinguish pro-resolution from anti-inflammation.11
In summary, our findings have uncovered new roles for AT-RvD1 in promoting resolution of gram-negative bacterial pneumonia. In this study, the DHA-derived LM AT-RvD1 was endogenously produced in a temporally regulated manner, enhanced bacterial clearance by direct actions on lung macrophages and lipocalin 2, and promoted the resolution of inflammation by directly increasing efferocytosis of apoptotic neutrophils by macrophage subsets in the alveolar space. These actions augmented antimicrobial therapy, thus offering a potential new host-directed therapeutic approach to the global threat of bacterial pneumonia that emphasizes endogenous resolution signaling pathways.
Methods
Materials
AT-RvD1 (7S,8R,17R-trihydroxy-4Z,9E,11E,13Z,15E,19Z-DHA) was obtained from Cayman Chemical (Ann Arbor, MI) and validated just prior to use with LC–tandem mass spectroscopy (MS/MS) and physical criteria reported earlier.15 E. coli (O6:K2:H1) strain ATCC 19138 and Pseudomonas aeruginosa (HER-1018) strain ATCC BAA-47 were obtained from the American Type Culture Collection (Manassas, VA). E. coli particles conjugated with pHrodo were purchased from Life Technologies (Carlsbad, CA). Lipopolysaccharide (LPS, E. coli O55:B5), ciprofloxacin, and hydrochloric acid were purchased from Sigma-Aldrich (St Louis, MO). Fluorescence-activated cell sorting (FACS) antibodies were obtained from eBioscience (San Diego, CA): F4/80 (BM8); Biolegend (San Diego, CA): CD11b (M1/70), and Ly6G (1A8); BD Biosciences (San Diego, CA): CD11c (HL3) and CD45 (30-F11). Permeabilization buffer was purchased from eBioscience.
Mice
C57BL/6 male mice (8–10-week-old, body weights 20–25 g; Charles River Laboratories, Wilmington, MA) were housed in isolation cages in pathogen-free conditions on a light–dark cycle with light from 0700 to 2000 hours at 25 °C. Mice were fed a standard diet (Laboratory Rodent Diet 5001; PMI Nutrition International, St Louis, MO) containing 4.5% total fat with 0.3% ω-3 fatty acids and <0.02% C20:4 and were provided water ad libitum. All studies were reviewed and approved by the Harvard Medical Area standing committee on animals.
Murine model of gram-negative bacterial pneumonia
Live gram-negative bacteria (E. coli or P. aeruginosa, 1 × 106 colony-forming unit (CFU) in 25 μl saline) were instilled selectively into the left mainstem bronchus of anesthetized mice as in ref. 30. Mice received AT-RvD1 (100 ng, IV) or vehicle (<0.1% (vol/vol) ethanol in saline) 1 h after IB E. coli or P. aeruginosa. In select experiments, mice received ciprofloxacin (0.2, 2, or 20 mg kg−1, intraperitoneal) or vehicle in addition to AT-RvD1, 1 h after IB E. coli. In separate experiments, animals were treated with AT-RvD1 (100 ng, IB) or vehicle via a repeat tracheostomy surgery 24 and 36 h after high-dose IB E. coli (2 × 106 CFU). A high dose of E. coli was used to induce neutrophil infiltration comparable to LPS-induced lung inflammation. In pathogen-mediated acute lung inflammation experiments, animals were treated with E. coli LPS (0.3 mg kg−1, 50 μl in saline, IB). In sterile acute lung inflammation experiments, hydrochloric acid (0.1 N, pH ∼1.0, 50 μl IB) was administered as in ref. 26.
Bacterial CFU determination
At pre-determined time points after infection (E. coli or P. aeruginosa, IB), lungs were collected aseptically and homogenized in 2.5 ml of ice-cold sterile saline. Aliquots of serial dilutions were plated on Lysogeny broth (LB)-agar plates. After incubation for 24 h at 37 °C, colonies were counted and results were expressed as CFU per whole lungs.
Lipid mediator metabololipidomics
Lung tissue isolates were disrupted gently in methanol then processed for liquid chromatography–MS/MS as in ref. 26. AT-RvD1 levels were monitored and quantified with multiple reaction monitoring developed using signature ion fragments for AT-RvD1 (m/z=375/141) and d5-RvD2 (m/z=380/141).15 Quantification was carried out on the basis of the peak areas obtained with multiple reaction-monitoring transitions and linear calibration curves for synthetic mediators, and deuterated standards for recoveries.15
Bronchoalveolar lavage
At timed intervals, mice were killed and the trachea exposed. A 20-gauge angiocatheter was inserted into the trachea and the lungs were lavaged with two separate 1 ml volumes of ice-cold PBS with 0.6 mM EDTA. The bronchoalveolar lavage (BAL) fluid was pooled, centrifuged at 500 g for 5 min at 4 °C to pellet the cell fraction. The cell pellet was resuspended in cold PBS.
FACS analysis and cell sorting
BAL cells were obtained after centrifugation of pooled BAL fluid, and the cell pellet was stained with fluorochrome-labeled antibodies to obtain a leukocyte differential—neutrophils (CD45+ F4/80− Ly6G+ CD11b+), infiltrating macrophages (iMacs; CD45+ SScHigh F4/80+ CD11cLow CD11b+), exudative macrophages (ExMacs; CD45+ SScHigh F4/80+ CD11cHigh CD11b+), and resident alveolar macrophages (rAM; CD45+ F4/80+ SScHigh CD11cHigh CD11b−). Cell counts were obtained by adding a known amount of fluorescent counting beads (CountBright, Life Technologies, Carlsbad, CA) to the BAL cell pellet prior to flow cytometry as per the manufacturer’s recommendations, and normalized to lavage. FACSCanto II (BD Biosciences) and FlowJo Ver. 10 software (Tree Star, Ashland, OR) were used for analyses. In some experiments, five bronchoalveolar lavage fluids (BALFs) were pooled and F4/80 positive macrophage subsets cells were sorted with >95% purity, using FACSaria (Becton Dickinson, Franklin Lake, NJ). The sorted populations were used for ex vivo efferocytosis assays. For sorting of macrophage subsets, BALF obtained 72 h after IB LPS was used, as LPS resulted in the highest maximal number of macrophages (see Results) allowing for a better yield of each macrophage subset using fluorescence-activated cell sorting.
Determination of bioactive molecule levels in BALF
Select cytokines were measured in aliquots of pooled BALF using a bead-based immunoassay as per the manufacturer’s instructions (LEGENDPlex by Biolegend). Lipocalin 2 levels in BAL fluid were determined by enzyme-linked immunosorbent assay (Lipocalin 2 Quantikine ELISA Kit, R&D Systems, Minneapolis, MN). LPS levels in BAL fluid were determined using a Limulus Amebocyte Lysate endotoxin quantitation kit (ThermoFisher Scientific, Cambridge, MA). All measurements were normalized to volume of pooled BAL fluid.
Ultra-thin lung sections
Lung sections were prepared as described previously.39 Briefly, after euthanasia of naive animals by isoflurane overdose, the lungs were exposed and slowly perfused free of blood with 5 ml HBSS. The lungs were then inflated with 1.2 ml agarose (low melting point, Life Technologies) in HBSS (1.5%) via a tracheal catheter (model 20G Intima, Becton Dickinson). After the agarose solidified, one lung lobe was separated and sectioned into 150-μm-thick slices using a tissue slicer (VF-300; Precisionary Instruments, Greenville, NC). Lung slices were incubated at 5% CO2, 37 °C in DMEM/F-12 supplemented with penicillin-streptomycin (Invitrogen, Cambridge, MA) overnight.
Phagocytosis assay
Ultra-thin lung sections were washed in HBSS then stained with anti-CD11c (FITC) and treated with AT-RvD1 (10 nM) for 30 min at 37 °C. The lung sections were then washed before addition of E. coli particles conjugated to pHrodo red dye (100 μg ml−1, Life Technologies). To maximize the sensitivity in detecting an increase in phagocytosis, a dose–response curve of E. coli particles per lung section was performed to have as little phagocytosis as possible by untreated lung sections. Sections were observed with Nikon Eclipse TS 100 epi-fluorescence microscopy (Nikon, Mellville, NY) with filter sets for indicator dye of Phrodo Red (green excitation) or FITC (blue excitation) at 0 and 60 min after addition of the E. coli particles. Images were then overlayed, and CD11c+ cells were enumerated. Phagocytosis was quantified as the ratio of CD11c+ E. coli+ cells over CD11c+ cells.
Macrophage efferocytosis
Macrophage efferocytosis was quantified as previously described.40 Briefly, neutrophils were obtained from peritoneal lavage fluid obtained 4 h after zymosan A (1 mg, intraperitoneal)—induced murine peritonitis, and labeled with carboxyfluorescein diacetate-succinimidyl ester (CFDA; 10 μM, 30 min, 37 °C) and cultured overnight (serum free media) to induce apoptosis (confirmed by >95% Annexin V+ 7AAD surface expression by flow cytometry, data not shown). Murine AM subsets were sorted from BALF obtained 72 h after IT LPS according to the gating strategy detailed above and allowed to adhere in a 96-well plate for 1 h at a density of 2.5 × 104 cells per well. The macrophages were then incubated with AT-RvD1 for 30 min at 37 °C before the addition of apoptotic neutrophils (1.25 × 105 cells per well, macrophage-to-neutrophil ratio of 1:5). After 60 min of co-incubation, trypan blue (1:2) was added to quench CFDA in extracellular neutrophils, and the number of phagocytosed cells (i.e., intracellular) was determined using a FLX-800 plate reader (Biotek, Minooski, VT) by monitoring fluorescence emission at 525 nm.
Histopathology and immunohistochemistry
Lung tissue was fixed by inflation with Zinc Fixative (BD Pharmingen, San Diego, CA) at a transpulmonary pressure of 20 cm H2O and embedded in paraffin. For histological analysis, lungs were collected 0, 24, and 48 h after E. coli instillation and paraffin-embedded 5-μm sections of lungs were cut and stained with hematoxylin and eosin for light microscopy. To detect bacteria, E. coli staining was performed using an anti-E. coli antibody (GeneTex, Irvine, CA) and an anti-rabbit secondary antibody (Abcam, Cambridge, MA). Leica DFC480 and Leica QWin Standard V3.4.0 (Leica Microsystems, Buffalo Grove, IL) were used for microscopic analysis.
Statistical analysis
All data are expressed as means±s.d. Comparisons between groups were conducted using analysis of variance and Student’s t test as appropriate. A level of P⩽0.05 was considered to indicate statistical significance. Statistics were performed using Prism 6.0 for Windows (GraphPad, San Diego, CA). For multivariate statistical analysis, principal component analysis (PCA) was performed using SIMCA 13.0.3 software (Umetrics, Umea, Sweden) following mean centering and unit variance scaling of LM amounts as in ref. 15. The score plot shows the systematic clusters among the observations (closer plots presenting higher similarity in the data matrix). Loading plots describe the magnitude and the manner (positive or negative correlation) in which the measured LM-SPM contribute the cluster separation in the score plot.
References
Serhan, C.N. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu. Rev. Immunol. 25, 101–137 (2007).
Matthay, M.A., Ware, L.B. & Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Invest. 122, 2731–2740 (2012).
Serhan, C.N. et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196, 1025–1037 (2002).
Sun, Y.P. et al. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J. Biol. Chem. 282, 9323–9334 (2007).
Serhan, C.N. & Petasis, N.A. Resolvins and protectins in inflammation resolution. Chem. Rev. 111, 5922–5943 (2011).
Eickmeier, O. et al. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal. Immunol. 6, 256–266 (2012).
Lozano, R. et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2095–2128 (2012).
Peleg, A.Y. & Hooper, D.C. Hospital-acquired infections due to gram-negative bacteria. N. Engl. J. Med. 19, 1804–1813 (2010).
Mizgerd, J.P. Acute lower respiratory tract infection. N. Engl. J. Med. 358, 716–727 (2008).
Eddens, T. & Kolls, J.K. Host defenses against bacterial lower respiratory tract infection. Curr. Opin. Immunol. 24, 424–430 (2012).
Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101 (2014).
Palmer, C.D., Mancuso, C.J., Weiss, J.P., Serhan, C.N., Guinan, E.C. & Levy, O. 17(R)-Resolvin D1 differentially regulates TLR4-mediated responses of primary human macrophages to purified LPS and live E. coli. J. Leukoc. Biol. 90, 459–470 (2011).
Duan, M., Li, W.C., Vlahos, R., Maxwell, M.J., Anderson, G.P. & Hibbs, M.L. Distinct macrophage subpopulations characterize acute infection and chronic inflammatory lung disease. J. Immunol. 189, 946–955 (2012).
Herold, S. et al. Exudate macrophages attenuate lung injury by the release of IL-1 receptor antagonist in gram-negative pneumonia. Am. J. Respir. Crit. Care Med. 183, 1380–1390 (2011).
Colas, R.A., Shinohara, M., Dalli, J., Chiang, N. & Serhan, C.N. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. Am. J. Physiol. Cell Physiol. 307, C39–C54 (2014).
Fiala, M. et al. Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer's disease patients are improved by bisdemethoxycurcumin. Proc. Natl Acad. Sci. USA 104, 12849–12854 (2007).
Flo, T.H. et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432, 917–921 (2004).
Kasuga, K. et al. Rapid appearance of resolvin precursors in inflammatory exudates: novel mechanisms in resolution. J. Immunol. 181, 8677–8687 (2008).
Miki, Y. et al. Lymphoid tissue phospholipase A2 group IID resolves contact hypersensitivity by driving antiinflammatory lipid mediators. J. Exp. Med. 210, 1217–1234 (2013).
Levy, B.D., Romano, M., Chapman, H.A., Reilly, J.J., Drazen, J. & Serhan, C.N. Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. J. Clin. Invest. 92, 1572–1579 (1993).
Kim, J.H., Sherman, M.E., Curriero, F.C., Guengerich, F.P., Strickland, P.T. & Sutter, T.R. Expression of cytochromes P450 1A1 and 1B1 in human lung from smokers, non-smokers, and ex-smokers. Toxicol. Appl. Pharmacol. 199, 210–219 (2004).
Divanovic, S. et al. Contributions of the three CYP1 monooxygenases to pro-inflammatory and inflammation-resolution lipid mediator pathways. J. Immunol. 191, 3347–3357 (2013).
Levy, B.D. & Serhan, C.N. Resolution of acute inflammation in the lung. Annu. Rev. Physiol. 76, 467–492 (2014).
Haworth, O., Cernadas, M., Yang, R., Serhan, C.N. & Levy, B.D. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat. Immunol. 9, 873–879 (2008).
El Kebir, D. et al. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 180, 311–319 (2009).
Abdulnour, R.E. et al. Maresin 1 biosynthesis during platelet-neutrophil interactions is organ-protective. Proc. Natl Acad. Sci. USA 111, 16526–16531 (2014).
Hsiao, H.M. et al. Resolvin D1 attenuates polyinosinic-polycytidylic acid-induced inflammatory signaling in human airway epithelial cells via TAK1. J. Immunol. 193, 4980–4987 (2014).
Spite, M. et al. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461, 1287–1291 (2009).
Gobbetti, T. et al. Nonredundant protective properties of FPR2/ALX in polymicrobial murine sepsis. Proc. Natl Acad. Sci. USA 111, 18685–18690 (2014).
Seki, H. et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J. Immunol. 184, 836–843 (2009).
Rogerio, A.P. et al. Resolvin D1 and aspirin-triggered resolvin D1 promote resolution of allergic airways responses. J. Immunol. 189, 1983–1991 (2012).
Chiang, N. et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 484, 524–528 (2012).
Bystrom, J. et al. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood 112, 4117–4127 (2008).
Schif-Zuck, S., Gross, N., Assi, S., Rostoker, R., Serhan, C.N. & Ariel, A. Saturated-efferocytosis generates pro-resolving CD11b low macrophages: modulation by resolvins and glucocorticoids. Eur. J. Immunol. 41, 366–379 (2011).
Poe, S.L. et al. STAT1-regulated lung MDSC-like cells produce IL-10 and efferocytose apoptotic neutrophils with relevance in resolution of bacterial pneumonia. Mucosal. Immunol. 6, 189–199 (2013).
World Health Organization Antimicrobial resistance: global report on surveillance 2014. World Health Organization, Geneva, Switzerland, 2014.
Meijvis, S.C.A. et al. Dexamethasone and length of hospital stay in patients with community-acquired pneumonia: a randomised, double-blind, placebo-controlled trial. Lancet 377, 2023–2030 (2011).
Van Den Berg, C., de Neeling, A.J., Schot, C.S., Hustinx, W.N., Wemer, J. & de Wildt, D.J. Delayed antibiotic-induced lysis of Escherichia coli in vitro is correlated with enhancement of LPS release. Scand. J. Infect. Dis. 24, 619–627 (1992).
Aven, L. et al. An NT4/TrkB-dependent increase in innervation links early-life allergen exposure to persistent airway hyperreactivity. FASEB J. 28, 897–907 (2014).
Dalli, J. & Serhan, C.N. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 120, e60–e72 (2012).
Acknowledgements
We thank Hiroyuki Seki and Iliyan K. Vlassakov for technical assistance. This research was supported in part by the US National Institutes of Health T32-HL07633 (R.-E.E.A. and B.D.L.), and P01-GM095467 (B.D.L. and C.N.S). The content is solely the responsibility of the authors and does not necessarily reflect the official views of NHLBI, NIGMS, or the National Institutes of Health. All authors concur with the submission and none of the data has been previously reported or is under consideration for publication elsewhere.
Author contribution
R.E.A. contributed to experimental design, carried out experiments, and data analyses and wrote the manuscript; H.P.S. and D.N.D. contributed to experimental design, carried out experiments, and data analysis and contributed to manuscript and figure preparation; R.A.C. and J.D. carried out lipid mediator metabololipidomics and data analysis; C.N.S. contributed to experimental design, data analysis, and manuscript preparation; B.D.L contributed to experimental design, carried out experiments and data analyses, contributed to manuscript preparation, and conceived the overall research plan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
C.N.S. is an inventor on patents (resolvins) assigned to BWH and licensed to Resolvyx Pharmaceuticals. C.N.S. was scientific founder of Resolvyx Pharmaceuticals and owns founder stock in the company. The interests of C.N.S. were reviewed and are managed by the Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies. B.D.L. is an inventor on patents (resolvins) assigned to BWH and licensed to Resolvyx Pharmaceuticals. The interests of B.D.L.s were reviewed and are managed by the Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies. The remaining authors declare no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Rights and permissions
About this article

Cite this article
Abdulnour, R., Sham, H., Douda, D. et al. Aspirin-triggered resolvin D1 is produced during self-resolving gram-negative bacterial pneumonia and regulates host immune responses for the resolution of lung inflammation. Mucosal Immunol 9, 1278–1287 (2016). https://doi.org/10.1038/mi.2015.129
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2015.129
This article is cited by
-
Characterization of a Murine Model System to Study MicroRNA-147 During Inflammatory Organ Injury
Inflammation (2021)
-
Dynamics of DHA and EPA supplementation: incorporation into equine plasma, synovial fluid, and surfactant glycerophosphocholines
Metabolomics (2021)
-
Lipid-Derived Mediators are Pivotal to Leukocyte and Lung Cell Responses in Sepsis and ARDS
Cell Biochemistry and Biophysics (2021)
-
Resolvin D1 enhances the resolution of lung inflammation caused by long-term Pseudomonas aeruginosa infection
Mucosal Immunology (2018)
